Introduction
Obstructive sleep apnea (OSA) is a commonly
occurring disease, which is characterized by repeated upper airway
collapse, increased breathing effort and decreased oxygen
saturation. Exposure to chronic intermittent hypoxia (CIH), as
observed in patients with OSA, has been found to be associated with
a variety of cardiovascular (CV) diseases, including coronary heart
disease, hypertension and chronic heart failure (1,2). On the
basis of previous results reported in human and animal research,
oxidative stress (3), inflammation
(4) and sympathetic activation may
be the key pathological processes involved in the pathogenesis of
OSA-related CV diseases. However, the optimal treatment for CIH
induced CV injuries has yet to be determined.
Complement regulatory protein, known as C1 inhibitor
(C1INH), a serine protease inhibitor, is considered to be the only
natural inhibitor of the complement pathway, and functions as a
negative regulator of the complement system (5). The use of a highly specific and potent
synthetic inhibitor of the activated C1 complement to block the
classical complement pathway may be an effective strategy for the
preservation of cells from hypoxic injury. Results of previous
studies conducted by the present authors have demonstrated that the
administration of C1INH protected cardiomyocytes against
ischemia/reperfusion (I/R) injury-induced apoptosis (5,6).
Therefore, the current study was conducted to investigate the
hypothesis that C1INH may also exert protective effects against
CIH-induced cardiomyocyte injury.
Materials and methods
Model of CIH in rats
Male Sprague-Dawley rats (n=60; weight, 200–250 g;
age, 8–10 weeks) were purchased from Wuhan University (Wuhan,
China). The rats were housed in an animal care center under a 12-h
dark:light cycle (temperature, 20–26°C; humidity, 40–70%) and were
allowed free access to standard chow and tap water. The rats were
randomly divided into three large groups (n=20/group) as follows:
i) Normal control (NC) group; ii) CIH group; and iii) CIH plus
C1INH (CIH + C1INH) group. The rats were then further subdivided
into six smaller groups (n=10). The CIH rat model employed in the
present study has been reported previously (7). Briefly, the rats were housed in a cage
placed in a chamber of CIH. The chamber of CIH was a glass cube
storehouse (width, 4 mm; length, 28×15×15 cm long), with a hole at
one end (10 cm diameter) through which the animals were placed or
removed, and two holes at the side (5 mm diameter) which was used
to input low oxygen gas and air respectively. The concentration of
the gas was regulated by an electromagnetic valve switch circuit
which was controlled by a single chip microcomputer program, so
that the input of low oxygen gas and air could be adjusted. The
inspired oxygen fraction was altered from 21 to 4–5% every 2 min
and sustained at the lower level for 20 sec. The intermittent
hypoxia events were carried out for 4 or 8 weeks. Rats in the NC
group were treated with 21% O2 in a separate chamber.
Rats in the CIH + C1INH group were also intravenously treated with
C1INH (40 U/kg, 1 U 0.15 mg) per treatment, administered twice a
week for 4 or 8 weeks (the effect of C1INH lasts for ≥3 days)
(5). An injection of saline (0.5 ml
per treatment) was administered to the NC and CIH groups. Data were
collected after 4 or 8 weeks. The rats were anesthetized with ether
inhalation, and the whole blood was collected prior to the
measurement of cardiac function. The rats were then sacrificed by
cervical dislocation. Tissue samples from the left ventricular were
obtained. Some tissue samples were fixed in 10% paraformaldehyde,
dehydrated, waxed, and embedded in paraffin, and others were
homogenized.
Analyses of neutrophil infiltration,
myocardial apoptosis, and cardiac C3 levels after CIH were
performed, as described below
Homogenates were centrifuged at 36,000 × g at 4°C
for 30 min. The supernatants were stored at −80°C.
