Introduction
Hepatic fibrosis is a type of repairing reaction to
chronic liver injuries caused by various pathogens, and is a
reversible pathological process (1).
The removal of relevant pathogenic factors or effective prevention
can improve the degree of hepatic fibrosis, otherwise hepatic
fibrosis can develop into decompensated liver cirrhosis at the
terminal stage. MicroRNA (miR) is a type of endogenous non-coding
single-stranded micromolecule RNA with a size of 19–22 nucleotides,
and through its complete or incomplete complementary pairing with
target mRNA, causes mRNA degradation or translation inhibition,
regulating the gene expression level after transcription (2). Previous findings showed that some miRs
have a relatively rich expression in liver, and levels of miRs were
markedly altered in liver diseases, thus affecting the genesis and
development of hepatic diseases (3).
miR-10a is involved in multiple physiological and pathological
processes including hematopoietic cell differentiation,
tumorigenesis and development, and immunoregulation (4,5).
In pulmonary fibrotic mouse lung tissues caused by
bleomycin, 161 miRs of different expressions were found, with
miR-10a participating in fibroblast activation and collagen
deposition by regulating the TGF-β signaling pathway (6). Activation of hepatic stellate cell
(HSC) or its phenotypic transformation into myofibroblast is a key
link for the formation of hepatic fibrosis (7). Transforming growth factor (TGF)-β1 is a
strong cell factor that induces hepatic fibrosis (8), and Smad protein is a key active
substrate of TGF-β1 (9). Smad and
TGF-β1 cause HSC activation, and initiate collagen gene expression,
resulting in the genesis of hepatic fibrosis. Activation of the
TGF-β1/Smads signal transduction pathway leads to pathological
changes, which play a significant role in the genesis and
development of hepatic fibrosis (10).
In the present study, the molecular mechanism of
miR-10a in regulating hepatic fibrosis was examined by using
miR-10a as a drug target of hepatic fibrosis to guide the clinical
diagnosis and treatment of hepatic fibrosis.
Materials and methods
Establishment of hepatic fibrosis
mouse model
Forty healthy 8-week-old C57BL6/J female mice,
weighing 18–22 g, were selected in the present study. The animals
were randomly divided into the control group (intraperitoneal
injection of 5 µl/g normal saline, twice per week for 8 weeks) and
hepatic fibrosis model group (intraperitoneal injection of 10% CCI4
olive oil, twice per week for 8 weeks), with 20 animals in each
group fed for 8 weeks and subsequently sacrificed.
The study was approved by the ethics committee of
Wenzhou Medical University (Zhenjiang, China).
Preparation of samples
The experimental animals were fasted for 12 h, and
sacrificed under ether anesthesia. Blood samples were obtained and
10% neutral formalin was fixed in hepatic tissues to prepare the
pathological sections. The remaining hepatic tissues were rapidly
frozen in liquid nitrogen, and then stored at −80°C.
Main reagents
Pronase E was purchased from Merck Millipore
(Darmstadt, Germany), collagenase II from Sigma-Aldrich (St. Louis,
MO, USA), lymphocyte separation medium from the Institute of
Biomedical Engineering of the Chinese Academy of Medical Sciences
(Beijing, China), Dulbeccos modified Eagles medium (DMEM) culture
solution dry powder from Gibco (Grand Island, NY, USA), fetal calf
serum (FCS) from Hangzhou Sijiqing Biological Engineering Materials
Co., Ltd. (Zhejiang, China), INTERFERin siRNA transfection reagent
from Polyplus-Transfection (New York, NY, USA), the miRNA analysis
system from Applied Biosystems Life Technologies (Foster City, CA,
USA), PrimeScript RT-PCR reverse transcription kit from Toyobo
(Osaka, Japan), miR-10 mimics from Guangzhou Ruibo Biotechnology
Co., Ltd. (Guangzhou, China), all McAbs from Abcam (Cambridge, MA,
USA), and the cell lysis solution from Cell Signaling Technology,
Inc. (Danvers, MA, USA).
Mouse HSC separation method
After an intraperitoneal injection of 2%
pentobarbital sodium (40 mg/kg), the abdominal skin was disinfected
with 75% ethyl alcohol, and enterocelia was opened using a
cross-shaped incision to expose heart and postcava. Subsequently,
no. 18 trochar was connected to an infusion flask and the perfusate
was injected into ventriculus sinister, blood was released from
postcava, hepatic perfusion was conducted at a flow velocity of 10
ml/min until the liver turned earthy yellow, followed by perfusion
with enzyme perfusate for 15–20 min until the liver became dark
brown.
