Introduction
Acute respiratory distress syndrome (ARDS) is a
common and severe lung disease that is associated with high rates
of mortality and morbidity, and leads to reduced levels of oxygen
in the blood (1,2). The characteristic features of ARDS
include excessively uncontrolled inflammation, hypoxemia and
non-cardiogenic pulmonary edema formation (3). These are caused by various increased
inflammatory cytokines and pulmonary microvascular permeability
(4).
The main sites of cell injury in ARDS are the
vascular endothelium and alveolar epithelium (4). Polymorphonuclear neutrophils (PMNs)
contribute towards lung inflammation and serve important roles in
the pathogenesis and progression of ARDS (3). Lung injures may result in the
infiltration and activation of PMNs, involving a complex process
including the recruitment, adhesion and chemotaxis of PMNs
(3,4). Bhatia and Moochhala (5) have demonstrated that a large number of
PMNs can accumulate in lung tissue and release inflammatory
cytokines, such as interleukin (IL)-8, IL-10 and tumor necrosis
factor-alpha (TNF-α), which serve important roles in activating and
maintaining the inflammatory response (6). IL-8 and the epithelial neutrophil
activating peptide (ENA-78) are critical for the accumulation of
PMNs in lung tissue (7); the
expression of adhesion molecules on the surface of PMNs increases
with appearance of these chemokines (5). Adhesion molecules and their ligands
provide a strong adhesion between PMNs and endothelial cells,
allowing PMNs to migrate to the vessel wall (8,9).
Cytokine-induced neutrophil chemoattractant (CINC), a member of the
IL-8 family, is a specific PMN chemokine that serves a crucial
function in the aggregation of PMNs in lung tissue (10,11).
Nuclear factor-κB (NF-κB) is a critical nuclear
transcription factor, able to activate the transcription of a
number of inflammatory cytokine genes and regulate the inflammatory
response and immunoreaction (12).
Typically, NF-κB exists as an inactive dimer in the cytoplasm and
directly combines with an inhibitor of nuclear-factor κB (IκB) to
produce a trimeric complex (13).
The P50/P65 heterodimer serves an important physiological function
during inflammation, and NF-κB P65 is the principal subunit
(14).
Lipopolysaccharide (LPS) is the main component of
gram-negative bacteria outer membranes, which is a common trigger
of sepsis, and is the initiation factor that activates the NF-κB
signaling pathway (15).
Phosphorylated (p)-NF-κB can enter into the nucleus and bind to
specific DNA sequences when stimulated by LPS (15). The activation of NF-κB is closely
associated with the overexpression of adhesion molecules,
chemokines and other cytokines involved in the migration of PMNs,
and therefore serves an important role in the regulation of
inflammation and ARDS (16).
However, IL-10 is a principal anti-inflammatory cytokine and can
inhibit the activation of NF-κB (17).
Pyrrolidine dithiocarbamate (PDTC), an inhibitor of
NF-κB, has been reported to inhibit the expression of inflammatory
cytokines, such as ILs and TNF-α, at the stage of transcription
(18). In addition, PDTC reduces the
expression of chemokines, such as CINC and ENA-78, and the
accumulation of inflammatory cells in lung tissue in order to
alleviate the pathological changes in the lung tissue of ARDS
(11,19). It is therefore important to reduce
the expression of CINC and ENA-78, and the accumulation of PMNs in
lung tissue, by regulating NF-κB phosphorylation and hindering
NF-κB activation. The present study aimed to investigate the
regulatory effect of CINC and ENA-78 on PMN aggregation mediated by
NF-κB, and the direct protective effects of PDTC on lung tissue in
LPS-induced ARDS mice.
Materials and methods
Animals
A total of 90 BALB/c mice (age, 8–10 weeks; weight,
20±2 g) were purchased from the Experimental Animal Center of
Shandong University (Jinan, China) and housed at room temperature
(24°C) with a 12-h light/dark cycle. The mice were allowed free
access to water and standard laboratory chow. The experimental
procedures were approved by the Ethics Review Committee for Animal
Studies at Qilu Hospital, Shandong University (Jinan, China) and
performed in accordance with animal welfare and animal experimental
guidelines.
Mice received an intraperitoneal (i.p.) injection of
20 mg/kg LPS (Escherichia coli O55:B5; Sigma-Aldrich, St.
Louis, MO, USA) and an i.p. injection of PDTC (0, 40, 120 or 160
mg/kg; L04358; USA Alikesi International Group (China), Ltd.). PDTC
(Beyotime Institute of Biotechnology, Haimen, China) was
administered 30 min prior the injection of LPS.
To further investigate the protective effect of PDTC
on LPS-induced ARDS mice, 90 mice were randomly divided into three
groups (n=30/group), as previously described (20): Control (20 ml/kg normal saline,
i.p.); LPS (20 mg/kg, i.p.); and PDTC (120 mg/kg, i.p) + LPS (20
mg/kg, i.p.).
Specimen collection
Blood, lung tissue and bronchoalveolar lavage fluid
(BALF) samples from each group of mice were collected
simultaneously after modeling for 2, 6, 12 or 24 h. The mice were
anesthetized by intraperitoneal injection with 10% chloral hydrate
(3.5 ml/kg; Sigma-Aldrich), prior to sacrifice via aortic
phlebotomy at the indicated time points. Subsequently, the lungs
were extracted and the left lung was prepared for hematoxylin and
eosin (HE) staining (Beyotime Institute of Biotechnology) and
immunohistochemistry, while the right lung was prepared for western
blot analysis. PMNs were isolated from BALF using Wright-Giemsa
staining (Beijing Leagene Biotech, Co., Ltd., Beijing, China).
After centrifugation at 1,200 × g for 10 min at 4°C, the
supernatant was collected and the expression of IL-8 and IL-10 was
detected using enzyme-linked immunosorbent assay (ELISA) kits.
Specifically, the expression of IL-8 was detected using the
Quantikine ELISA kit from R&D Systems Europe, Ltd. (Abingdon,
UK), whereas the expression of IL-10 was detected using the LEGEND
MAX™ Mouse IL-10 ELISA kit from BioLegend, Inc. (San Diego, CA,
USA).
Histopathological analysis
The left lung was fixed with 4% paraformaldehyde
(Beijing CellChip Biotechnology, Co., Ltd., Beijing, China) for 24
h, embedded in paraffin and cut into 4 µm sections. Once stained
with hematoxylin and eosin, an evaluation was performed to
characterize the degree of lung injury. Briefly, the lung injury
score was calculated by assessing the degree of inflammatory cell
infiltration, hemorrhage, interstitial and alveolar edema and the
thickness of the alveolar septum in five random fields in a blind
manner using a light microscope (Olympus BX43; Olympus
Corportation, Tokyo, Japan).
Determination of the difference
between alveolar and arterial oxygen partial pressure
[P(A-a)O2)]
PaO2 and PaCO2 were analyzed
in 150-µl arterial blood samples and the oxygen partial pressure
(alveolar oxygen partial pressure) was calculated according to the
results of a blood gas analysis: PaO2 = (atmospheric
pressure − 47) × FiO2 - PaCO2 / R (R, the
exchange rate; R=0.8). Alveolar - arterial oxygen partial pressure
difference (A-a) O2 = (atmospheric pressure − 47) ×
FiO2 - PaCO2 / R - arterial blood oxygen
partial pressure. The linear correlation coefficient was calculated
to study the efficacy of gas exchange.
Extraction of cytoplasm and nuclear
proteins
Lung tissue samples weighing ~100 mg were washed
with 0.01 M phosphate-buffered saline (PBS) supplemented with 1.5
ml nuclear protein extract lysis buffer A (BioTeke Corporation,
Beijing, China). Subsequently, the samples were placed on ice for
15–30 min and homogenized using an electric homogenizer following
the addition of 0.5 ml ice-cold NP-40 (10%; BioVision, Inc.,
Milpitas, CA, USA). Then, the samples were vortexed for 10 sec and
centrifuged at 4°C and 12,000 × g for 30 sec; the supernatant
produced was the cytoplasmic protein extract. The precipitate was
washed once with cold PBS and then centrifuged at 4°C and 12,000 ×
g for 30 sec, after which the supernatant was discarded.