Cardiac function
Echocardiographic examination was performed in
conscious rats at 4 or 8 weeks after the initiation of CIH using a
Sequoia 512 Echocardiographic System (Siemens Healthcare, Mountain
View, CA, USA) equipped with an 8-MHz linear transducer. The
anterior chest area of rats was shaved, and 2D images and M-mode
tracings were obtained. The following parameters were measured:
Left ventricular end-diastolic dimension (LVDd, in cm) and left
ventricular end-systolic dimension (LVDs, in cm), and the results
were used to calculate the left ventricular end-systolic volume
[LVESV; 7.0/(2.4 + LVDs) × LVDs3], left ventricular
end-diastolic volume [LVEDV; 7.0/(2.4 + LVDd) × LVDd3],
left ventricular ejection fraction [LVEF (%); (LVEDV - LVESV)/LVEDV
× 100] and left ventricular fractional shortening [LVFS (%); (LVDd
- LVDs)/LVDd × 100].
Detection of myeloperoxidase (MPO)
activity
As a marker of neutrophil infiltration, MPO activity
in the myocardium and serum was determined using an MPO detection
kit (Nanjing Jiancheng Bioengineering Institute, Nanjing, China)
according to the manufacturer's protocol.
Terminal deoxynucleotidyl
transferase-mediated dUTP nick end labeling (TUNEL)
TUNEL analysis was employed to evaluate myocardial
apoptosis in the rats of each group. Briefly, myocardial tissue
samples (5 µm) were rinsed in 1X phosphate-buffered saline (PBS; pH
7.4) and fixed for 30 min in 4% paraformaldehyde in 1X PBS (pH 7.4)
at room temperature. Subsequent to rinsing with 1X PBS (pH 7.4),
the cells were permeabilized in 0.1% Triton X-100 and 0.1% sodium
citrate, then rinsed twice in PBS (pH 7.4). After washing with 1X
PBS (pH 7.4), the cells were mounted in Gummi (Sinopharm Chemical
Reagent, Co., Ltd.), an antifade agent, and visualized with an
Olympus light microscope (Leica Microsystems GmbH, Wetzlar,
Germany). Further characterization of apoptosis was achieved using
a commercially available in situ cell death detection kit
(Roche Diagnostics, Indianapolis, IN, USA) to detect DNA strand
breaks using the TUNEL reagent, according to the manufacturer's
protocol.
Western blot analysis
Left ventricular free wall tissue sample was
obtained, and 100 mg myocardial tissue was placed into 1 ml lysis
buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% NP-40, 0.1% SDS, 1
mmol/l EDTA, 0.5 mmol/l PMSF, 4 g/ml peptide enzyme) prior to
homogenization on ice. The sample was then centrifugated at 4°C
(12,000 × g for 20 min), to obtain the supernatant, and the
concentration of protein was detected by the Lowry method. The
concentration of the total protein was adjusted to ~50 µg/15 µl.
Protein samples (50 µg)were subjected to electrophoresis using a
10% sodium dodecyl sulfate-Tris-glycine polyacrylamide gel. The
proteins were electrophoretically transferred to nitrocellulose
membranes and then blocked with 5% fat-free milk in 1X PBS (pH
7.4), containing 0.05% Tween at 4°C overnight. The blocked
membranes were incubated with anti-C3 (cat. no. sc-20137; Santa
Cruz Biotechnology, Inc., Dallas, TX, USA), B-cell lymphoma 2
(Bcl-2; cat. no. 2870-P; Cell Signaling Technology, Inc., Danvers,
MA, USA), Bcl-2-associated X protein (Bax; cat. no. BS2538;
Bioworld Technology, Inc., St. Louis Park, MN, USA), caspase-3
(cat. no. 19677-1-AP; Bioworld Technology, Inc.), β-actin (Wuhan
Boster Biological Engineering Co., Ltd., Wuhan, China; cat. no.
BM0627), or GAPDH (cat. no. AB-P-R001; Hangzhou Greensky Biological
Tech. Co., Ltd., Hangzhou, China) antibodies (all diluted 1:2,000)
in 5% fat-free milk for 2 h at room temperature, washed with 1X PBS
(pH 7.4) for 20 min, and incubated with a 1:10,000 dilution of
Immunopure anti-goat IgG (H+L) conjugated with horseradish
peroxidase (cat. no. BA1054; Wuhan Boster Biological Technology,
Ltd.) for 2 h at room temperature. Development of protein bands was
performed using a SuperSignal Chemiluminescent Substrate kit
(Thermo Fisher Scientific, Inc., Waltham, MA, USA). The blots were
quantified by grey value using analysis software (Bandscan 4.3;
BioMarin Pharmaceuticals Inc., Novato, CA, USA).
