Introduction
Statins are widely used for the treatment of
hypercholesterolemia and to decrease the risk of cardiovascular
disease (1). Statins act by
inhibiting the conversion of HMG-CoA to mevalonate, which is a
rate-limiting step in the cholesterol biosynthesis pathway and thus
lowering the LDL-cholesterol in the blood (2). Previous clinical trials have
demonstrated that statins are able to substantially decrease
cardiovascular-associated morbidity and mortality in patients with
and without coronary disease (3–5). Since
serum cholesterol levels have been closely associated with coronary
artery disease (6), it was initially
hypothesized that the ability of statins to lower cholesterol
levels was the predominant mechanism underlying the observed
reduction in the risk of cardiovascular diseases (7). However, it has since emerged that the
therapeutic benefits of statins may not solely be explained by
their cholesterol-lowering effects (8,9); it was
reported that patients treated with statins were less susceptible
to cardiovascular incidents, independent of cholesterol lowering
(10). Therefore, mechanisms other
than cholesterol-lowering may underlie the ability of statins to
protect against cardiovascular incidents.
Statins have been shown to directly improve
endothelial function and promote vascular relaxation by increasing
the synthesis of nitric oxide by endothelial nitric oxide synthase
(eNOS) (11), and by reducing the
levels of angiopoietin-2 (12),
which are important regulatory factors in endothelial cells for
protection against the development of arteriosclerosis (13,14). In
addition, statins promote atherosclerotic plaque stability
(15) and decrease the production of
reactive oxygen species from monocytes and macrophages (16). Furthermore, statins reduce
pro-inflammatory cytokine expression and oxygen radical formation
(17–19), while they are also associated with a
reduced risk of Alzheimer's disease (1). Impairment of vascular endothelial
cells, which may result from increased production of reactive
oxygen species and decreased synthesis or secretion of
endothelial-derived nitric oxide (20–22), has
been shown to be involved in the pathogenesis of arteriosclerosis
and other cardiovascular diseases (23). Therefore, maintenance of vascular
endothelial function is important to prevent cardiovascular disease
(24,25). However, to the best of our knowledge,
no previous study has systematically investigated the effects of
statins on endothelial cells, and thus it is difficult to
mechanistically illustrate their beneficial clinical effects.
Therefore, the present study used cDNA microarrays
to investigate the effect of lovastatin on global gene expression
patterns in human umbilical vein endothelial cells (HUVECs), in
order to identify unknown intracellular targets of statins and
potential mediators of the pleiotropic effects observed for this
drug. The results of the present study may provide further insights
into the mechanisms underlying the pleiotropic effects of statins
and their anti-atherogenic potential.
Materials and methods
Reagents
Lovastatin (Sigma-Aldrich, St. Louis, MO, USA)was
converted to an active form as described previously (26). Briefly, inactive lovastatin was
dissolved in warm (55°C) ethanol, followed by the addition of 0.6 M
NaOH and H2O, and was incubated at room temperature for
30 min. Subsequently, HCl was used to adjust the final solution to
pH 8.0, and the lovastatin solution was stored as a 10 mM stock at
−20°C. Rabbit polyclonal anti-SGK3 (1:1,000, ab153981), rabbit
monoclonal anti-ATP2B1 (1:1000, ab190355), rabbit monoclonal
anti-PTK2B (1:1,000, ab32571), rabbit polyclonal anti-eNOS
(1:1,000, ab5589), rabbit monoclonal anti-MAP3K3 (1:2,000, ab40756)
and rabbit polyclonal MAP4K3 (1:2,000, ab103481) antibodies were
purchased from Abcam (Cambridge, MA, USA). In addition, rabbit
polyclonal anti-BCL2 (1:1,000, 2872), rabbit monoclonal anti-BAX
(1:1,000, 5023), mouse monoclonal anti-caspase-3 (1:500, 9668),
mouse monoclonal anti-AKT(1:2,000, 2920), rabbit monoclonal
anti-p-AKT (1:1,000, 4060) and mouse monoclonal β-actin (1:5,000,
3700) antibodies were purchased from Cell Signaling Technology,
Inc. (Danvers, MA, USA).
Cell culture
HUVECs were purchased from ATCC (Manassas, VA, USA)
and maintained in Dulbecco's modified Eagle's medium (Invitrogen;
Thermo Fisher Scientific, Inc., Waltham, MA, USA) supplemented with
10% fetal bovine serum (Gibco; Thermo Fisher Scientific, Inc.).
Cells were cultured at 37°C in a 5% CO2 atmosphere until
further use, and were seeded in 6-well plates at a density of
4×105 cells per well. For dose-response experiments, the
cells were treated with various concentrations of lovastatin for 24
h (0.1, 0.5, 2.5 and 12.5 µM). For time-course experiments, the
cells were treated with 0.5 µM lovastatin for various durations (2,
6, 12 and 24 h). Control cells were treated with 0.1% dimethyl
sulfoxide (vehicle).
Cellular cholesterol measurement
Total cholesterol was extracted from the cells
according to a previously described method (27), with minor modifications. Briefly,
cells were harvested and washed twice with phosphate-buffered
saline (PBS). Next, extraction was performed with 1 ml isopropanol,
followed by sonication for 20 min at 4°C, and centrifugation at
10,000 × g for 10 min at 4°C. The supernatant was evaporated under
a vacuum and the pellet was resuspended in 20 µl isopropanol for
the determination of cellular cholesterol. Cholesterol levels were
measured using a Total Cholesterol Detection kit (1021; Jiemen
Biotechnology Corp., Shanghai, China). Subsequently, the sediment
fractions were lysed using a Mammalian Cell Extraction kit
(K269-500; BioVision, Inc., Milpitas, CA, USA), and total protein
levels were quantified using the Bradford method (P0006; Beyotime
Institute of Biotechnology).