The liver was then washed with normal saline,
enveloped and connective tissues were removed and sectioned. The
tissue were digested in solution at 37°C for 20 min, followed by
centrifugation at 6 × g for 20 min and the cell suspension was
filtered through 200-mesh steel mesh to collect filtrate in two 50
ml centrifugal tubes. The cell suspension was centrifuged at 340 ×
g for 5 min and the supernatant was discarded. D-Hanks solution was
used for re-suspension and deposition, and then centrifuged at 50 ×
g for 2 min, prior to depositing hepatic cells, and the supernatant
was removed to conduct gradient separation. The lymphocyte
separation medium was used to pave the gradient, and fully digested
single-cell suspension was added to the upper layer and centrifuged
at 1,400 × g for 20 min (25°C). Horizontal projection
centrifugation method was used to separate the cells, a small
amount of white liquid was carefully adsorbed in the middle layer
of HSC. DMEM was centrifuged at 340 × g for 5 min, and washed
twice, prior to inoculation of 1×106 cells in 50 ml
plastic culture flask under static culture for 24 h. After 72 h,
the culture was replaced by DMEM culture solution containing 10%
FCS, and the culture solution was replaced once every three days.
Trypan blue staining was used to calculate the survival rate of
cells, with HSC survival rates of mice in the two groups reaching
>90%.
Real-time PCR
Expression of miR-10a in the control and hepatic
fibrosis groups were measured using RT-PCR. The TRIzol reagent was
used to extract total RNA in the two groups. A SYBR Premix ExTaq
fluorescent quantitative PCR kit and LightCycler instrument were
used to conduct the operation and analysis.
Inverse transcriptional primer sequence of miR-10a:
5-GTCGTATC-CAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACACAAA-3;
quantitative upstream primer sequence of miR-10a:
5-ACGTACCCTGTAGATCCG-3 and downstream sequence:
5-GTG-CAGGGTCCGAGGT-3. Inverse transcriptional primer sequence of
U6: 5-CGCTCACGAATTTGCGTGTCAT-3; quantitative upstream primer
sequence of U6: 5-CTCGCTTCGGCAGCACA-3 and downstream sequence:
5-GTG-CAGGGTCCGAGG-3. DNA amplification was conducted at 94°C for
15 sec, 94°C for 20 sec, 55°C for 10 sec, 72°C for 10 sec for total
of 40 cycles, and final extension at 72°C for 10 min. The Ct value
of the tested sample with PCR was quantified and the U6Ct value was
subtracted in the corresponding sample, thereby obtaining the ΔCt
value. The expression quantity of miR-10a in hepatic fibroblasts
was calculated using 2−ΔΔCt, and that in hepatic
fibrosis tissue samples was calculated by log2 transformed, i.e.,
log22−ΔΔCt.
Cell culture and transfection of
miR-10a mimics
DMEM culture solution containing 10% FCS was used to
subculture mouse hepatic fibroblasts in a 5% CO2
saturated humidity incubator at 37°C. Fibroblasts in logarithmic
growth were inoculated in a 12-well culture plate (3×105
cells/well), when the degree of cell growth was at 50%. The cells
were divided into the miR-10a mimics transfection and control
groups. Opti-MEM culture medium containing miR-10a mimics and
contrast miRNA, respectively, was used to transfect cells.
Subsequently, interferon was added to improve transfection
efficiency. Final concentrations of the miR-10a mimics and contrast
miRNA in each well during transfection were 20 nmol/l, with
interferin at 4 µl, and transfection lasting 72 h and repeated
three times.
Cell Counting Kit-8 (CCK-8)
The CCK-8 kit was used to test the hepatic
fibroblast proliferation capacity according to the protocol.
Following transfection of hepatic fibroblasts with miR-10a mimics
or contrast to miRNA for 72 h, the cells in the two groups were
inoculated into a 96-well culture plate (100 µl/well) at a density
of 2×104 cells/well, and three parallel wells were
established. When the cells were adhered to the walls after 3–4 h,
100 µl RPMI-1640 culture solution and 10 µl CCK-8 was added, and
placed in a 5% CO2 incubator at 37°C to be cultured for
2 h. Subsequently, the microplate reader was used to test the
optical density (OD) value, and this step was repeated three
times.