Reverse transcription-polymerase chain
reaction (RT-qPCR)
The tissue samples were homogenized with liquid
nitrogen, and total RNA was extracted in 1 ml TRIzol (Invitrogen;
Thermo Fisher Scientific, Inc.). The concentration of the total RNA
was detected by spectrophotometer. A First Chain Synthesis kit for
cDNA (Fermentas) was used for reverse transcription. SYBR
Green/Fluorescein qPCR Master Mix(2X) (Fermentas; Thermo Fisher
Scientific, Inc., Pittsburg, PA, USA) and Ex Taq™ (Takara
Bio, Inc., Otsu, Japan) were used for RT-qPCR. RT-qPCR was employed
to determine mRNA expression levels using the following primers:
5′-CTGCCTGTCCTTCAAAGTCCACC-3′ and 5′-TCAGCATTCCATCGTCCTTCTCC-3′ for
C3; and 5′-CACGATGGAGGGGCCGGACTCATC-3′ and
5′-TAAAGACCTCTATGCCAACACAGT-3′ for β-actin. The reaction was
carried out on an ABI Real-Time qPCR instrument. The reaction
conditions were as follows: 94°C for 4 min, 94°C for 30 sec, 56°C
for 30 sec, 72°C for 25 sec, and 30 cycles at 72°C for 4 min.
RT-PCR experiments were performed with 1 g of total RNA, followed
by 40 cycles of PCR amplification. The expression levels were
quantified using changes in the fluorescence signal through the
analysis of the Cq value and standard curve; the starting template
was quantitatively analyzed with RUO ViiA™ 7 software (Thermo
Fisher Scientific, Inc.).
Statistical analysis
Data were expressed as the mean ± standard error of
the mean, and analyzed using one-way analysis of variance followed
by post-hoc analysis to compared the means. P<0.05 was
considered to indicate a statistically significant difference.
Results
C1INH preserves cardiac function
following CIH
C1INH administration significantly preserved cardiac
function in rats in the CIH + C1INH group, compared with those in
the untreated (CIH) group, as reflected by a significant increase
in ejection fraction and fractional shortening, as well as
decreased LVDd, LVDs, LVEDV and LVESV after C1INH treatment at 4
weeks (P<0.05) and 8 weeks (P<0.05) after intervention
(Table I).
 | Table I.Echocardiographic data. |
Table I.
Echocardiographic data.
| Variable | Normal | CIH (4 weeks) | CIH + C1INH (4
weeks) | CIH (8 weeks) | CIH + C1INH (8
weeks) |
|---|
| LVDd (cm) |
0.556±0.050 |
0.580±0.048 |
0.563±0.476a |
0.595±0.474 |
0.580±0.445b |
| LVDs (cm) |
0.345±0.046 |
0.396±0.082 |
0.391±0.048a |
0.427±0.064 |
0.394±0.044b |
| LVEDV (ml) |
0.095±0.050 |
0.103±0.018 |
0.098±0.019a |
0.144±0.031 |
0.133±0.025b |
| LVESV (ml) |
0.504±0.036 |
0.537±0.046 |
0.527±0.048a |
0.572±0.064 |
0.557±0.061b |
| LVEF (%) |
82.04±3.11 |
77.49±3.50 |
78.88±2.53a |
75.74±3.09 |
76.74±3.13b |
| LVFS (%) |
41.14±2.39 |
38.98±2.36 |
39.78±2.49a |
38.16±2.16 |
38.81±2.13b |
C1INH reduces inflammation in the
myocardium following the induction of CIH
In the rat model of CIH, administration of C1INH was
able to suppress the overactivated inflammatory response at 4 and 8
weeks, compared with that in the CIH group. This was revealed by
the significant reduction of myocardial and serum MPO activity
observed in rats in the C1INH intervention group at 4 and 8 weeks
(P<0.05; Table II).
| Table II.Effect of C1INH on MPO activity. |
Table II.