cDNA microarray
Total RNA was extracted from the cells using TRIzol
reagent (Invitrogen; Thermo Fisher Scientific, Inc.), followed by
purification on an RNeasy column (74104; Qiagen, Inc., Valencia,
CA, USA) and quantification by UV absorption using a 2000 Nanodrop
spectrophotometer (Thermo Fisher Scientific, Inc., Wilmington, DE,
USA). RNA quality was assessed using an Agilent 2100 Bioanalyzer
(Agilent Technologies, Inc., Santa Clara, CA, USA). A cDNA
microarray analysis was conducted according to a previous study
(28). Briefly, a home-made
microarray was used and reverse transcription was done using an
Ambion RETROscript reverse transcription kit (AM1710). Samples from
three wells of the vehicle-treated group were pooled and used as a
reference to label Cy3 (GE Healthcare Life Sciences). RNA from the
drug treated cells was isolated in triplicate and labeled with Cy5
individually. Cy3- and Cy5-labeled cDNA pools were mixed to
hybridize. Hybridization was performed at 56°C for 16 h and washes
were performed and followed by scanning on an Axon 4000B Scanner
(Axon Instruments, Sunnyvale, CA, USA). In total 8,583
genes/expressed sequence tags were selected according to signal
intensity. Gene expression profiling for each time point was
conducted with Significance Analysis of Microarrays software,
version 2.20 (http://statweb.stanford.edu/~tibs/SAM/), with One
Class response type (false discovery rate <0.01). Genes were
considered as differentially expressed when their fold-change was
>1.5-fold. The function of these differentially-expressed genes
was searched with bioinformatics tools including KEGG (http://www.genome.jp/kegg/) and DAVID (https://david.ncifcrf.gov/) pathway analysis.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
A total of 2 µg of total RNA was used for reverse
transcription using an Ambion RETROscript reverse transcription kit
(AM1710). Next, the cDNA was diluted 1:20 for RT-qPCR. RT-qPCR was
conducted on an ABI 7300 Real-Time PCR system (Applied Biosystems;
Thermo Fisher Scientific, Inc.) with SYBR Green I fluorescent dye
(Toyobo Co., Ltd., Osaka, Japan). The PCR reaction contained 1 µl
of cDNA, 1 µl of primers (5 µM), 8 µl of H2O and 10 µl
of reaction mixture with a total volume of 20 µl. The reactions
were performed in triplicate at 94°C for 15 s and 60°C for 1 min
and 40 cycles. GAPDH was used as an internal control. Gene
expression was evaluated with the 2−∆∆Ct method
(27). The primer sequences of the
genes used for qPCR were designed with primer express 2.0 and are
listed in Table I.
 | Table I.Gene names and polymerase chain
reaction primer sequences. |
Table I.
Gene names and polymerase chain
reaction primer sequences.
| Gene name | Forward primer
(5′-3′) | Reverse primer
(5′-3′) |
|---|
| BCL2 |
AAAATTTCCTGCATCTCATG |
TATTGGATGTGCTTTGCATT |
| PTK2B |
GCCCAGCCGACCTAAGTACAG |
CGTAGGAGAGCTGGCACACA |
| MAP3K3 |
GAGAGCGTGACCCGAAAGTA |
GGATGTTGGCTCCCTTAATG |
| MAP4K2 |
CAACTCCCAAGGTGCATATG |
CGAGTAACAGGGTGAATCCA |
| CDC7 |
AAGACGGAAAGGAGGGATCT |
GCAGGGCTCTCATGTGAAAT |
| SNPH |
GCCAGCAACACCTATGAGAA |
GGTACTGCACGAAGTCTGTCT |
| RAB25 |
GCTGCAGAAGTCCCCTTACC |
GGCGACAAATCTGTGTGTTG |
| OVGP1 |
TTGATGGTCTTGACCTTTTC |
GCAAACAGGAGCTCTTCAAT |
| ATP2B1 |
CATGTGTTAGGGCCCAGATT |
TCTTTGCTTGGTCAGCACTCT |
| SGK3 |
TGAAATGCTGTATGGATTGC |
ACTCACTCCTGGCCTCAAAC |
Cell counting kit (CCK)-8 assay
Following the treatment of HUVECs with various
concentrations of lovastatin for 24 h or 0.5 µM lovastatin for
various durations, cell viability was determined by adding 10 µl
CCK-8 solution (Dojindo Molecular Technologies, Inc., Kumamoto,
Japan), followed by incubation at 37°C for 2 h. Absorbance at 450
nm was measured using a SpectraMax 190 Microplate Reader (Molecular
Devices, LLC, Sunnyvale, CA, USA). In each assay, four parallel
wells were run and experiments were conducted in triplicate. In
addition, cells were treated with oxidized (ox)-low-density
lipoprotein (LDL),lovastatin or both for 24 h, and then measured
with CCK-8 as described above, to assess the protective effect of
lovastatin on injury induced by ox-LDL.
Cell apoptosis assay
To investigate the apoptosis of HUVECs, the cells
were seeded at a density of 2×105 cells/well in a 6-well
plate and incubated at 37°C overnight. Subsequently, the cells were
treated with ox-LDL, lovastatin or both for 24 h, detached with
0.25% trypsin digestion and washed with cooled PBS. Next, cells
were collected and resuspended in binding buffer containing
Annexin-V and propidium iodide (Beyotime Institute of
Biotechnology) and incubated for 15 min in the dark at room
temperature. Apoptosis analyses were performed on a FACSCalibur
flow cytometer (BD Biosciences, San Jose, CA, USA).
Western blot analysis
HUVECs were washed twice with ice-cold PBS and lysed
in radioimmunoprecipitation assay buffer (50 mM Tris, 150 mM NaCl,
1% Triton X-100, 0.1% SDS and 1% sodium deoxycholate; Beyotime
Institute of Biotechnology, Haimen, China) containing protease and
phosphatase inhibitors. Next, equal volumes of protein (10 µg) were
separated by 12% SDS-PAGE and transferred to Hybond-C
nitrocellulose membranes (GE Healthcare Life Sciences, Little
Chalfont, UK) in transfer buffer (192 mM glycine, 25 mM Tris, 2.5
mM SDS, 10% methanol). The blots were blocked with 5% nonfat milk
in Tris-buffered saline supplemented with Tween-20 for 1 h at room
temperature, followed by incubation with primary antibodies
overnight at 4°C. Subsequently, the membranes were incubated with
horseradish peroxidase-conjugated secondary antibody (anti-rabbit
IgG, 1:2,000; 7074; anti-mouse IgG, 1:2,000, 7076; Cell Signaling
Technology, Inc.). The blots were developed with ECL Plus Western
Blotting Detection Reagent (GE Healthcare Life Sciences) and
analyzed using ImageJ software (1.43b; National Institutes of
Health, Bethesda, MD, USA).
RNA silencing
Accell small interfering (si)RNA targeting
PTK2B was purchased from GE Dharmacon (GE Healthcare Life
Sciences), with a scrambled siRNA used as the control (GE
Healthcare Life Sciences). Transfection of HUVECs with PTK2B
siRNA or scrambled siRNA was performed using Lipofectamine RNAiMAX
(Thermo Fisher Scientific, Inc.), according to the manufacturer's
instructions.
Statistical analysis
Data are presented as the mean ± standard deviation.
Comparisons between groups were conducted by one-way analysis of
variance and least significant difference using SPSS software,
version 19.0 (IBM SPSS, Armonk, NY, USA). P<0.05 were considered
to indicate a statistically significant difference.