Western blotting
Following the transfection of hepatic fibroblast for
72 h, the cells in the transfection and control groups were
collected and divided using cell lysis solution. The BCA method was
used to test the protein concentration, 50 µg protein was taken and
separated by 8% SDS-PAGE and transferred to a PVDF membrane at room
temperature for 1 h. The primary antibodies, TGF-β1 antibody
(1:2,000), (Abcam, Cambridge, MA, USA, catalog no.: ab92486) Smad7
antibody (1:2,000) (Santa Cruz Biotechnology, CA, USA, catalog no.:
sc-11392) and β-actin antibody (1:1,000) (Santa Cruz Biotechnology,
catalog no.: sc-7210) were added and incubated overnight at 4°C.
The secondary antibody [polyclonal goat-anti-rabbit-HRP (Santa Cruz
Biotechnology, catalog no.: sc-2030)] was added with peroxidase
after membrane washing and incubated at room temperature for 1 h.
ECL was then used to develop it after membrane washing. GeneGnome
(Fremont, CA, USA) was used to collect images and ImageJ software
(Bethesda, MD, USA) was used for quantification of bands intensity
for three replicates.
Statistical analysis
SPSS 18.0 statistical software (Chicago, IL, USA)
was used for data analysis. Data were presented as mean ± standard
deviation. Matched or non-matched t-test analysis was used for
comparisons between groups. P<0.05 was considered to indicate a
statistically significant difference.
Results
miR-10a is highly expressed in mouse
hepatic fibrotic tissues
RT-PCR data showed that miR-10a in fibrotic tissues
was significantly higher than that in the control group (−7.84±1.38
vs. −9.97±1.59, P<0.05).
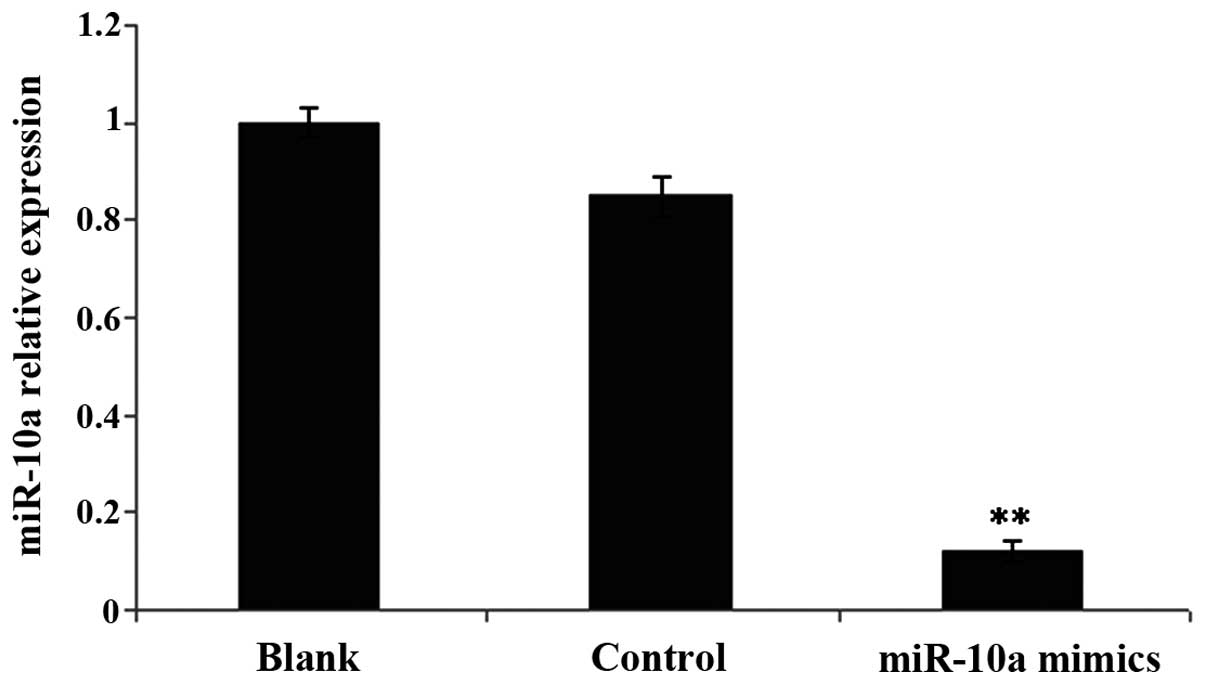
Hepatic fibroblasts had a high
expression in miR-10a following transfection of miR-10a mimics
miR-10a expression quantity in cells in the
transfection group was approximately 1/16 of that in the control
group (Fig. 1).