Subsequently, 1.5 ml join nucleoprotein extract lysis buffer B
(BioTeke Corporation) was added and the samples were placed on ice
for 30 min, prior to centrifugation at 4°C and 12,000 × g for 2 min
in order to produce the aspirate supernatant (nucleoprotein). Using
the Bradford Protein Assay, the nuclear protein concentration was
adjusted to 0.5–1.0 µg/µl and the samples were placed and stored at
−70°C, according to a previous study (20).
Measurement of NF-κB protein
expression by western blot analysis
The total protein quantity was analyzed using a
Pierce BCA Protein Assay kit (#23250; Thermo Fisher Scientific,
Inc., Waltham, MA, USA). Equal quantities of total protein (30 µg)
were separated on a 10% Bis-Tris gel in NuPAGE MOPS SDS Running
Buffer (all Invitrogen; Thermo Fisher Scientific, Inc.) and
transferred to a polyvinylidene difluoride (PVDF) membrane
(Immobilon; EMD Millipore, Billerica, MA, USA). The PVDF membrane
was then blocked using 5% skimmed milk in Tris-buffered saline
(TBS; Tris-Cl, 50 mM; NaCl, 150 mM; pH 7.5; Thermo Fisher
Scientific, Inc.) for 60 min at room temperature. Next, the
membrane was incubated for 15 h at 4°C with mouse anti-pNF-κB
monoclonal antibody (1:200; cat. no. sc-166748; Santa Cruz
Biotechnology, Inc., Dallas, TX, USA), rabbit anti-pNF-κB p65
polyclonal antibody (1:200; cat. no. ab16502; Abcam, Cambridge, UK)
and mouse anti-β-actin monoclonal antibody (1:500; #BA2305; Boster
Systems, Inc., Pleasanton, CA, USA). The membrane was washed three
times for 5 min each with 1X TBS containing 0.15 Tween-20, and then
incubated for 1 h with horseradish peroxidase (HRP)-conjugated
secondary antibodies (1:1,000; sc-2370; Santa Cruz Biotechnology,
Inc.) at room temperature. The membrane was exposed to high
performance autoradiography film (#87897; Fuji XR film; Fujifilm
Corporation, Tokyo, Japan) and visualized using enhanced
chemiluminescence reagents and the ChemiScope 2850
Fluorescence/Chemiluminescence Imaging system (Clinx Science
Instruments Co., Ltd., Shanghai, China). The integrated density
value of the band intensities on the film was analyzed using
ImageQuant version 5.2 software (Molecular Devices, Sunnyvale, CA,
USA).
Evaluation of CINC mRNA expression
level in lung tissue using reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
RNA was isolated from lung tissue using TRIzol
(Invitrogen; Thermo Fisher Scientific, Inc.) and quantified using a
NanoDrop 2000/2000c (Thermo Fisher Scientific, Inc.). Subsequently,
1 µg total RNA from each sample was denatured at 70°C for 10 min
and chilled on ice for 10 min. RT reactions were performed in a
volume of 20 µl containing 4 µl 5X RT buffer (Toyobo Co., Ltd.,
Osaka, Japan), 1 µl RT Enzyme Mix (Toyobo Co., Ltd.) and 1.0 µl (5
pmol) of the sense and antisense primers (BGI, Shenzhen, China) in
the presence of PCR buffer (Toyobo, Co., Ltd.). qPCR was performed
in a volume of 20 µl containing 2 µl cDNA, 8 µl forward and reverse
primers (10 pmol/µl, 10 µM) and 10 µl QuantiTect SYBRs Green PCR
kit (QIAGEN, Inc., Toronto, Canada), which consisted of DNA
polymerase, deoxynucleotide mix, buffer, MgCl2 and
fluorescent dyes. The gene-specific primer sequences were as
follows: CINC forward, 5′-TATTGGGAGACCATTAGGTG-3′ and reverse,
5′-CATAAAATGTCCAAGGGAAG-3′; and GAPDH forward,
5′-GAACATCCAGAGTTTGAAGG-3′ and reverse, 5′-TCGGTGCAATCTATCTTCTT-3′.
The PCR protocol consisted of three stages: Denaturation,
amplification and melting curve analysis for product
identification. The denaturation and amplification conditions were
95°C for 20 min, followed by 40 cycles of 30 sec denaturation at
95°C, 10 sec annealing at 60°C and 15 sec elongation at 72°C. The
temperature transition rate was 20°C/sec, except when heating at
72°C, when it was 5°C/sec. Fluorescence was measured at the end of
every cycle to allow quantification of cDNA. Relative mRNA
expression levels were calculated using the 2−ΔΔCq
method (21). This value was then
used to determine the relative amount of amplification in each
sample by interpolating from a standard curve. The mRNA expression
level of CINC was normalized to that of GAPDH. Nuclease-free water
was used as a RT-minus control.
Immunohistochemical analysis NF-κB,
CINC and ENA-78 expression in lung tissue
The lobes of lungs from mice were dissected, fixed
in 10% formaldehyde and processed in preparation for
immunohistochemistry. Slides were dewaxed and rehydrated, and
antigen retrieval was performed using 10 mM sodium citrate (pH 6.1;
Beyotime Institute of Biotechnology) and blocked using 5% bovine
serum albumin (Sigma-Aldrich) for 60 min at room temperature.
Sections of lung tissue (4 µm) were incubated with primary
antibodies anti-NF-κB (1:300; Santa Cruz Biotechnology, Inc.),
anti-CINC (1:300; Santa Cruz Biotechnology, Inc.) and anti-ENA-78
(1:200; Santa Cruz Biotechnology, Inc.) overnight at 4°C and then
with polyclonal immunoglobulins/HRP (1:100; Beyotime Institute of
Biotechnology) for 1 h at room temperature. Nuclei were
counterstained with HE. Control samples were incubated with the
same antibodies. Cover slips were mounted with 80% glycerol
(Beijing Zhongshan Golden bridge Biotechnology, Co., Ltd., Beijing,
China). Samples were examined under a microscope equipped with a
digital camera (BX51 TRF; Olympus Corporation). ENA-78 positive
areas were quantified by densitometry using Image-Pro Plus software
6.0 (Media Cybernetics, Inc., Rockville, MD, USA).
Measurement of leukocytes and
proportion of PMNs in BALF
A total of 0.5 ml BALF was collected and total cells
and neutrophils were counted using a hemocytometer in a
double-blind manner for the measurement of leukocytes and the
proportion of PMNs.
Statistical analysis
The results are expressed as the mean ± standard
deviation. Statistical analyses consisted of a one-way analysis of
variance and a Student's t-test was used to compare groups.
P<0.05 was considered to indicate a statistically significant
difference. The statistical analysis was conducted using SPSS 19.0
software (IBM SPSS, Armonk, NY, USA).