Results
Effects of lovastatin on the
cholesterol content of HUVECs
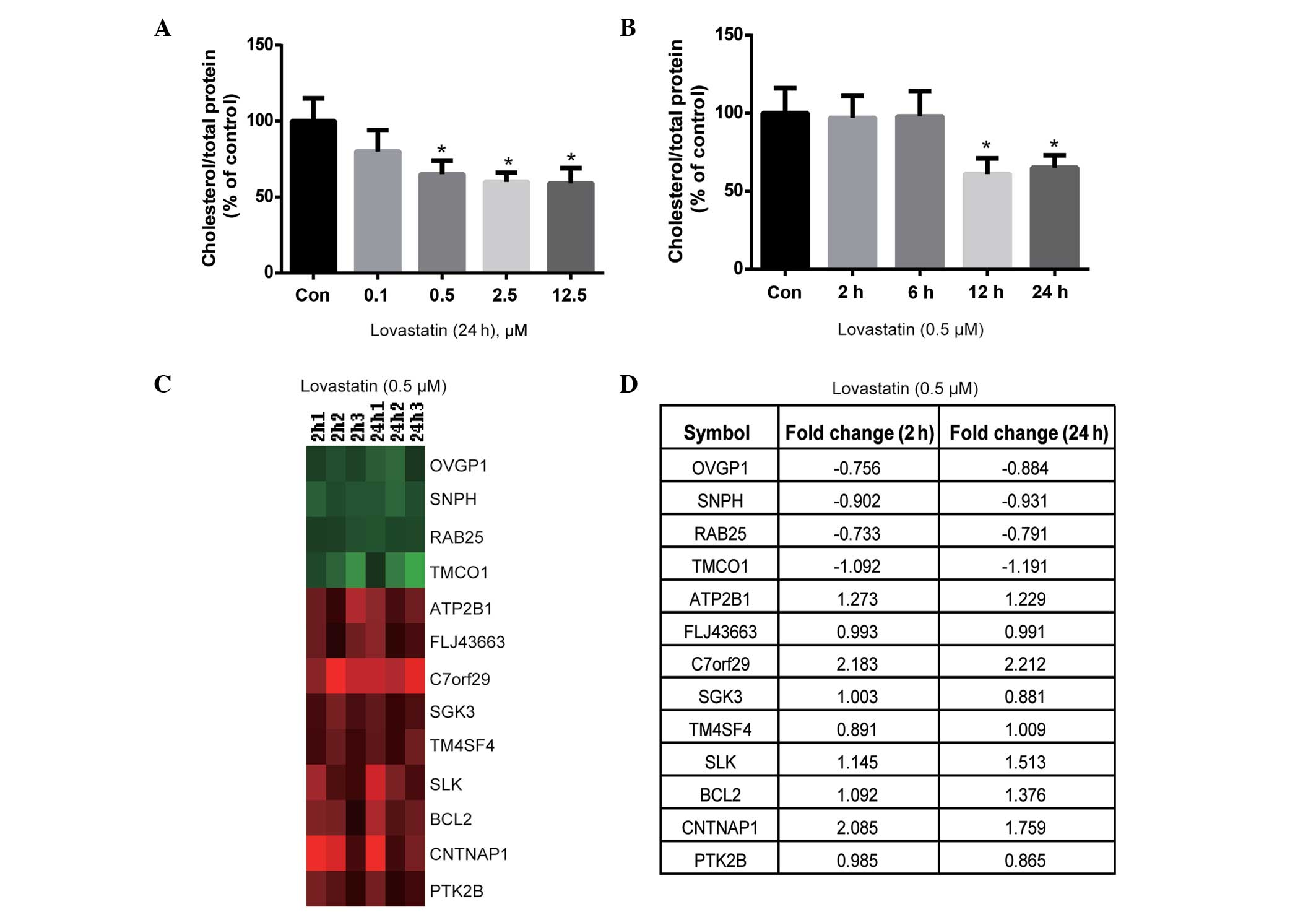
The effect of lovastatin on intracellular
cholesterol content was evaluated. Lovastatin (0.5, 2.5 and 12.5
µM) significantly decreased the cholesterol content of HUVECs
following 24 h of incubation (P<0.05; Fig. 1A). Notably, the cholesterol content
was not significantly different between the 0.5, 2.5 and 12.5 µM
lovastatin-treated cells (P>0.05; Fig. 1A); therefore, 0.5 µM lovastatin was
used for the time-course experiment. As shown in Fig. 1B, lovastatin significantly decreased
the cholesterol content of HUVECs following 12 and 24 h of
treatment (P<0.05), but not after 2 or 6 h of treatment
(P>0.05). Therefore, the duration of treatment with 0.5 µM
lovastatin was set at 2 and 24 h, in order to observe short- and
long-term gene expression profiles, which may be dependent or
independent of cholesterol lowering.
Effects of lovastatin on the gene
expression profile of HUVECs
cDNA microarrays were used to analyze the effects of
0.5 µM lovastatin on the gene expression profile of HUVECs after 2
and 24 h of treatment. The results indicated that the expression
levels of a number of genes were altered by lovastatin. After 2 h
of treatment with 0.5 µM lovastatin, 12 genes were downregulated
and 178 genes were upregulated, with fold-changes of >1.5 (data
not shown). Similarly, after 24 h of treatment with 0.5 µM
lovastatin, 33 genes were downregulated and 77 genes were
upregulated, presenting fold-changes of >1.5 (data not shown).
These genes may represent unknown intracellular targets of
lovastatin and require further clarification.
Subsequently, the genes that were differentially
expressed at 2 and 24 h of treatment were compared to identify
which genes were altered at both treatment durations, since these
may serve important roles in the effects of lovastatin on
endothelial cells. As shown in Fig.
1C, 4 genes were downregulated at both 2 and 24 h of treatment,
including OVGP1, SNPH, RAB25 and TMCO1,
whereas the ATP2B1, FLJ43663, C7orf29,
SGK3, TM4SF4, SLK, BCL2, CNTNAP1
and PTK2B genes were upregulated at 2 and 24 h of treatment
(the fold change is summarized in Fig.
1D). According to the known functions of these genes, such as
SGK3, ATP2B1 and PTK2B involved in cell proliferation and survival,
(29–31), it may be hypothesized that they are
independent of the cholesterol-lowering effects of lovastatin and
likely respond to lovastatin as early as 2 h after treatment.
Therefore, further investigation into the biological significance
of these genes may be important.