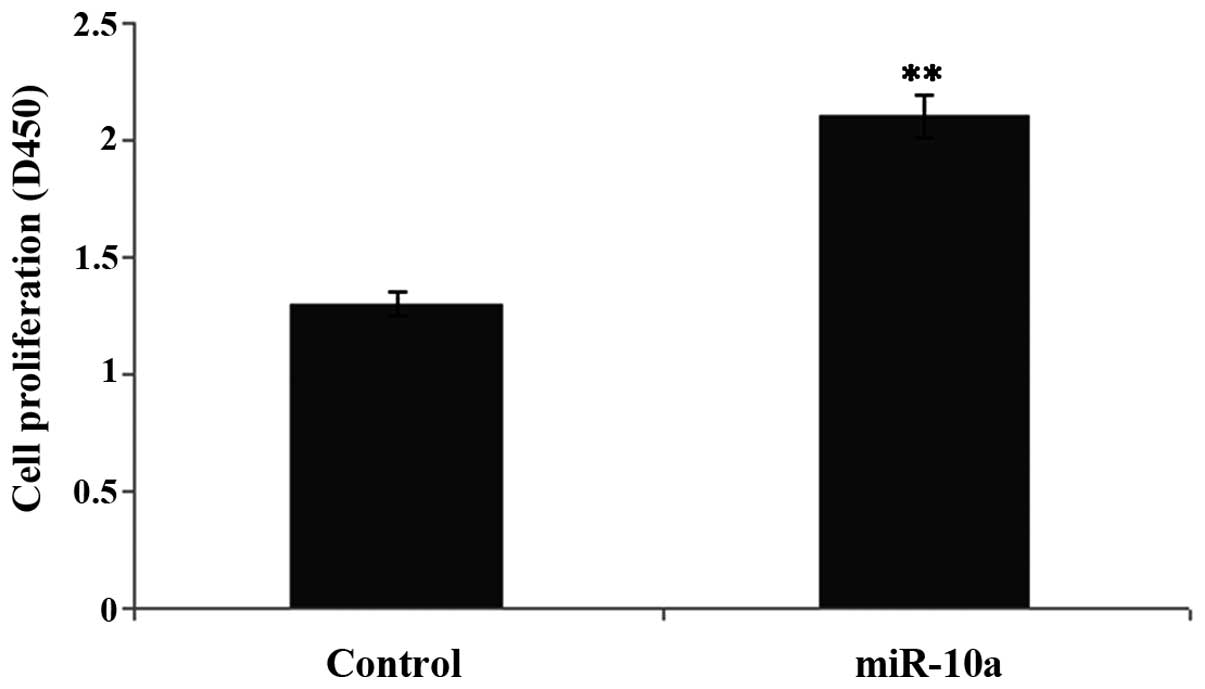
miR-10a high expression promotes
proliferation of hepatic fibroblasts
To examine the effect of a high expression of
miR-10a on the proliferation of hepatic fibroblasts, CCK-8 was used
to test the proliferation levels in the transfection and control
groups. Compared to the control group, proliferation of hepatic
fibroblasts with a high expression of miR-10a was significantly
increased (P<0.05) (Fig. 2).