Results
Animal phenotype
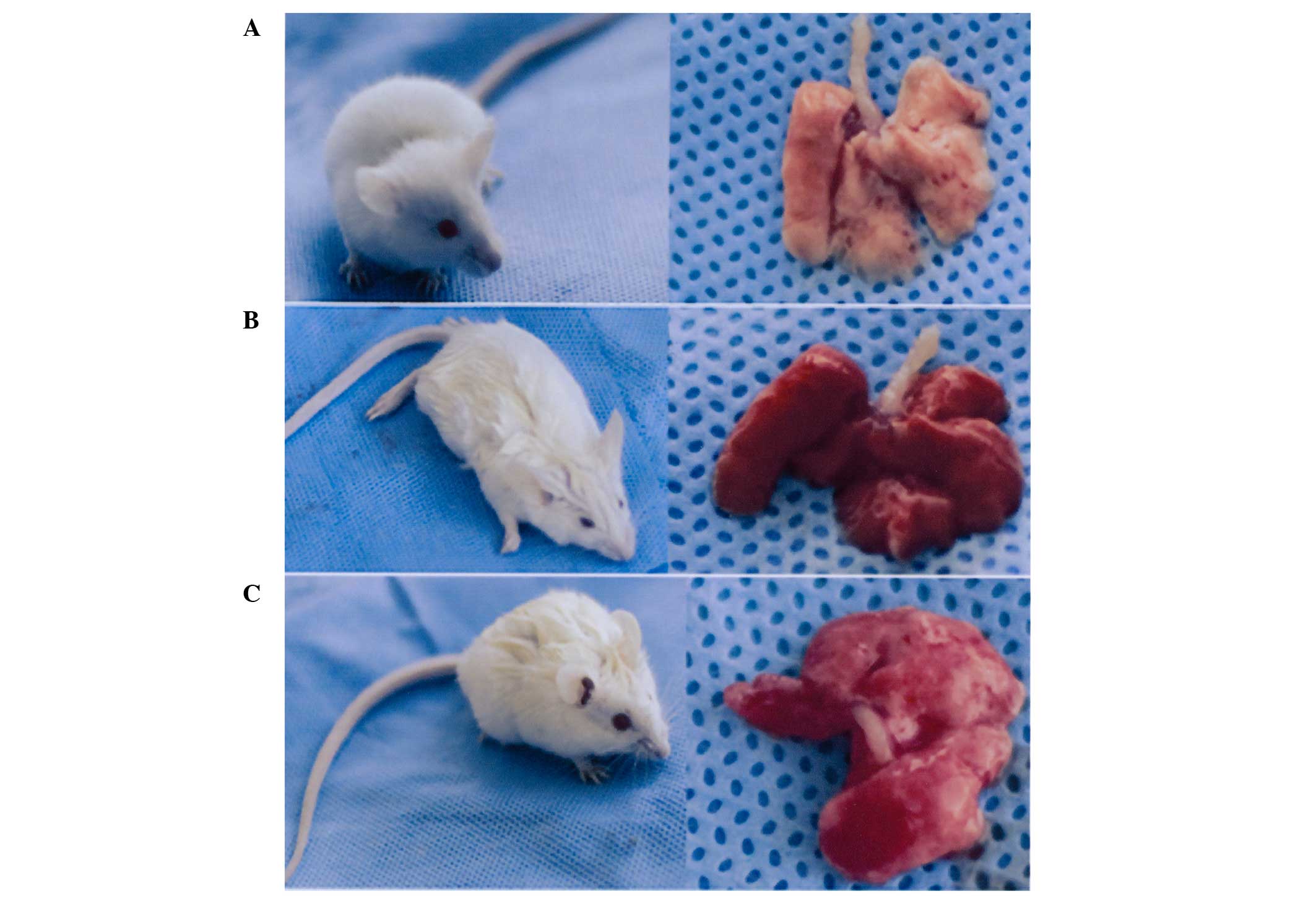
The mice in the control group were breathing easily
and their lungs appeared pink upon dissection. In the ARDS mice,
after 6 h induction with LPS, shortness of breath with oral
cyanosis and blood-like liquid in the nose was observed. In
addition, the lung volume was increased, the lungs were deep purple
in appearance and flake bleeding was observed under the visceral
pleura. In the PDTC + LPS group, the shortness of breath was
attenuated and the lung was less inflamed and reddened. The
scattered bleeding on the surface of the lung and the capsular
tension was reduced on the lungs of mice in the PDTC + LPS group in
comparison with the LPS group (Fig.
1).
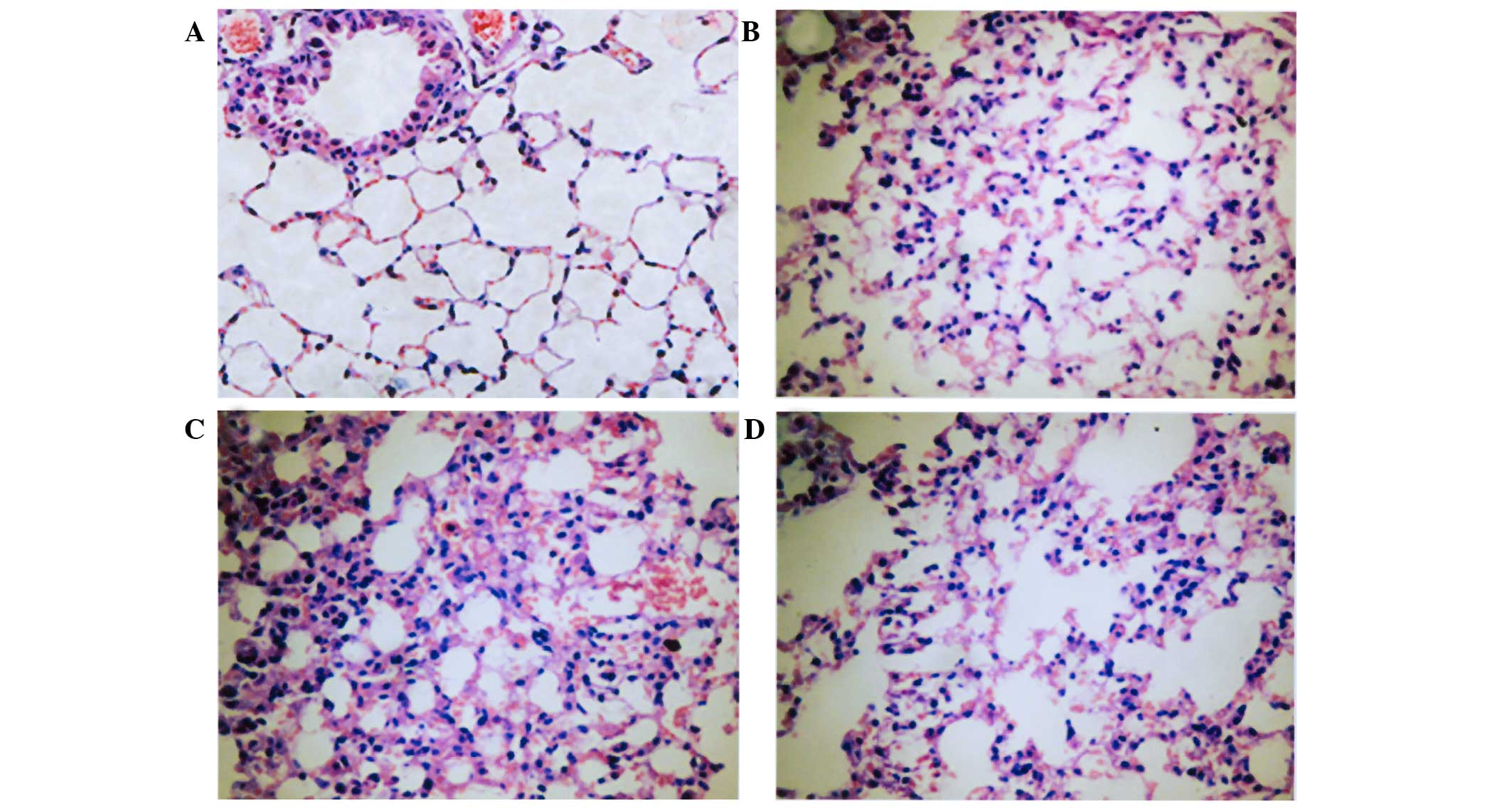
Pathology of lung tissue determined by
HE staining
In the control group, the structure of the lung
tissue was normal and no inflammatory cells were detected. However,
in the LPS group after 12 and 24 h, the alveoli septum was
thickened, the pulmonary interstitial was highly congested, the
alveolar wall was fractured and a large number of infiltrative
inflammatory cells were detected. In addition, as time progressed
there was an increase in inflammatory cell infiltration and
aggravated lung tissue damage. Similar symptoms were detected in
the PDTC + LPS group; however, the lung tissue injury was milder
compared with LPS group, as determined by HE staining (Fig. 2).