Critical genes regulated by lovastatin
in HUVECs
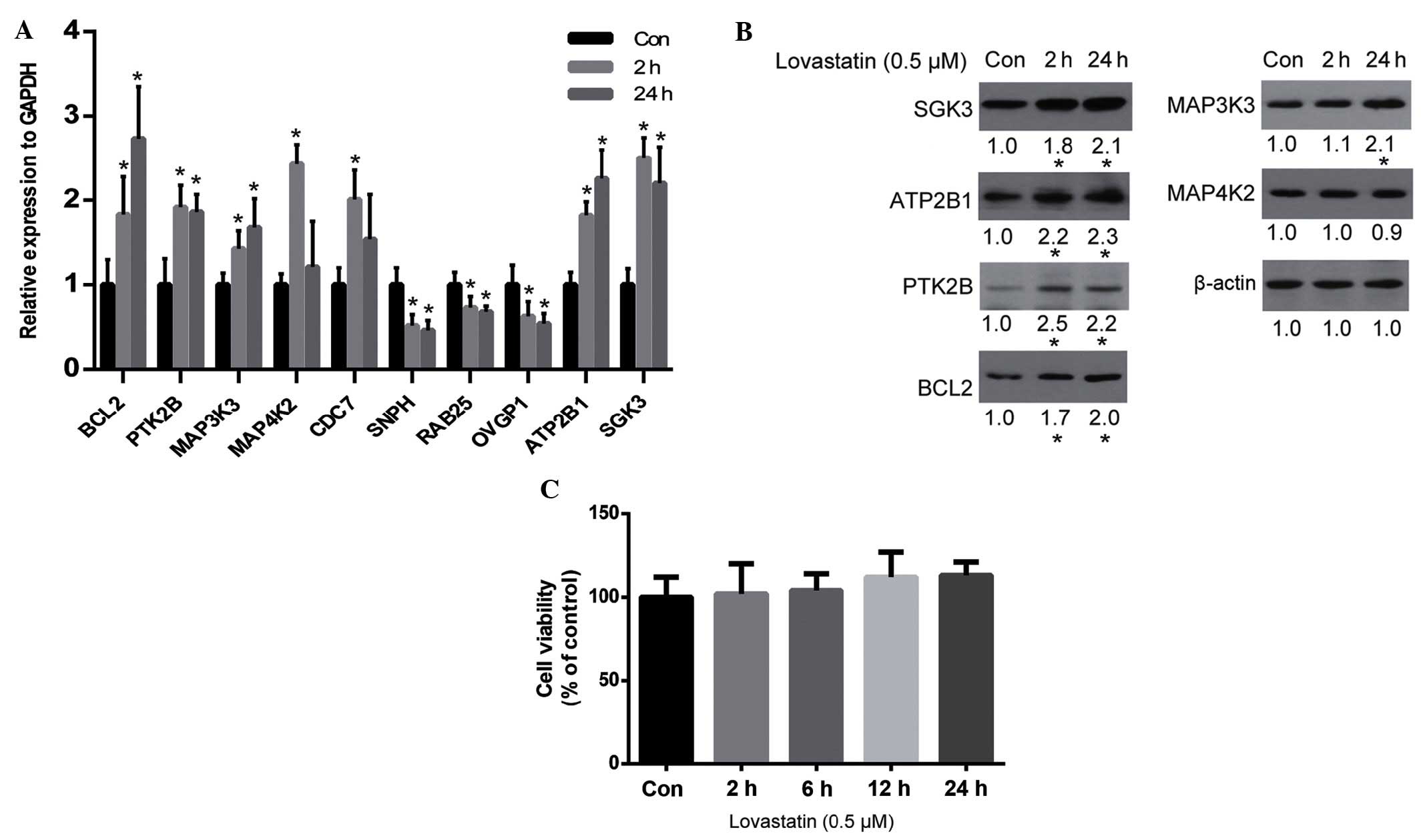
The results of the cDNA microarray analysis were
verified by qPCR. As is shown in Fig.
2A, the qPCR results for the majority of genes were consistent
with the microarray results, including the upregulation of
BCL2, PTK2B, MAP3K3, MAP4K2,
ATP2B1 and SGK3, and the downregulation of
SNPH, RAB25 and OVGP1. Whether alterations in
the expression levels of these genes resulted in alterations in
protein expression was unclear. Therefore, the protein expression
levels of certain important genes (according to their known
function) were analyzed by western blot analysis. The results
demonstrated that SGK3, ATP2B1, PTK2B, BCL2 and MAP3K3 were all
upregulated by lovastatin, which was consistent with the alteration
of their gene expression levels, with the exception of MAP4K2
(Fig. 2B). Since SGK2, ATP2B1, PTK2B
and MAP3K3 are associated with cell proliferation and survival
(29–32), and BCL2 is an anti-apoptosis protein
(33), it may be hypothesized that
lovastatin may affect cell proliferation. However, the viability of
HUVECs was not significantly altered by lovastatin following 24 h
of treatment (P>0.05; Fig.
2C).
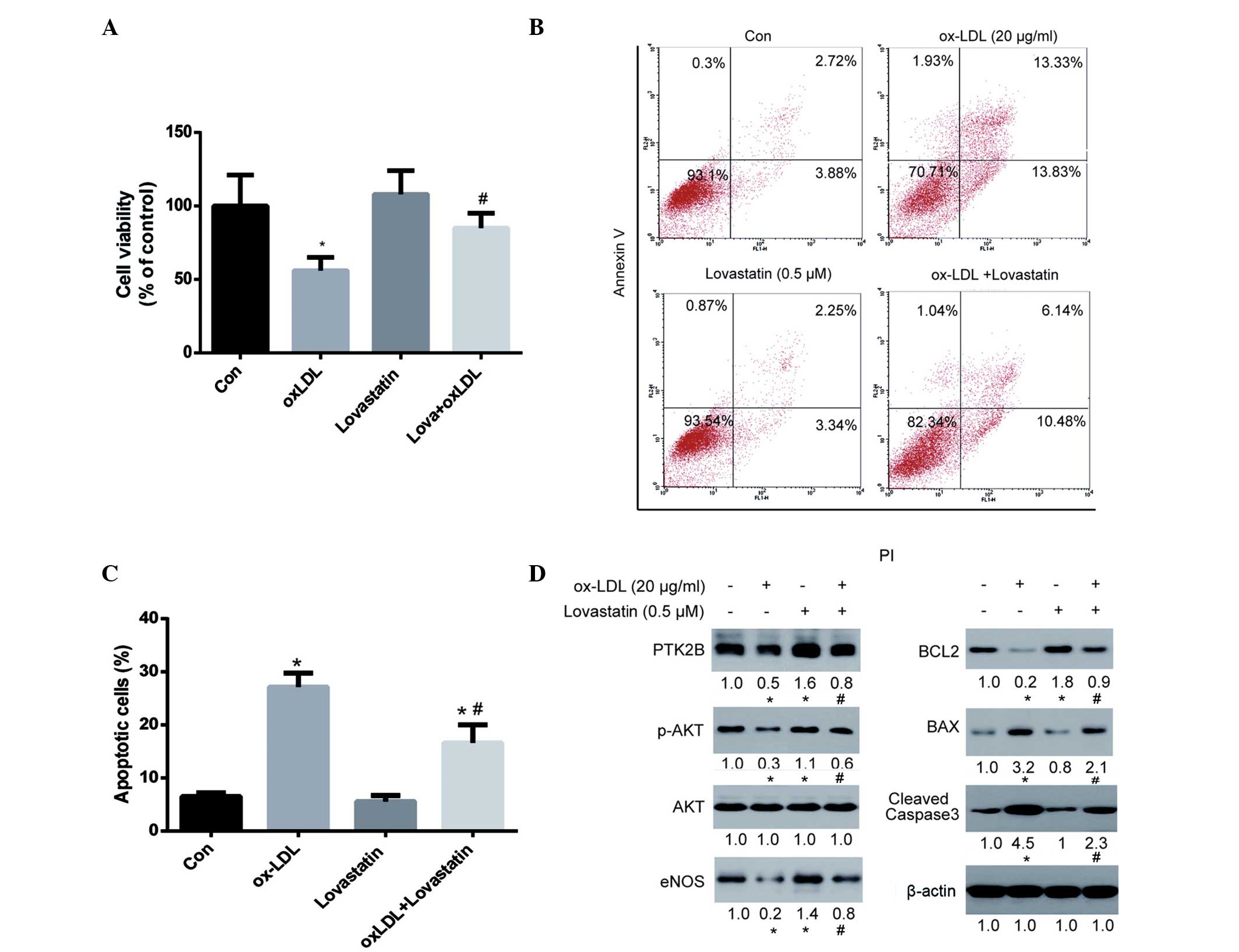
Lovastatin protects HUVECs against
ox-LDL-induced cytotoxicity via the PTK2B/AKT signaling
pathway
Since lovastatin did not affect the viability of
HUVECs after 24 h of treatment, the ability of lovastatin to
protect HUVECs against ox-LDL-induced cytotoxicity was
investigated. As is shown in Fig.