Effect of C1INH on MPO activity.
| Group | MPO in myocardium
(U/l wet weight) | MPO in blood (U/l wet
weight) |
|---|
| Normal control |
1.583±0.059 |
63.58±3.476 |
| 4 weeks |
|
|
| CIH |
2.580±0.128 |
90.69±4.048 |
| CIH +
C1INH |
2.210±0.062a |
88.76±4.019a |
| 8 weeks |
|
|
| CIH |
4.014±0.11 |
161.22±3.00 |
| CIH +
C1INH |
3.286±0.041b |
131.98±4.53b |
C1INH attenuates CIH-induced
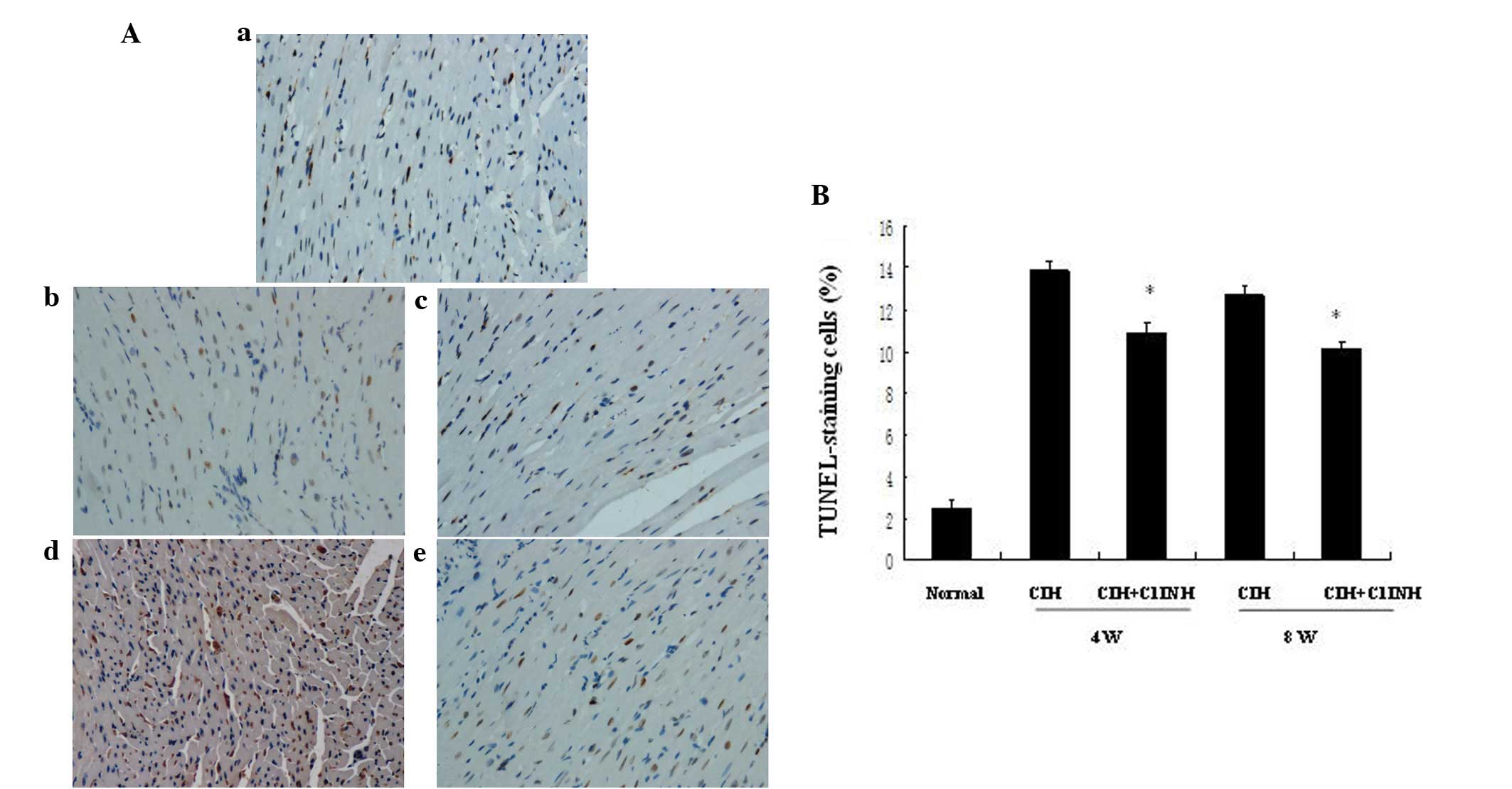
cardiomyocyte apoptosis
Cardiomyocyte apoptosis may be induced by numerous
stimuli, including hypoxia. Administration of C1INH has been
reported to be a potential cardioprotective strategy for
hypoxia/reoxygenation-induced tissue injury (5). The presence of increased numbers of
apoptotic cells in the myocardium was observable following 4 and 8