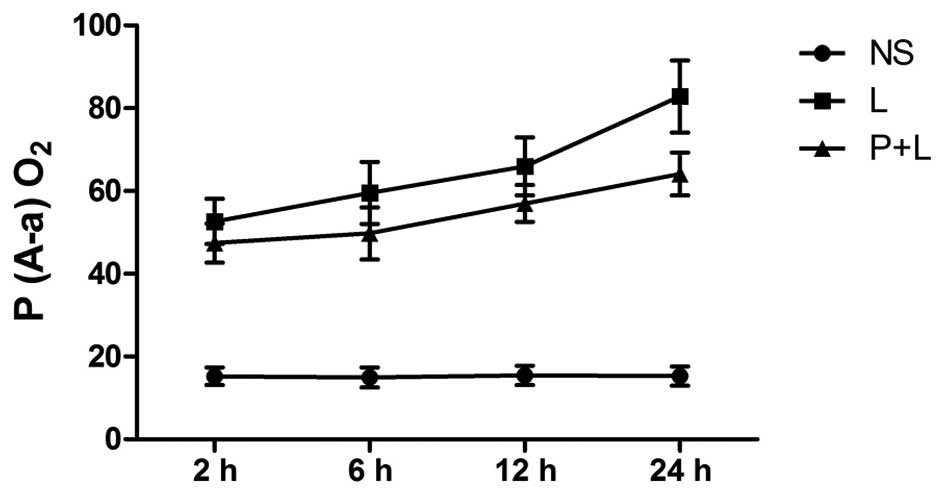
Effect of PDTC on P(A-a)O2
in LPS-induced ARDS mice
P(A-a)O2 was significantly higher in the
LPS group in comparison with the control group at each time point
(P<0.01; Fig. 3). Following
treatment with PDTC, the level of oxygenation improved in lung
tissue as time progressed, and the P(A-a)O2 gradually
decreased in comparison with the LPS group (P<0.05). However,
P(A-a)O2 in the PDTC + LPS group was significantly
higher compared with the control group (Fig. 3).
Differences in p-NF-κB and NF-κB P65
protein expression in the cytoplasm and nucleus in lung tissue
The expression levels of p-NF-κB were significantly
higher in the LPS group in comparison with the control group
(P<0.01). However, p-NF-κB protein expression in the lung tissue
of the PDTC + LPS group was significantly reduced, as compared with
the LPS group (P<0.05), although it remained increased, as
compared with the control group. This suggests that PDTC can
effectively inhibit the phosphorylation of NF-κB (Fig. 4A and B). NF-κB P65 cytoplasmic
protein expression was significantly lower in the LPS group in
comparison with the control group (P<0.01); however, the
production of P65 was significantly higher in the pulmonary nucleus
of the LPS group in comparison with the control group (P<0.01;
Fig. 4C and D). P65 protein
expression in the cytoplasm of lung tissue was significantly
increased in the PDTC + LPS group in comparison with the LPS group
(P<0.05), and the expression of P65 was significantly decreased
in the nuclei of the PDTC+LPS group in comparison with the LPS
group (P<0.01; Fig. 4E and
F).
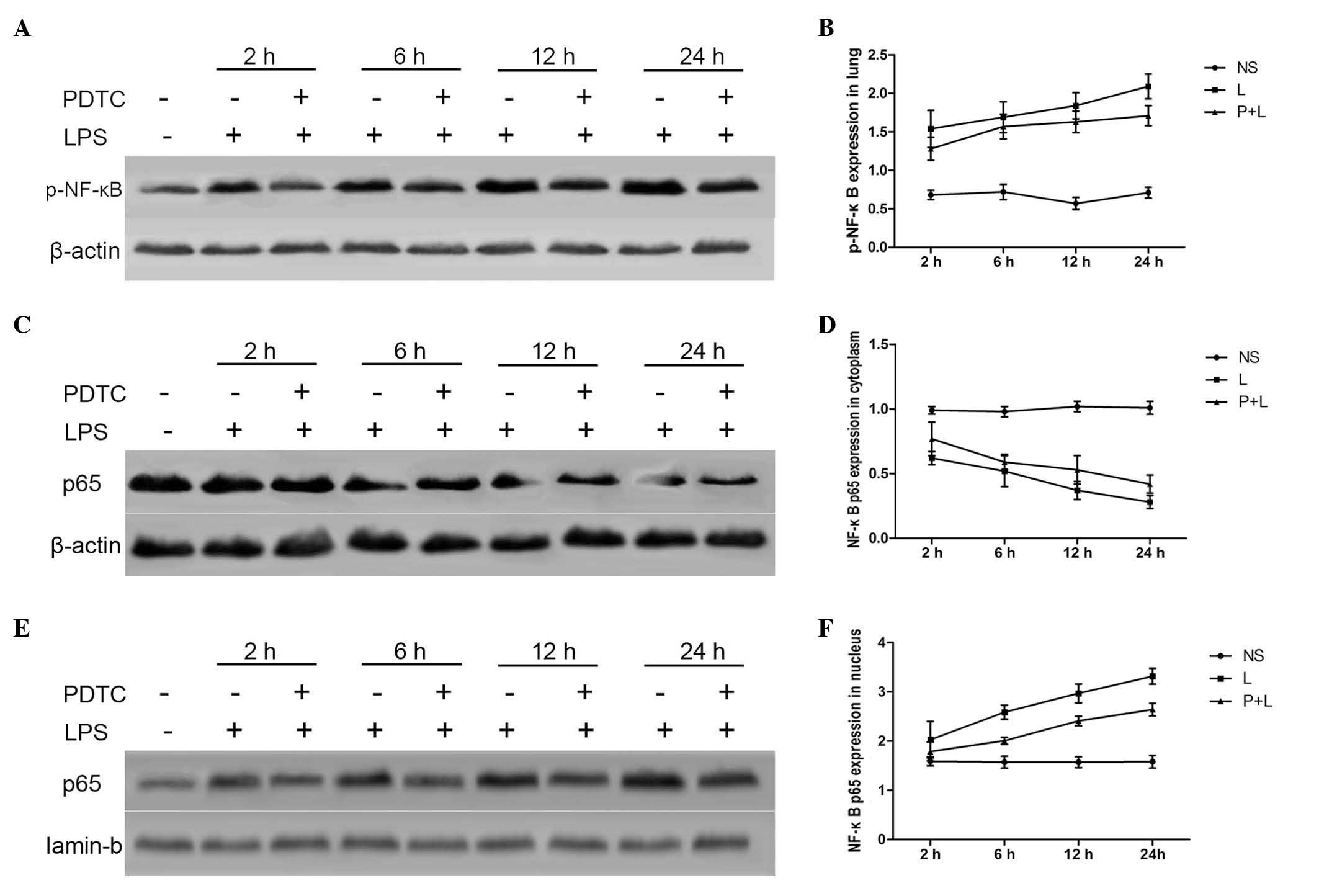 | Figure 4.Effect of PDTC on p-NF-κB and NF-κB
P65 protein expression in lung tissue and the cytoplasm. (A and B)
The expression of p-NF-κB was significantly increased in the LPS
group (P<0.01) and significantly decreased in the PDTC + LPS
group (P<0.05), demonstrating that PDTC can inhibit the
activation of NF-κB. (C and D) NF-κB P65 protein expression was
reduced in the cytoplasm of the LPS group (P<0.01); the
production of P65 was significantly increased in nuclei of the LPS
group in comparison with the NS group (P<0.01). (E and F) P65
expression was significantly increased in the cytoplasm
(P<0.05), and significantly decreased in the nucleus, of the
PDTC + LPS group in comparison with the LPS group (P<0.01).