3A, 20 µg/ml ox-LDL significantly reduced the viability of
HUVECs after 24 h of treatment (P<0.05). Although lovastatin did
not significantly affect the viability of HUVECs after 24 h of
treatment, it was able to significantly protect HUVECs against
ox-LDL-induced injury (P<0.05). In addition, the ability of
lovastatin to protect HUVECs against ox-LDL-induced apoptosis was
evaluated (Fig. 3B and C). Annexin
V-positive cells were significantly decreased in the lovastatin
plus ox-LDL group, as compared with the ox-LDL group (P<0.05;
Fig. 3C), thus further supporting
the protective role of lovastatin against ox-LDL-induced HUVEC
cytotoxicity.
In the western blot analysis, both PTK2B and BCL2
were upregulated in the lovastatin-treated HUVECs. Since PTK2B
serves an important role in cell survival via the AKT pathway
(34), and BCL2 is a major
anti-apoptotic protein in mitochondrial-dependent apoptosis, it may
be hypothesized that this pathway plays an important role in
lovastatin-mediated protection against ox-LDL-induced cell injury.
As is shown in Fig. 3D, ox-LDL
treatment was associated with a downregulation of PTK2B and its
downstream p-AKT and eNOS, as well as downregulation of BCL2 and
upregulation of the pro-apoptotic protein BAX and cleaved
caspase-3. These results suggested that ox-LDL exerted
pro-apoptotic effects on mitochondrial-dependent apoptosis.
However, treatment with lovastatin was able to reverse these
alterations in protein expression, including upregulating PTK2B,
p-AKT, eNOS and BCL2, and downregulating BAX and cleaved caspase-3.
These alterations in protein expression levels were in parallel
with cell viability and apoptosis assays, thus suggesting that
these proteins may serve critical roles in lovastatin-mediated
protection against ox-LDL-induced cell injury.
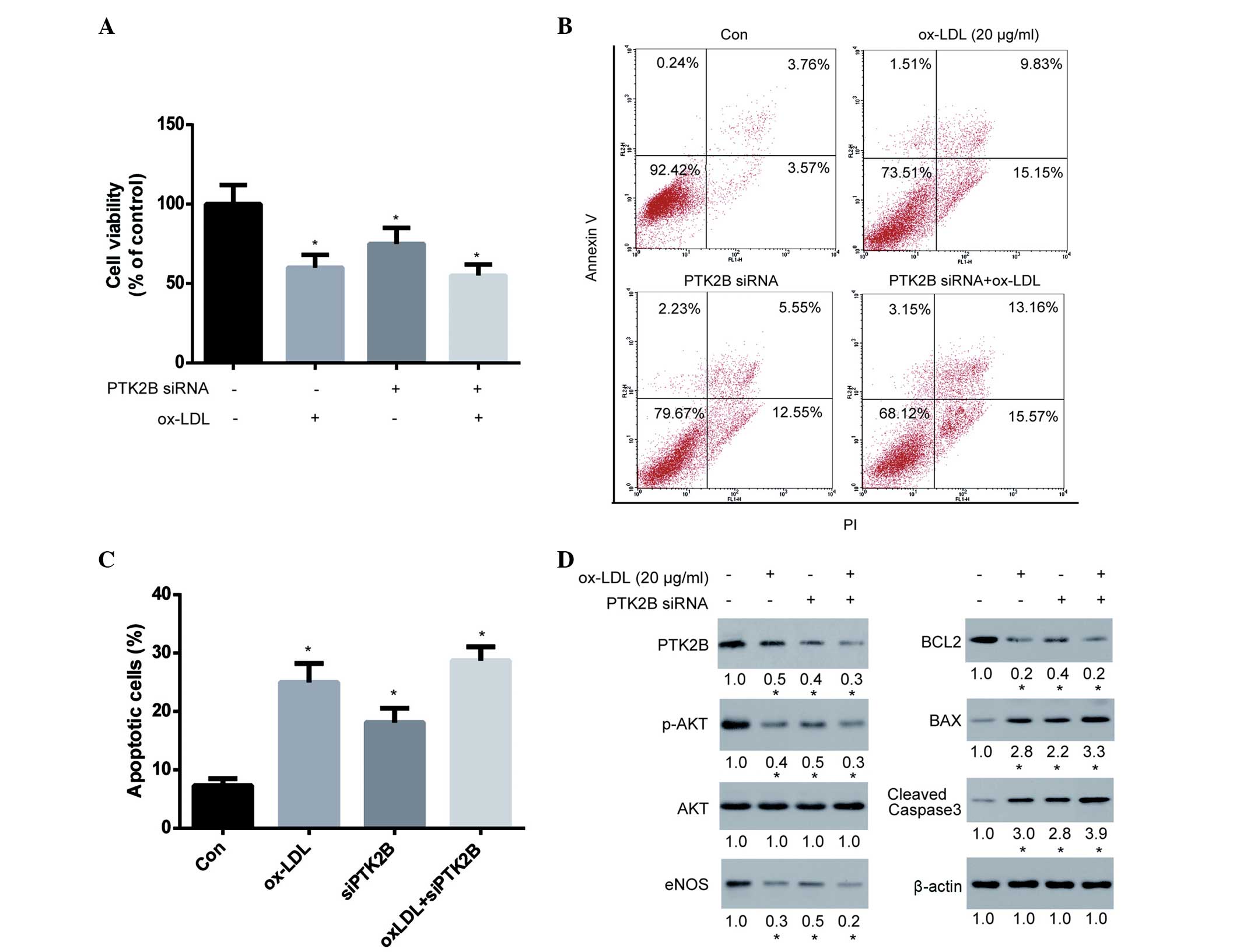
Knockdown of PTK2B attenuates
ox-LDL-induced HUVEC injury
AKT is known to promote cell survival by
upregulating the expression of BCL2 and downregulating the
expression of BAX (35). In
addition, PTK2B has been shown to be an upstream protein of AKT and
eNOS (36). Therefore, it may be
hypothesized that PTK2B promotes the AKT/BCL2 signaling cascade. To
test this hypothesis, PTK2B expression in HUVECs was transiently
knocked down for 48 h using siRNA, after which HUVECs were treated
with ox-LDL for 24 h to observe the effect of PTK2B on cell
viability and apoptosis. The results demonstrated that silencing of
PTK2B expression significantly decreased cell viability,
thus indicating its importance for cell survival in HUVECs
(Fig. 4A). Notably, knockdown of
PTK2B was able to markedly reduce ox-LDL-induced cytotoxicity.