weeks of CIH (Fig. 1). C1INH, when
administered at concentrations of 100–150 µg/ml (within the
physiological concentration range in human plasma), markedly
attenuated CIH-induced myocardial cellular apoptosis at both 4 and
8 weeks (Fig. 1). Furthermore, the
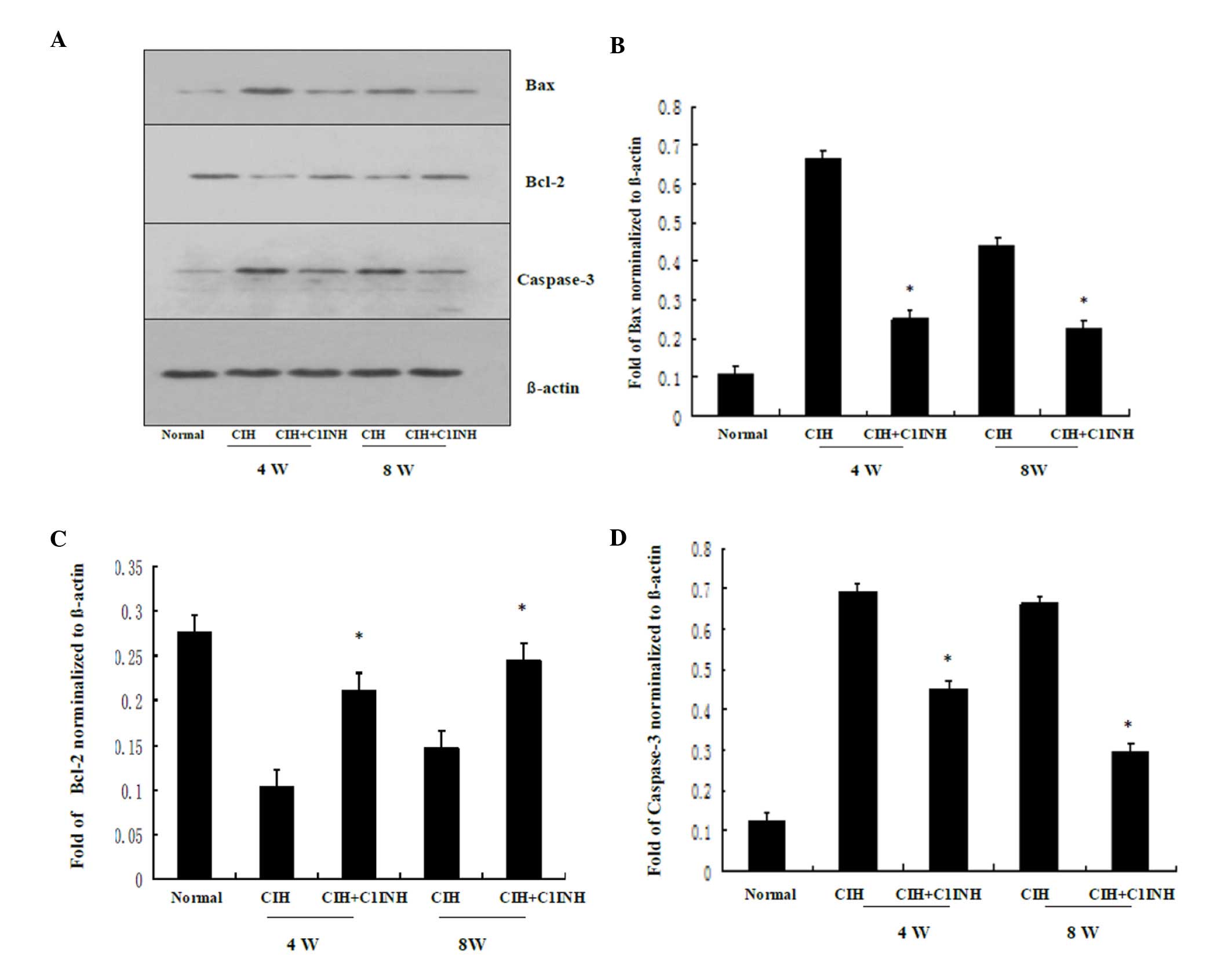
protein expression levels of Bax induced by CIH were downregulated
following treatment with C1INH, and Bcl-2 protein expression
levels, which were reduced by CIH were upregulated by treatment
with C1INH (Fig. 2). In addition,
CIH-induced increased in myocardial caspase-3 protein expression
were attenuated by treatment with C1INH (Fig. 2). The aforementioned data demonstrate
that C1INH exerts an anti-apoptotic effect during CIH-induced
myocardial injury.