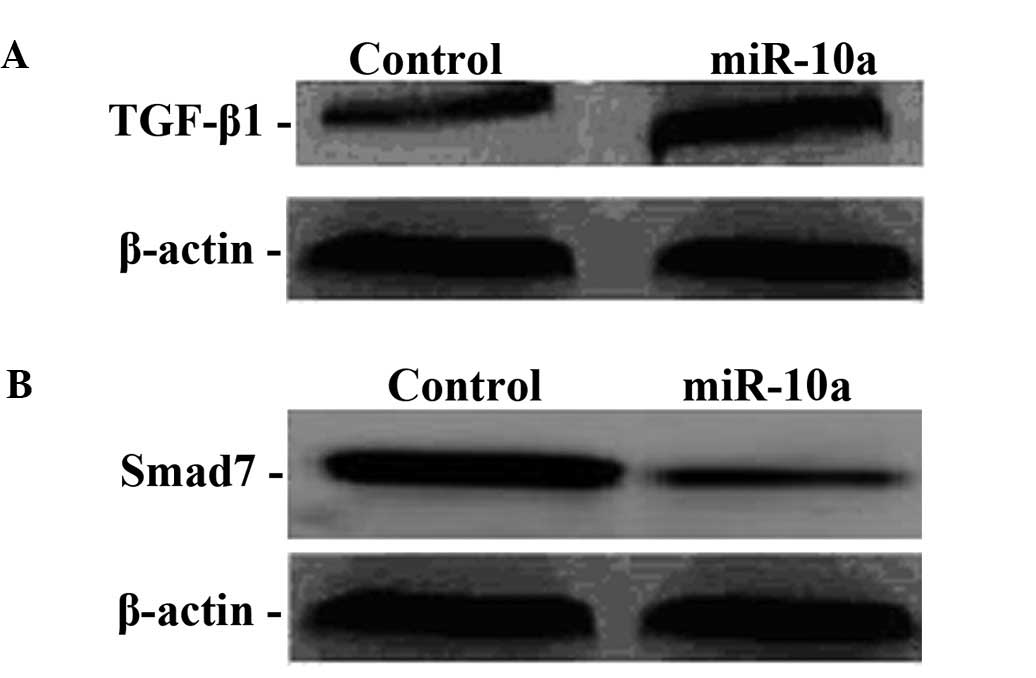
High miR-10a expression regulates the
TGF-βl/Smads signal transduction pathway
To examine the molecular mechanism of miR-10a in the
genesis and development of hepatic fibrosis, the protein expression
of TGF-βl and Smad7 was determined in hepatic fibroblasts after
miR-10a mimics were transfected. Compared to the control group,
TGF-βl protein expression in the transfection group was
significantly increased, indicating that miR-10a upregulated TGF-βl
expression while Smad7 protein expression in the transfection group
significantly was decreased, indicating that miR-10a downregulated
Smad7 expression (Fig. 3).
Discussion
Previous findings have shown that TGF-βl and its
Smad signal transduction pathway are closely associated with the
genesis and development of hepatic fibrosis (11,12),
whereby TGF-βl activates hepatic fibroblasts and is the strongest
hepatic fibrosis accelerator (13).
Smad protein is a key active substrate of the TGF-β family receptor
kinase. According to difference in structure and function, it is
divided into three types, Smad7 as an inhibitory type of Smads
mainly suppresses the TGF-β transduction pathway, Smad7 inhibits
Smads phosphorylation by binding activated TGF-β1 receptor, and
enters Smad7 cytoplasm, making it impossible for Smads to bind
receptors (14). Smad protein
constitutes the negative feedback loop in TGF-β signal transduction
and exerts its anti-fibrosis effect (14,18).
miRNA is involved in the genesis and development of
hepatic fibrosis, although the molecular mechanism of
miRNA-regulating hepatic fibrosis remains to be elucidated
(15–17). As a member of the miRNA family,
miR-10a is involved in the genesis and development of hepatic
fibrosis. We found that miR-10a had a high expression in mouse
hepatic fibrotic tissues, and this expression was capable of
promoting the proliferation of hepatic fibroblasts (19). The results of the present study
showed that miR-10a exerted a fibrotic factor-promoting effect in
hepatic fibrosis.
We also found that compared to the control group,
TGF-βl protein expression in the transfection group significantly
increased, indicating that miR-10a downregulated Smad7 expression
and exerted a hepatic fibrosis-promoting effect by regulating the
TGF-βl/Smads signal transduction pathway, and this provided a
potential therapeutic target for the treatment of hepatic
fibrosis.
In conclusion, miR-10a expression in mouse hepatic
fibrosis tissues increased, whereas a low miR-10a expression
promoted the proliferation of hepatic fibroblasts. This
proliferative effect was exerted by upregulating TGF-βl expression
and downregulating Smad7 expression following the regulation of hte
expression of TGF-βl and Smad7 in the TGF-βl/Smads signal
transduction pathway. The present study has found a new molecular
mechanism for the genesis and development of hepatic fibrosis.
Acknowledgements
The present study was supported by the Wenzhou
Public Welfare Science and Technology Planning Project (grant no.
Y20150023).