Subsequently, the ox-LDL-induced apoptosis of HUVECs following
knockdown of PTK2B was analyzed. The ability of ox-LDL to
induce apoptosis of HUVECs was markedly compromised following
knockdown of PTK2B (Fig. 4B and
C). Furthermore, we examined the signaling cascade involved in
this pathway. As is shown in Fig.
4D, PTK2B was downregulated following treatment of HUVECs with
ox-LDL and siRNA, although the extent of ox-LDL-induced PTK2B
downregulation was markedly reduced in PTK2B knockdown
cells. Accordingly, the ox-LDL-induced inhibition of p-AKT and
eNOS, downregulation of BCL2 and upregulation of BAX and cleaved
caspase-3 were greatly compromised following knockdown of
PTK2B. These results suggest that the PTK2B-mediated
signaling pathway is upregulated by lovastatin and serves an
important role in the lovastatin-mediated protection against
ox-LDL-induced cytotoxicity.
Discussion
In the present study, cDNA microarrays were used to
identify unknown intracellular targets of lovastatin and potential
mediators of the previously observed pleiotropic effects of
lovastatin (37). In addition, the
gene expression profiles for HUVECs treated with lovastatin at
various concentrations and durations were presented.
The present study compared the genes regulated after
2 and 24 h of lovastatin treatment. The results demonstrated that
13 genes were consistently altered following lovastatin treatment
for the two different durations, including 4 genes that were
downregulated (OVGP1, SNPH, RAB25 and
TMCO1) and 9 genes that were upregulated, such as
ATP2B1, FLJ43663, C7orf29, SGK3,
TM4SF4, SLK, BCL2, CNTNAP1 and
PTK2B. Subsequently, the functions of these genes were
searched in the Uniprot and PubMed databases in order to obtain
useful information for further study; however, only a few of the
genes were associated with cell proliferation or survival,
including SGK3, BCL2 and PTK2B, which were
upregulated. SGK3 is serine/threonine-protein kinase that is
involved in the regulation of numerous ion channels, cell
proliferation, survival and migration (38,39).
BCL2 is a well-known anti-apoptotic protein in the
mitochondrial-dependent apoptosis signaling pathway. PTK2B
encodes a cytoplasmic protein tyrosine kinase that is involved in
calcium-induced regulation of ion channels and activation of
phosphatidylinositol 3-kinase and the AKT signaling cascade
(40,41), as well as activation of the MAPK
signaling cascade (42,43). Therefore, the authors of the present
study hypothesized that lovastatin may promote cell viability via
the upregulation of SGK3, PTK2B and BCL2.
However, cell apoptosis and viability assays demonstrated that
lovastatin did not affect HUVEC viability. These results suggested
that lovastatin may exert its beneficial effects via a different
mechanism. Therefore, the protective effect of lovastatin against
ox-LDL-induced HUVEC cytotoxicity was then investigated.
ox-LDL serves a major role during atherogenesis
(44), impairs endothelial
vasodilator function (45,46) and stimulates apoptosis (47,48). In
the present study, lovastatin was able to protect HUVECs against
ox-LDL-induced cytotoxicity and cell apoptosis, thus suggesting
that some unidentified mechanisms may exist underlying this effect.
PTK2B is known to activate the AKT survival pathway to promote BCL2
upregulation, while AKT is an upstream regulator of eNOS, which
serves an important role in the protection against ox-LDL (49,50).
Based on these previous observations, and since lovastatin
upregulated the expression of PTK2B and BCL2 in the
current study, the AKT/BCL-2 signaling pathway was selected for
further analysis. The results demonstrated that p-AKT was
upregulated in HUVECs following treatment with lovastatin, as well
as the upregulation of eNOS and downregulation of BAX. The
expression levels of PTK2B, p-AKT and eNOS were downregulated in
HUVECs treated with ox-LDL, and this was associated with
upregulation of the mitochondrial-dependent apoptosis. Notably,
this signaling cascade was inhibited or reversed by lovastatin,
thus suggesting that the effects of lovastatin may be mediated via
this pathway.
In the present study, PTK2B expression was
silenced using siRNA in order to observe whether this would alter
the effects of lovastatin. Notably, knockdown of PTK2B
decreased the downstream expression of p-AKT, eNOS and BCL2, and
increased the expression of BAX and cleaved caspase-3, thus
confirming that PTK2B plays an important role in this cascade. In
addition, knockdown of PTK2B markedly diminished the effect of
ox-LDL on this cascade, including the extent to which p-AKT, eNOS
and BCL2 were downregulated, and BAX and cleaved caspase-3 were
upregulated. These results suggested that PTK2B may exert
anti-apoptotic effects against ox-LDL-induced cytotoxicity.
Consistent with this, the effects of ox-LDL on HUVEC viability and
apoptosis were markedly reduced following knockdown of
PTK2B. Therefore, the upregulation of PTK2B served an
important role in the ability of lovastatin to protect against
ox-LDL-induced cell injury. This result is consistent with the
findings of previous studies, which demonstrated that PTK2B
mediates anti-apoptotic AKT signaling (40), and that upregulation of PTK2B
increased cell viability and mobility (51–53).
However, the present study had certain limitations.