PDTC, pyrrolidine dithiocarbamate; NF-κB, nuclear factor-κB,
p-NF-κB, phosphorylated-NF-κB; LPS, lipopolysaccharide; NS, normal
saline; h, hours. |
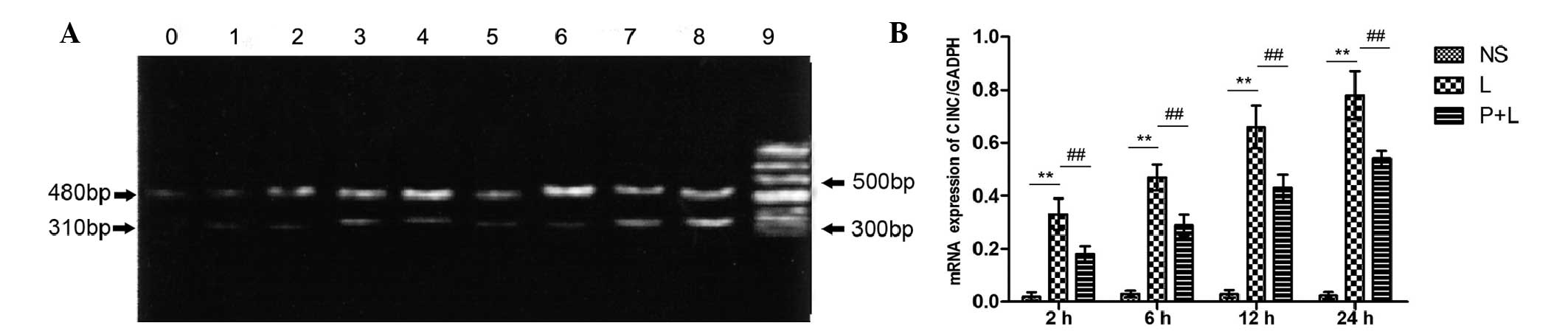
CINC mRNA expression in lung
tissue
The expression of CINC mRNA in lung tissue was
significantly increased in the mice that received an i.p. injection
of LPS in comparison with the control group (P<0.01), and
continued to increase over time. In the PDTC + LPS group, CINC mRNA
expression in lung tissue was significantly decreased in comparison
with the LPS group (P<0.05; Fig.
5).
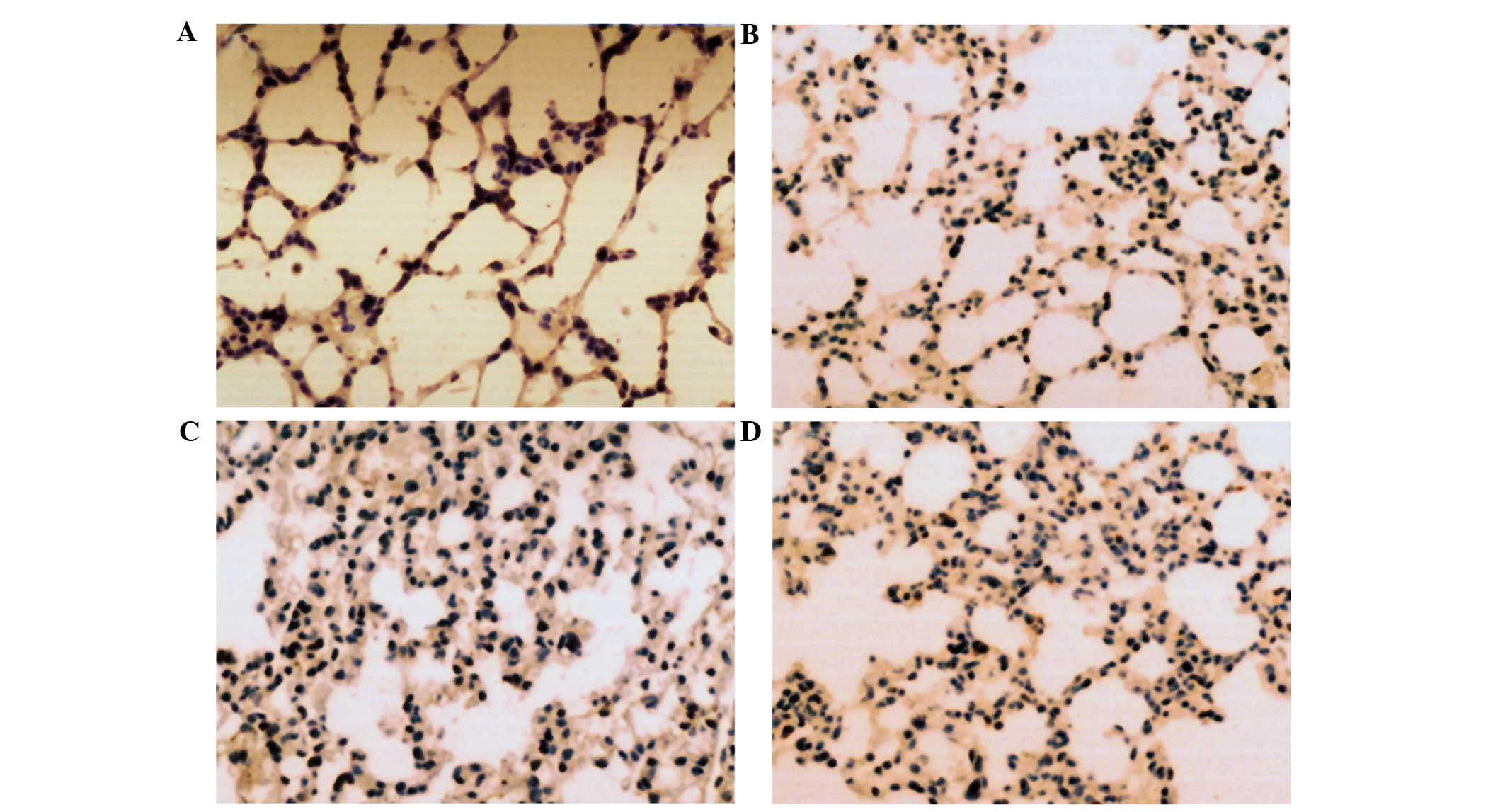
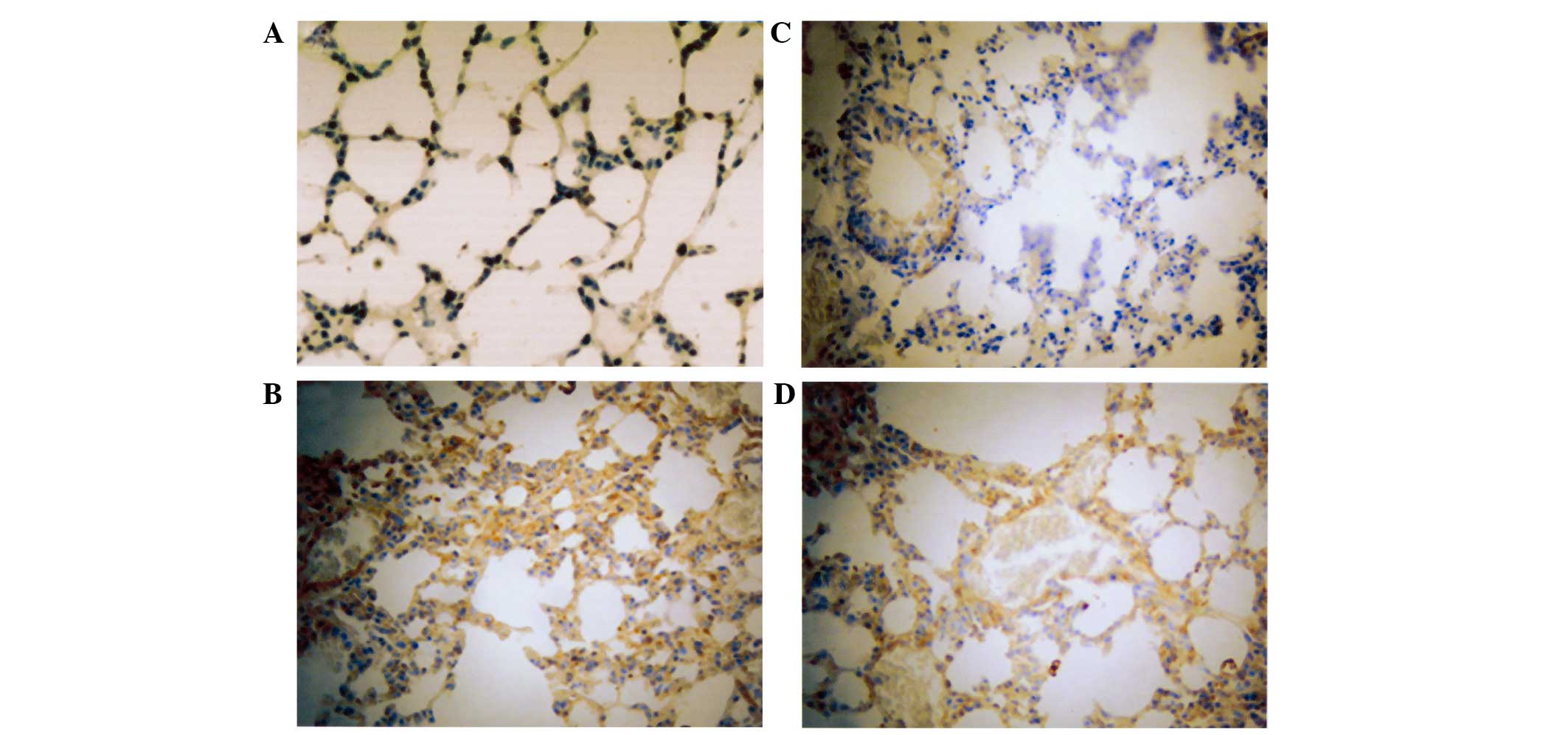
Expression of NF-κB, CINC and ENA-78
in lung tissue
The immunohistochemistry results demonstrated that
the expression levels of NF-κB, CINC and ENA-78 were markedly
increased in the lung tissue of LPS-induced ARDS mice. Cell
morphological analysis revealed that the majority of cells in the
LPS lung tissue were infiltrated PMNs and endothelial cells.