References
|
1
|
Lee YA and Friedman SL: Reversal,
maintenance or progression: what happens to the liver after a
virologic cure of hepatitis C? Antiviral Res. 107:23–30. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
van der Ree MH, de Bruijne J, Kootstra NA,
Jansen PL and Reesink HW: MicroRNAs: role and therapeutic targets
in viral hepatitis. Antivir Ther. 19:533–541. 2014. View Article : Google Scholar
|
|
3
|
Kim KM and Kim SG: Autophagy and microRNA
dysregulation in liver diseases. Arch Pharm Res. 37:1097–1116.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Havelange V, Ranganathan P, Geyer S,
Nicolet D, Huang X, Yu X, Volinia S, Kornblau SM, Andreeff M, Croce
CM, et al: Implications of the miR-10 family in chemotherapy
response of NPM1-mutated AML. Blood. 123:2412–2415. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ohuchida K, Mizumoto K, Lin C, Yamaguchi
H, Ohtsuka T, Sato N, Toma H, Nakamura M, Nagai E, Hashizume M, et
al: MicroRNA-10a is overexpressed in human pancreatic cancer and
involved in its invasiveness partially via suppression of the HOXA1
gene. Ann Surg Oncol. 19:2394–2402. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Xie T, Liang J, Guo R, Liu N, Noble PW and
Jiang D: Comprehensive microRNA analysis in bleomycin-induced
pulmonary fibrosis identifies multiple sites of molecular
regulation. Physiol Genomics. 43:479–487. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Uemura M, Swenson ES, Gaça MD, Giordano
FJ, Reiss M and Wells RG: Smad2 and Smad3 play different roles in
rat hepatic stellate cell function and alpha-smooth muscle actin
organization. Mol Biol Cell. 16:4214–4224. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tomita K, Tamiya G, Ando S, Ohsumi K,
Chiyo T, Mizutani A, Kitamura N, Toda K, Kaneko T, Horie Y, et al:
Tumour necrosis factor alpha signalling through activation of
Kupffer cells plays an essential role in liver fibrosis of
non-alcoholic steatohepatitis in mice. Gut. 55:415–424. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Heldin CH, Miyazono K and ten Dijke P:
TGF-β signalling from cell membrane to nucleus through SMAD
proteins. Nature. 390:465–471. 1997. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Schnabl B, Kweon YO, Frederick JP, Wang
XF, Rippe RA and Brenner DA: The role of Smad3 in mediating mouse
hepatic stellate cell activation. Hepatology. 34:89–100. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Martínez Fernández EM, López-Cortés LF,
Regordan C and Cordero Matía E: Meningitis by Cryptococcus
neoformans in patients with HIV infection. Neurologia. 14:218–223.
1999.(In Spanish). PubMed/NCBI
|
|
12
|
Paradis V, Dargere D, Bonvoust F, Vidaud
M, Segarini P and Bedossa P: Effects and regulation of connective
tissue growth factor on hepatic stellate cells. Lab Invest.
82:767–774. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Long J, Wang G, Matsuura I, He D and Liu
F: Activation of Smad transcriptional activity by protein inhibitor
of activated STAT3 (PIAS3). Proc Natl Acad Sci USA. 101:99–104.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kavsak P, Rasmussen RK, Causing CG, Bonni
S, Zhu H, Thomsen GH and Wrana JL: Smad7 binds to Smurf2 to form an
E3 ubiquitin ligase that targets the TGF beta receptor for
degradation. Mol Cell. 6:1365–1375. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Roderburg C, Luedde M, Vargas Cardenas D,
Vucur M, Mollnow T, Zimmermann HW, Koch A, Hellerbrand C,
Weiskirchen R, Frey N, et al: miR-133a mediates TGF-β-dependent
derepression of collagen synthesis in hepatic stellate cells during
liver fibrosis. J Hepatol. 58:736–742. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kumar V and Mahato RI: Delivery and
targeting of miRNAs for treating liver fibrosis. Pharm Res.
32:341–361. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chen SL, Zheng MH, Shi KQ, Yang T and Chen
YP: A new strategy for treatment of liver fibrosis: letting
microRNAs do the job. BioDrugs. 27:25–34. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Padda RS, Gkouvatsos K, Guido M, Mui J,
Vali H and Pantopoulos K: A high-fat diet modulates iron metabolism
but does not promote liver fibrosis in hemochromatotic
Hjv−/− mice. Am J Physiol Gastrointest Liver
Physiol. 308:G251–G261. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Rieger JK, Klein K, Winter S and Zanger
UM: Expression variability of absorption, distribution, metabolism,
excretion-related microRNAs in human liver: Influence of nongenetic
factors and association with gene expression. Drug Metab Dispos.
41:1752–1762. 2013. View Article : Google Scholar : PubMed/NCBI
|