A number of genes were altered by lovastatin treatment; however, it
is difficult to distinguish the ‘driver’ from the ‘passenger’
genes, since certain alterations may not be associated with the
pharmacological effect of lovastatin. In addition, full integration
of the gene profile according to a limited functional study of
these genes was challenging, although certain bioinformatics tools
were used, including KEGG and DAVID pathway analysis. Furthermore,
due to the inherent limitations of microarray and bioinformatics
analyses, certain observed alterations may not actually be
significant. Therefore, further studies are required to fully
analyze the microarray data and to present a comprehensive
mechanistic view of the effects of lovastatin on endothelial
cells.
In conclusion, the present study investigated the
effects of lovastatin on gene expression in HUVECs, and
demonstrated that the expression levels of a number of genes were
regulated by lovastatin. In addition, certain genes were found to
be closely associated with cell survival, in particular
PTK2B and BCL2, which were upregulated. Thus, it was
hypothesized that this effect of lovastatin may be beneficial
against endothelial cell injury induced by ox-LDL, a known risk
factor for endothelial dysfunction and atherosclerosis. Lovastatin
was shown to protect against ox-LDL-induced cytotoxicity, and this
protective effect was exerted via the regulation of the signaling
cascade involving PTK2B, p-AKT, eNOS, BCL2, BAX and caspase-3. The
present study demonstrated that high-throughput screening
technology may be considered a useful tool for mechanistic analyses
of drugs.
References
|
1
|
Liao JK and Laufs U: Pleiotropic effects
of statins. Annu Rev Pharmacol Toxicol. 45:89–118. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Goldstein JL and Brown MS: Regulation of
the mevalonate pathway. Nature. 343:425–430. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ganesh SK, Nass CM and Blumenthal RS:
Anti-atherosclerotic effects of statins: Lessons from prevention
trials. J Cardiovasc Risk. 10:155–159. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
LaRosa JC: Statins and risk of coronary
heart disease. JAMA. 283:2935–2936. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Clemenson ND: Statins and risk of coronary
heart disease. JAMA. 283:29352000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Klag MJ, Ford DE, Mead LA, He J, Whelton
PK, Liang KY and Levine DM: Serum cholesterol in young men and
subsequent cardiovascular disease. N Engl J Med. 328:313–318. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Goldstein JL and Brown MS: A century of
cholesterol and coronaries: From plaques to genes to statins. Cell.
26:161–72. 2015. View Article : Google Scholar
|
|
8
|
Massy ZA, Keane WF and Kasiske BL:
Inhibition of the mevalonate pathway: Benefits beyond cholesterol
reduction? Lancet. 347:102–103. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wierzbicki AS, Poston R and Ferro A: The
lipid and non-lipid effects of statins. Pharmacol Ther. 99:95–112.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Influence of pravastatin and plasma lipids
on clinical events in the West of Scotland Coronary Prevention
Study (WOSCOPS). Circulation. 97:1440–1445. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Laufs U: Beyond lipid-lowering: Effects of
statins on endothelial nitric oxide. Eur J Clin Pharmacol.
58:719–731. 2003.PubMed/NCBI
|
|
12
|
Hilbert T, Poth J, Frede S, Klaschik S,
Hoeft A, Baumgarten G and Knuefermann P: Anti-atherogenic effects
of statins: Impact on angiopoietin-2 release from endothelial
cells. Biochem Pharmacol. 86:1452–1460. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Moncada S, Palmer RM and Higgs EA: Nitric
oxide: Physiology, pathophysiology and pharmacology. Pharmacol Rev.
43:109–142. 1991.PubMed/NCBI
|
|
14
|
Rabelink TJ and Luscher TF: Endothelial
nitric oxide synthase: Host defense enzyme of the endothelium?
Arterioscler Thromb Vasc Biol. 26:267–271. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sukhova GK, Williams JK and Libby P:
Statins reduce inflammation in atheroma of nonhuman primates
independent of effects on serum cholesterol. Arterioscler Thromb
Vasc Biol. 22:1452–1458. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Giroux LM, Davignon J and Naruszewicz M:
Simvastatin inhibits the oxidation of low-density lipoproteins by
activated human monocyte-derived macrophages. Biochim Biophys Acta.
1165:335–338. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Arnaud C, Braunersreuther V and Mach F:
Toward immunomodulatory and anti-inflammatory properties of
statins. Trends Cardiovasc Med. 15:202–206. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kumai T, Matsumoto N, Koitabashi Y, Takeba
Y, Oonuma S, Sekine S, Tadokoro M and Kobayashi S: Pleiotropic
effects of 3-hydroxy-3-methylglutaryl-coenzyme A reductase
inhibitors: Candidate mechanisms for anti-lipid deposition in blood
vessels. Curr Med Chem Cardiovasc Hematol Agents. 3:195–201. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Rasmussen LM, Hansen PR, Nabipour MT,
Olesen P, Kristiansen MT and Ledet T: Diverse effects of inhibition
of 3-hydroxy-3-methylglutaryl-CoA reductase on the expression of
VCAM-1 and E-selectin in endothelial cells. Biochem J. 360:363–370.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liles WC and Kain KC: Endothelial
activation and dysfunction in the pathogenesis of microvascular
obstruction in severe malaria-a viable target for therapeutic
adjunctive intervention. J Infect Dis. 210:163–164. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Alvarez De Sotomayor M, Herrera MD,
Marhuenda E and Andriantsitohaina R: Characterization of
endothelial factors involved in the vasodilatory effect of
simvastatin in aorta and small mesenteric artery of the rat. Br J
Pharmacol. 131:1179–1187. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bonetti PO, Lerman LO and Lerman A:
Endothelial dysfunction: A marker of atherosclerotic risk.
Arterioscler Thromb Vasc Biol. 23:168–175. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Matthys KE and Bult H: Nitric oxide
function in atherosclerosis. Mediators Inflamm. 6:3–21. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ross R: The pathogenesis of
atherosclerosis: A perspective for the 1990s. Nature. 362:801–809.
1993. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Noll G, Tschudi M, Nava E and Lüscher TF:
Endothelium and high blood pressure. Int J Microcirc Clin Exp.
17:273–279. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kita T, Brown MS and Goldstein JL:
Feedback regulation of 3-hydroxy-3-methylglutaryl coenzyme A
reductase in livers of mice treated with mevinolin, a competitive
inhibitor of the reductase. J Clin Invest. 66:1094–1100. 1980.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Huang TH, Peng G, Li GQ, Yamahara J,
Roufogalis BD and Li Y: Salacia oblonga root improves postprandial
hyperlipidemia and hepatic steatosis in Zucker diabetic fatty rats:
Activation of PPAR-alpha. Toxicol Appl Pharmacol. 210:225–235.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hitosugi T, Kang S, Vander Heiden MG,
Chung TW, Elf S, Lythgoe K, Dong S, Lonial S, Wang X, Chen GZ, et
al: Tyrosine phosphorylation inhibits PKM2 to promote the Warburg
effect and tumor growth. Sci Signal. 2:ra732009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lang F, Henke G, Embark HM, Waldegger S,
Palmada M, Böhmer C and Vallon V: Regulation of channels by the
serum and glucocorticoid-inducible kinase - implications for
transport, excitability and cell proliferation. Cell Physiol
Biochem. 13:41–50. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Husain M, Jiang L, See V, Bein K, Simons
M, Alper SL and Rosenberg RD: Am J Physiol. 272:C1947–C1959.