However, in the control group, the expression of NF-κB, CINC and
ENA-78 was not detected in the lung tissue of mice. The number of
positive cells stained with NF-κB, CINC and ENA-78 in PDTC + LPS
group was markedly decreased in comparison with the LPS group
(Figs. 6–8).
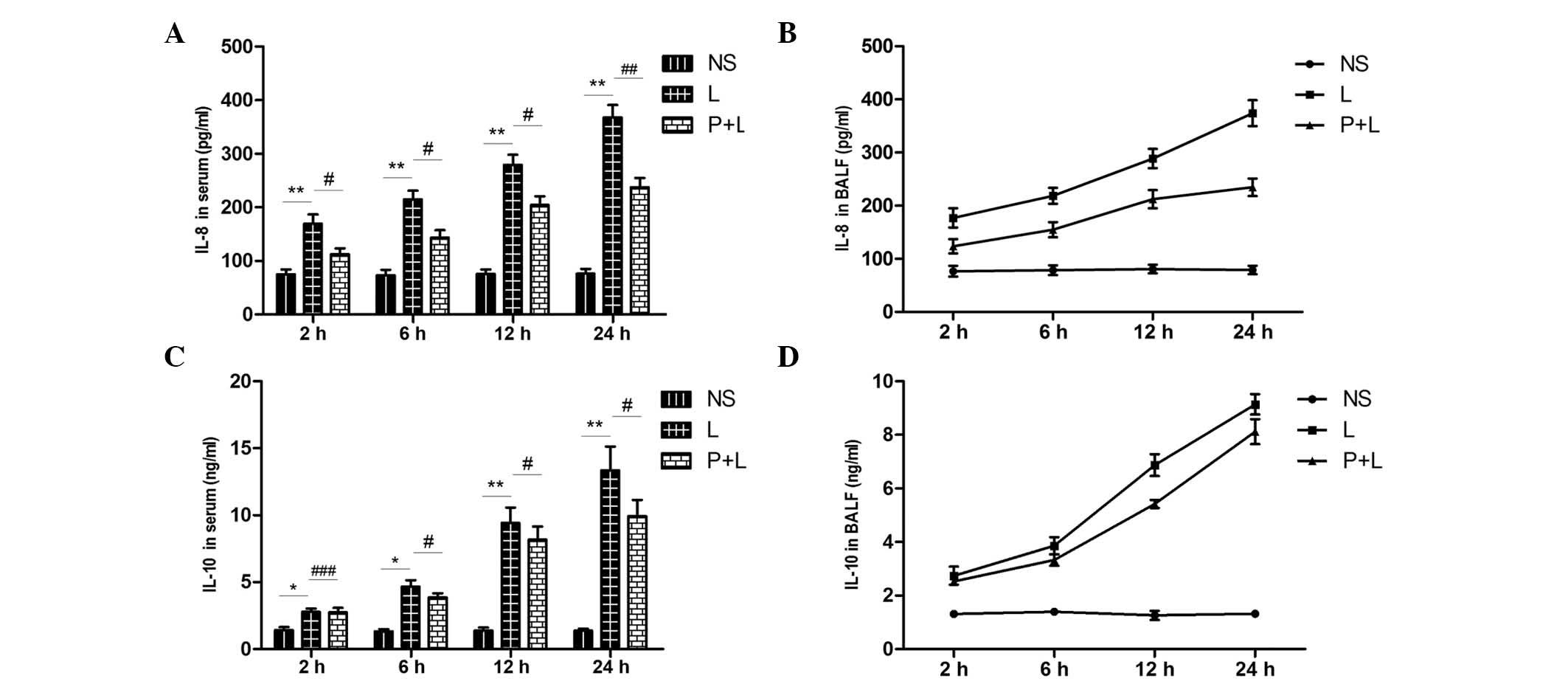
Expression of IL-8 and IL-10 in serum
and BALF
The expression levels of IL-8 and IL-10 in the serum
and BALF from the LPS group was significantly increased, as
compared with the control group (P<0.01). In the PDTC + LPS
group, IL-8 expression was significantly decreased, as compared
with the LPS group (P<0.05), whereas it was increased in
comparison with the control group. Over the time period, however,
the production of IL-10 in the serum and BALF from the LPS group
was increased. In the PDTC + LPS group, the expression level of
IL-10 was substantially higher than that in the control group,
whereas it was significantly reduced in comparison with the LPS
group (P<0.05; Fig. 9).
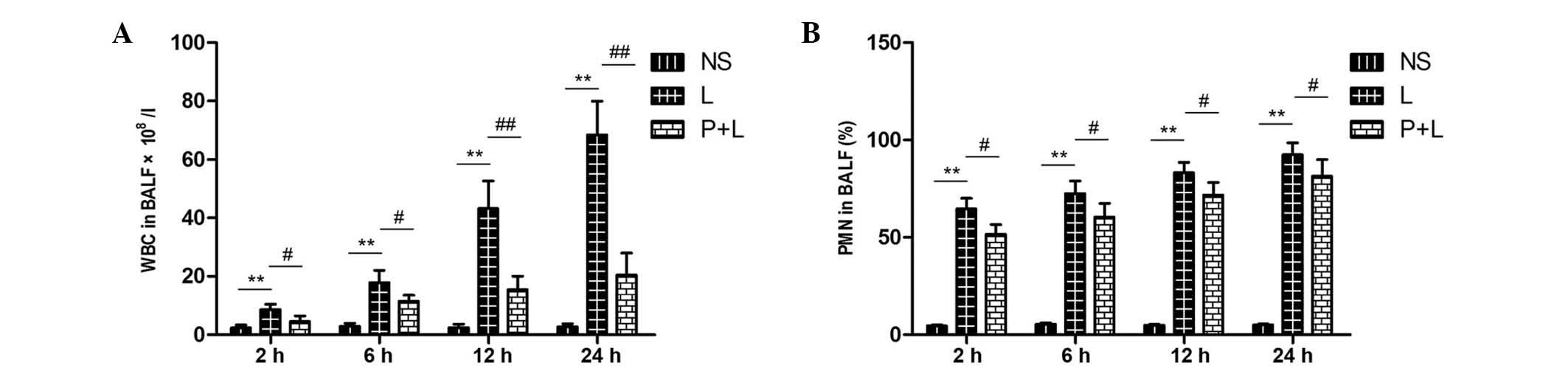
Number of leukocytes and proportion of
PMNs in BALF
The total number of leukocytes was significantly
increased in the LPS group, as compared with the control group
(P<0.01). In the PDTC + LPS group, the number of leukocytes was
significantly decreased, as compared with the LPS group (P<0.05)
at each time point, but was significantly increased, as compared
with the control group (P<0.05). The proportion of PMNs in BALF
was significantly higher in the LPS and PDTC + LPS groups in
comparison with the control group (P<0.01), but the proportion
of PMNs was significantly reduced in the PDTC + LPS group in
comparison with the LPS group (P<0.05; Fig. 10).