1997.PubMed/NCBI
|
|
31
|
Gadepalli R, Singh NK, Kundumani-Sridharan
V, Heckle MR and Rao GN: Novel role of proline-rich nonreceptor
tyrosine kinase 2 in vascular wall remodeling after balloon injury.
Arterioscler Thromb Vasc Biol. 32:2652–2661. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Slattery ML, Lundgreen A and Wolff RK: MAP
kinase genes and colon and rectal cancer. Carcinogenesis.
33:2398–2408. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Vogler M: Targeting BCL2-proteins for the
treatment of solid tumours. Adv Med. 2014:9436482014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Tse KW, Lin KB, Dang-Lawson M,
Guzman-Perez A, Aspnes GE, Buckbinder L and Gold MR: Small molecule
inhibitors of the Pyk2 and FAK kinases modulate
chemoattractant-induced migration, adhesion and Akt activation in
follicular and marginal zone B cells. Cell Immunol. 275:47–54.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Maddika S, Ande SR, Panigrahi S,
Paranjothy T, Weglarczyk K, Zuse A, Eshraghi M, Manda KD, Wiechec E
and Los M: Cell survival, cell death and cell cycle pathways are
interconnected: implications for cancer therapy. Drug Resist Updat.
10:13–29. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Nemoto S, Kobayashi T, Taguchi K,
Matsumoto T and Kamata K: Losartan improves aortic
endothelium-dependent relaxation via proline-rich tyrosine kinase
2/Src/Akt pathway in type 2 diabetic Goto-Kakizaki rats. Am J
Physiol Heart Circ Physiol. 301:H2383–H2394. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hilbert T, Poth J, Frede S, Klaschik S,
Hoeft A, Baumgarten G and Knuefermann P: Anti-atherogenic effects
of statins: Impact on angiopoietin-2 release from endothelial
cells. Biochem Pharmacol. 86:1452–1460. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wang Y, Zhou D, Phung S, Masri S, Smith D
and Chen S: SGK3 is an estrogen-inducible kinase promoting
estrogen-mediated survival of breast cancer cells. Mol Endocrinol.
25:72–82. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Maier G, Palmada M, Rajamanickam J,
Shumilina E, Böhmer C and Lang F: Upregulation of HERG channels by
the serum and glucocorticoid inducible kinase isoform SGK3. Cell
Physiol Biochem. 18:177–186. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Burdick AD, Ivnitski-Steele ID, Lauer FT
and Burchiel SW: PYK2 mediates anti-apoptotic AKT signaling in
response to benzo[a]pyrene diol epoxide in mammary epithelial
cells. Carcinogenesis. 27:2331–2340. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Cao J, Chen Y, Fu J, Qian YW, Ren YB, Su
B, Luo T, Dai RY, Huang L, Yan JJ, et al: High expression of
proline-rich tyrosine kinase 2 is associated with poor survival of
hepatocellular carcinoma via regulating phosphatidylinositol
3-kinase/AKT pathway. Ann Surg Oncol. 20(Suppl 3): S312–S323. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Daou GB and Srivastava AK: Reactive oxygen
species mediate Endothelin-1-induced activation of ERK1/2, PKB and
Pyk2 signaling, as well as protein synthesis, in vascular smooth
muscle cells. Free Radic Biol Med. 37:208–215. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Sun CK, Man K, Ng KT, Ho JW, Lim ZX, Cheng
Q, Lo CM, Poon RT and Fan ST: Proline-rich tyrosine kinase 2 (Pyk2)
promotes proliferation and invasiveness of hepatocellular carcinoma
cells through c-Src/ERK activation. Carcinogenesis. 29:2096–2105.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Ross R: Atherosclerosis - an inflammatory
disease. N Engl J Med. 340:115–126. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Kugiyama K, Kerns SA, Morrisett JD,
Roberts R and Henry PD: Impairment of endothelium-dependent
arterial relaxation by lysolecithin in modified low-density
lipoproteins. Nature. 344:160–162. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Simon BC, Cunningham LD and Cohen RA:
Oxidized low density lipoproteins cause contraction and inhibit
endothelium-dependent relaxation in the pig coronary artery. J Clin
Invest. 86:75–79. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Dimmeler S, Haendeler J, Galle J and
Zeiher AM: Oxidized low-density lipoprotein induces apoptosis of
human endothelial cells by activation of CPP32-like proteases. A
mechanistic clue to the ‘response to injury’ hypothesis.
Circulation. 95:1760–1763. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Harada-Shiba M, Kinoshita M, Kamido H and
Shimokado K: Oxidized low density lipoprotein induces apoptosis in
cultured human umbilical vein endothelial cells by common and
unique mechanisms. J Biol Chem. 273:9681–9687. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Chavakis E, Dernbach E, Hermann C, Mondorf
UF, Zeiher AM and Dimmeler S: Oxidized LDL inhibits vascular
endothelial growth factor-induced endothelial cell migration by an
inhibitory effect on the Akt/endothelial nitric oxide synthase
pathway. Circulation. 103:2102–2107. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Kotamraju S, Hogg N, Joseph J, Keefer LK
and Kalyanaraman B: Inhibition of oxidized low-density
lipoprotein-induced apoptosis in endothelial cells by nitric oxide:
Peroxyl radical scavenging as an antiapoptotic mechanism. J Biol
Chem. 276:17316–17323. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Sun CK, Ng KT, Lim ZX, Cheng Q, Lo CM,
Poon RT, Man K, Wong N and Fan ST: Proline-rich tyrosine kinase 2
(Pyk2) promotes cell motility of hepatocellular carcinoma through
induction of epithelial to mesenchymal transition. PLoS One.
6:e188782011. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Lim ST, Miller NL, Nam JO, Chen XL, Lim Y
and Schlaepfer DD: Pyk2 inhibition of p53 as an adaptive and
intrinsic mechanism facilitating cell proliferation and survival. J
Biol Chem. 285:1743–1753. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Yoon H, Choi YL, Song JY, Do I, Kang SY,
Ko YH, Song S and Kim BG: Targeted inhibition of FAK, PYK2 and
BCL-XL synergistically enhances apoptosis in ovarian clear cell
carcinoma cell lines. PLoS One. 9:e885872014. View Article : Google Scholar : PubMed/NCBI
|