Discussion
NF-κB is a target for anti-inflammatory ARDS
treatment and serves an important role in initiating and developing
inflammatory reactions (16,22). The association between NF-κB,
cytokines and chemokines has become a topic of interest with
regards to the pathogenesis of ARDS (12). The expression of inflammatory
mediators can be downregulated by delaying the activation of the
NF-κB signal pathway, thereby suppressing PMN accumulation in the
lungs, effectively repressing the excessive activation of the
inflammatory response and reducing the damage inflicted on lung
tissue (23).
A previous study observed that the activation of
NF-κB in ARDS could increase the production of inflammatory
cytokines, including adhesion molecules (such as CD11b/CD18,
ICAM-1), chemokines (such as IL-8), TNF-α and IL-1β, involving a
number of downstream gene transcriptions (5). NF-κB was demonstrated to be a central
transcription factor that promotes the production of cytokines,
adhesion molecules and chemokines by specifically binding to κB
promoters in the nucleus (24,25). The
P50/P65 NF-κB heterodimer serves a primary physiological function
during inflammation (12,13). As the principal subunit, NF-κB P65
exists in an inactive dimer form in the cytoplasm and directly
combines with the inhibitory protein IκB to form a trimeric complex
(26,27). Previous studies have demonstrated
that when NF-κB is excessively activated, IL-8 and TNF-α are
overexpressed, whereas the expression of IL-10 is decreased, in
ARDS (20). An imbalance of the
inflammatory response is the principal feature in ARDS (28,29).
Therefore, inhibiting the activation of the NF-κB signal pathway
and reducing the expression of inflammatory cytokines and
chemokines may inhibit the inflammatory response at the source,
leading to novel strategies in treating ARDS.
In the present study, the widened lung interval,
highly congested pulmonary interstitial, fractured alveolar wall
and increased infiltrative inflammatory cell in the LPS group
aggravated lung tissue damage, gradually increased
P(A-a)O2 and resulted in oxygenation obstacles over
time. In addition, the results of the NF-κB western blot
demonstrated that p-NF-κB protein expression was significantly
increased in the lung cells of the ARDS mice at each time point in
comparison with the control group. The expression of the P65
protein in the nucleus was significantly enhanced, suggesting that
NF-κB may be activated by phosphorylation and degradation. These
results are consistent with previous studies (20).
IL-8, a proinflammatory cytokine, is a promoter for
ARDS and serves a crucial role in the course of PMN migration from
circulating blood to sites of inflammation (30). In patients with ARDS, the body can
prevent excessive activation of the inflammatory response by
activating an anti-inflammatory response and releasing
anti-inflammatory cytokines, such as IL-10, in order to attenuate
the extent of tissue damage (31).
IL-10 is a crucial anti-inflammatory cytokine in the immune
response and can inhibit the production of IL-8 and TNF-α in
monocytes and macrophages, and repair injured tissue by regulating
the immune response; however, overexpression can aggravate the
immune response (17,32). Previous studies demonstrated that the
genes of cytokines such as IL-8 and IL-10 encode the binding site
for NF-κB (20), and that
LPS-activated NF-κB binds to these genes, resulting in their
expression (5,33). In the present study, it was observed
that the production of IL-8 was rapidly increased after 2 h in the
serum and BALF of ARDS mice; however, the expression of IL-10 was
not significantly increased during the first 6 h following
injection of LPS. This indicates that there is an imbalance of
proinflammatory and anti-inflammatory molecules during the early
stage of ARDS. Although the level of IL-8 and IL-10 were increased
simultaneously over time, the pro- and anti-inflammatory cytokines
were imbalanced, and a large number of proinflammatory cytokines
released from inflammatory cells further aggravated lung tissue
damage. Thus, a cascade of inflammatory responses commenced.
The results from the current study demonstrated that
chemokines (in particular IL-8, ENA-78 and CINC) released by
damaged cells in ARDS stimulated PMN extravasation and their
migration to sites of inflammation. CINC, a member of the IL-8
family, is a specific PMN chemokine (11). CINC-1 and −3 serve important roles in
PMN recruitment to the lung in ARDS induced by LPS (10). The enhanced expression of CINC mRNA
and protein inhibits PMN apoptosis and activates a large number of
PMNs, causing them to accumulate in the lung and result in lung
injury (11). Endogenous TNF-α is
associated with the increased expression of ENA-78. ENA-78, a
member of CXC chemokine superfamily, can activate PMNs, induce
secretion of numerous cytokines in PMNs and aggravate inflammation
(19). In the present study, the
results demonstrated that the expression of CINC mRNA was higher
than that in the control group, and the immunohistochemical results
demonstrated that there were a large number of positive
immunoreactive cells with CINC and ENA-78 in the cytoplasm of
macrophages and pulmonary interstitial cells. The number of
leukocytes and PMNs in BALF were increased in comparison to the
control group, which indicated that the high expression of
chemokines (CINC and ENA-78) is closely related to the accumulation
of PMNs.
As a dithiocarbamate of the pyrrole derivatives,
PDTC can inhibit the activation of NF-κB by hindering the
dissociation of IκB from the NF-κB complex, decrease the production
of CINC and ENA-78, and prevent the aggregation of PMNs in the lung
tissue of ARDS (18,34). The results from the present study
suggest that PDTC can attenuate the mRNA expression of p-NF-κB,
CINC and ENA-78 by inhibiting the activation of NF-κB, reducing the
expression of IL-8 and IL-10, and inhibiting the activation of PMNs
in BALF. Pretreatment with PDTC may have partially reduced
lethality in LPS-induced mice and attenuated lung tissue edema,
damage, the production of inflammatory cytokines and chemokines,
the infiltration of PMNs in the lung and pulmonary capillary
permeability. This suggests that the NF-κB signaling pathway may
become an important target in regulating ARDS in the lung, and that
it may be useful in investigating the pathogenesis of ARDS and
exploring more effective and targeted clinical therapies.
In conclusion, in the lung tissue of mice with ARDS
induced by LPS, the degradation of NF-κB was activated by
phosphorylation and NF-κB p65 migration from the cytoplasm to the
nucleus. This was followed by NF-κB activation that commenced the
synthesis and release of CINC and ENA-78, and resulted in the
imbalanced expression of proinflammatory and anti-inflammatory
cytokines, such as IL-8 and IL-10. These cytokines then stimulated
the extravasation of PMNs and their direct migration to sites of
inflammation, and resulted in a widened lung interval, highly
congested pulmonary interstitial, fractured alveolar wall,
increased inflammatory cell infiltration, the P(A-a)O2
and oxygenation obstacles. Therefore, it can be hypothesized that
PDTC, a specific inhibitor of NF-κB, can reduce the release of
chemokines and cytokines via the NF-κB signal pathway, thereby
decreasing PMN accumulation in lung tissue, lung tissue damage and
improved oxygenation. These results indicate that strategies to
regulate the expression of chemokines, cytokines and the NF-κB
signal pathway in ARDS should focus on restricting the duration of
PMN infiltration and the subsequent effects at inflammatory sites
in ARDS.
Acknowledgements
The present study was supported by the Project of
Science and Technology Department of Liaoning Province (grant no.
2013225305).
References
|
1
|
Ware LB and Matthay MA: The acute
respiratory distress syndrome. N Engl J Med. 342:1334–1349. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Malhotra A: Low-tidal-volume ventilation
in the acute respiratory distress syndrome. N Engl J Med.
357:1113–1120. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Meliton AY, Muñoz NM, Meliton LN, Binder
DC, Osan CM, Zhu X, Dudek SM and Leff AR: Cytosolic group IVa
phospholipase A2 mediates IL-8/CXCL8-induced transmigration of
human polymorphonuclear leukocytes in vitro. J Inflamm (Lond).
7:142010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Williams AE and Chambers RC: The mercurial
nature of neutrophils: Still an enigma in ARDS? Am J Physiol Lung
Cell Mol Physiol. 306:L217–L230. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bhatia M and Moochhala S: Role of
inflammatory mediators in the pathophysiology of acute respiratory
distress syndrome. J Pathol. 202:145–156. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Giebelen IA, van Westerloo DJ, LaRosa GJ,
de Vos AF and van der Poll T: Local stimulation of alpha7
cholinergic receptors inhibits LPS-induced TNF-alpha release in the
mouse lung. Shock. 28:700–703. 2007.PubMed/NCBI
|
|
7
|
Nakayama S, Mukae H, Ishii H, Kakugawa T,
Sugiyama K, Sakamoto N, Fujii T, Kadota J and Kohno S: Comparison
of BALF concentrations of ENA-78 and IP10 in patients with
idiopathic pulmonary fibrosis and nonspecific interstitial
pneumonia. Respir Med. 99:1145–1151. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Marcus BC, Wyble CW, Hynes KL and Gewertz
BL: Cytokine-induced increases in endothelial permeability after
adhesion molecule expression. Surg. 2006.120
|
|
9
|
Nooteboom A, van der Linden CJ and
Hendriks T: Modulation of adhesion molecule expression on
endothelial cells after induction by lipopolysaccharide-stimulated
whole blood. Scand J Immunol. 59:440–448. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Guo YL, Huang H, Zeng DX, Zhao JP, Fang HJ
and Lavoie JP: Interleukin (IL)-4 induces production of
cytokine-induced neutrophil chemoattractants (CINCs) and
intercellular adhesion molecule (ICAM)-1 in lungs of asthmatic
rats. J Huazhong Univ Sci Technol. 33:470–478. 2013. View Article : Google Scholar
|
|
11
|
Ulich TR, Howard SC, Remick DG, Wittwer A,
Yi ES, Yin S, Guo K, Welply JK and Williams JH: Intratracheal
administration of endotoxin and cytokines. VI. Antiserum to CINC
inhibits acute inflammation. Am J Physiol. 268(2 Pt 1): L245–L250.
1995.PubMed/NCBI
|
|
12
|
Lawrence T: The nuclear factor NF-kappaB
pathway in inflammation. Cold Spring Harb Perspect Biol.
1:a0016512009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
May MJ and Ghosh S: Signal transduction
through NF-kappa B. Immunol Today. 19:80–88. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lawrence T, Gillroy DW, Colville-Nash PR
and Willoughby DA: Possible new role for NF-kappaB in the
resolution of inflammation. Nat Med. 7:1291–1297. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kupfner JG, Arcaroli JJ, Yum HK, Nadler
SG, Yang KY and Abraham E: Role of NF-kappaB in endotoxemia-induced
alterations of lung neutrophil apoptosis. J Immunol. 167:7044–7051.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
DeDiego ML, Nieto-Torres JL, Regla-Nava
JA, Jimenez-Guardeño JM, Fernandez-Delgado R, Fett C,
Castaño-Rodriguez C, Perlman S and Enjuanes L: Inhibition of
NF-κB-mediated inflammation in severe acute respiratory syndrome
coronavirus-infected mice increases survival. J Virol. 88:913–924.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Németh ZH, Haskó G and Vizi ES:
Pyrrolidine dithiocarbamate augments IL-10, inhibits TNF-alpha,
MIP-1alpha, IL-12, and nitric oxide production and protects from
the lethal effect of endotoxin. Shock. 10:49–53. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Eren G, Cukurova Z, Hergunsel O, Demir G,
Kucur M, Uslu E, Dalo E, Uhri M and Tugcu V: Protective effect of
the nuclear factor kappa B inhibitor pyrrolidine dithiocarbamate in
lung injury in rats with streptozotocin-induced diabetes.
Respiration. 79:402–410. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Keane MP, Belperio JA, Burdick MD, Lynch
JP, Fishbein MC and Strieter RM: ENA-78 is an important angiogenic
factor in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med.
164:2239–2242. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang H, Xu L, Zhao J, Wang D, Guo R, Wang
J, Gong W, Liu T, Zhang Y and Dong L: Regulatory mechanism of
pyrrolidine dithiocarbamate is mediated by nuclear factor-κB and
inhibits neutrophil accumulation in ARDS mice. Exp Ther Med.
8:614–622. 2014.PubMed/NCBI
|
|
21
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2-ΔΔCt method. Methods. 25:402–408. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Christman JW, Lancaster LH and Blackwell
TS: Nuclear factor kappa B: A pivotal role in the systemic
inflammatory response syndrome and new target for therapy.
Intensive Care Med. 24:1131–1138. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chen X, Yang X, Liu T, Guan M, Feng X,
Dong W, Chu X, Liu J, Tian X, Ci X, et al: Kaempferol regulates
MAPKs and NF-κB signaling pathways to attenuate LPS-induced acute
lung injury in mice. Int Immunopharmacol. 4:209–216. 2012.
View Article : Google Scholar
|
|
24
|
Kang JL, Lee HW, Lee HS, Pack IS, Chong Y,
Castranova V and Koh Y: Genistein prevents nuclear factor-kappa B
activation and acute lung injury induced by lipopolysaccharide. Am
J Resp Crit Care Med. 164:2206–2212. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chignard M and Balloy V: Neutrophil
recruitment and increased permeability during acute lung injury
induced by lipopolysaccharide. Am J Physiol Lung Cell Mol Physiol.
279:L1083–L1090. 2000.PubMed/NCBI
|
|
26
|
Rahman A and Fazal F: Blocking NF-κB: An
inflammatory issue. Proc Am Thorac Soc. 8:497–503. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Leverence JT, Medhora M, Konduri GG and
Sampath V: Lipopolysaccharide-induced cytokine expression in
alveolar epithelial cells: Role of PKCζ-mediated p47phox
phosphorylation. Chem Biol Interact. 189:72–81. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Park WY, Goodman RB, Steinberg KP,
Ruzinski JT, Radella F II, Park DR, Pugin J, Skerrett SJ, Hudson LD
and Martin TR: Cytokine balance in the lungs of patients with acute
respiratory distress syndrome. Am J Respir Crit Care Med. 164(10 Pt
1): 1894–1903. 2001.
|
|
29
|
Suratt BT and Parsons PE: Mechanisms of
acute lung injury/acute respiratory distress syndrome. Clin Chest
Med. 27:579–589, abstract viii. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kunkel SL, Standiford T, Kasahara K and
Strieter RM: Interleukin-8 (IL-8): The major neutrophil chemotactic
factor in the lung. Exp Lung Res. 17:17–23. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Shen Y, Wang D and Wang X: Role of CCR2
and IL-8 in acute lung injury: A new mechanism and therapeutic
target. Expert Rev Respir Med. 5:107–114. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Foulds KE, Rotte MJ and Seder RA: IL-10 is
required for optimal CD8 T cell memory following Listeria
monocytogenes infection. J Immunol. 177:2565–2574. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lin CH, Cheng HW, Ma HP, Wu CH, Hong CY
and Chen BC: Thrombin induces NF-kappaB activation and IL-8/CXCL8
expression in lung epithelial cells by a Rac1-dependent PI3K/Akt
pathway. J Biol Chem. 286:10483–10494. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lauzurica P, Martínez-Martínez S,
Marazuela M, del Gómez Arco P, Martínez C, Sánchez-Madrid F and
Redondo JM: Pyrrolidine dithiocarbamate protects mice from lethal
shock induced by LPS or TNF-α. Eur J Immunol. 29:1890–1900. 1999.
View Article : Google Scholar : PubMed/NCBI
|