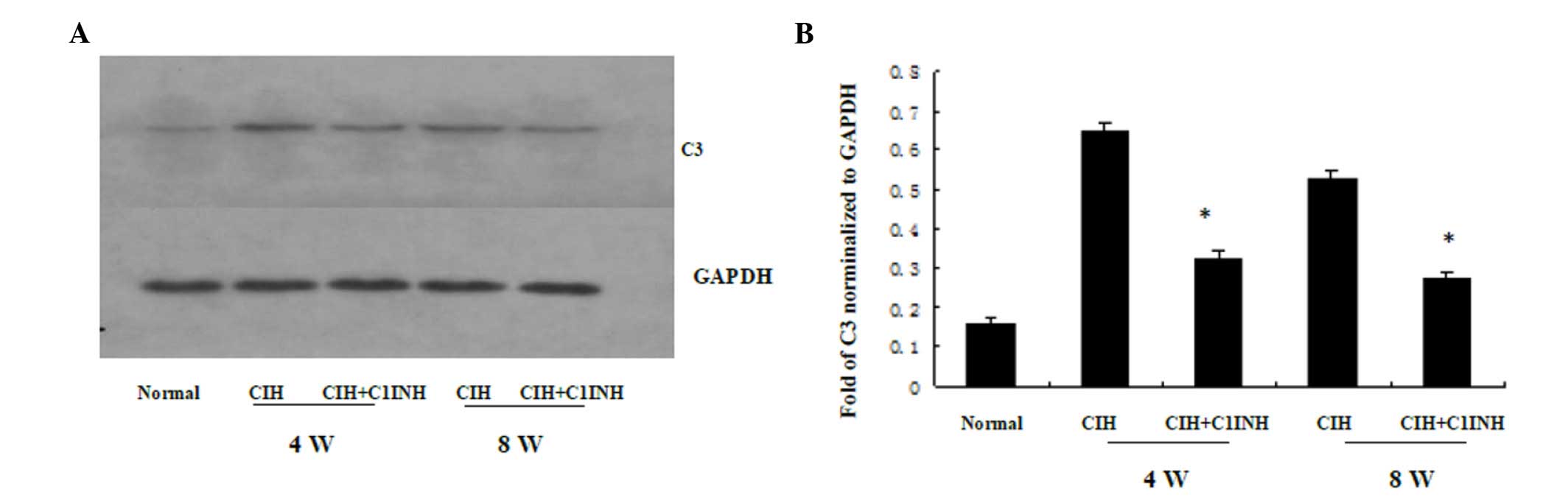
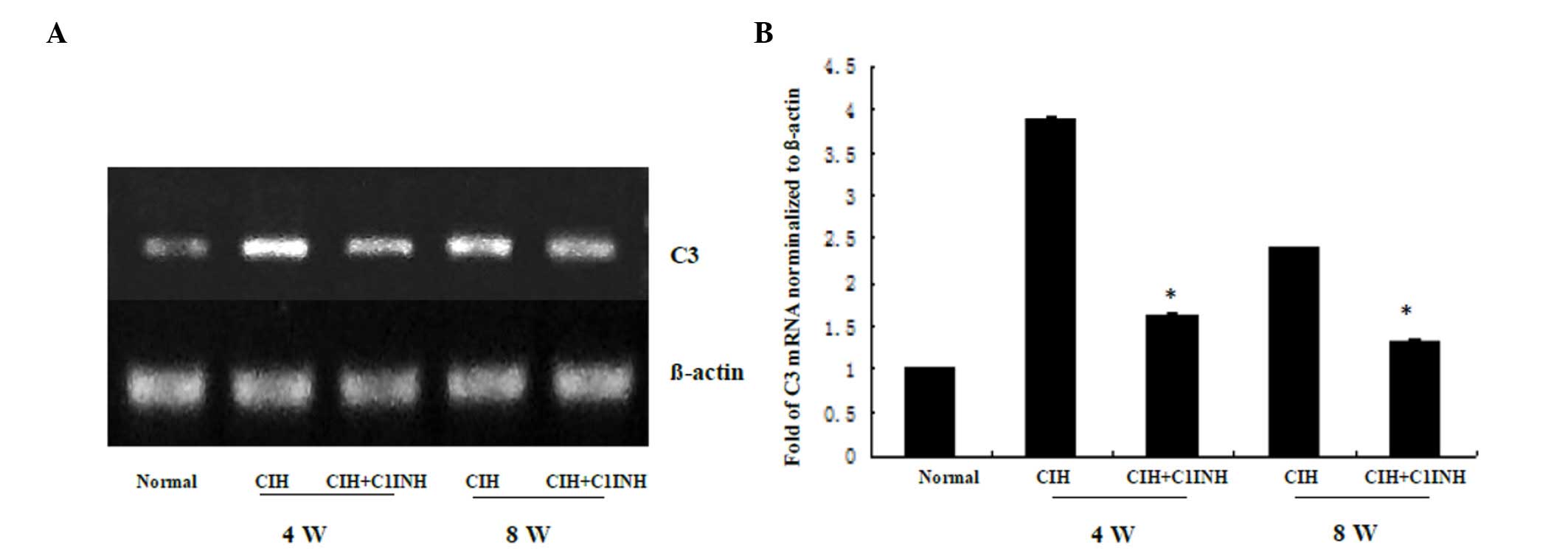
C1INH inhibits CIH-induced cytoplasmic
C3 synthesis in cardiac myocytes
The results of western blotting and RT-PCR analyses
revealed that CIH at 4 and 8 weeks was associated with increased
cytoplasmic C3 protein and mRNA expression levels (Figs. 3 and 4, respectively). A single dose of C1INH
(100 µg/ml) markedly reduced the C3 protein expression and
cytoplasmic C3 mRNA expression induced by CIH. These results
indicate that, in addition to its physiological function as the
only inhibitor of the classical complement pathway proteases (C1r
and C1s), C1INH may also exert a novel protective function against
CIH-induced C3 synthesis in the myocardium.
Discussion
CIH has been suggested to negatively affect left
ventricular function, and oxidative stress has been recognized as
an important mediator for myocardial damage during this
pathophysiological process (8). The
major observation made within the current study was that C1INH
supplementation during CIH preserved myocardial function, and
attenuated myocardial apoptosis through the inhibition of
complement activity.
Chronic intermittent hypoxia/reoxygenation is
characteristic of CIH, and several studies support the notion that
complement activation is involved in the pathogenesis of
hypoxia/reoxygenation injury, and that complement inhibition
reduces the extent of injury (5,8,9). In support of this hypothesis, studies
have demonstrated that the localization/deposition of complement
components (such as C1q, C3, C4 and C5) occurs in hypoxic
myocardium (10–13). Furthermore, mice deficient in C3, C5
or C6 have less severe I/R-induced myocardial injury compared with
those of the wild-type (11). In
addition, depletion (with cobra venom factor) or inhibition (with
C1INH or soluble complement receptor 1) of complement activation
attenuates cardiomyocyte injury during the hypoxia/reoxygenation
process (12). C1INH has been
demonstrated to block the classical complement pathway by binding
to the activated C1 complex (C1q, C1s and C1r) (10), which results in dissociation of the
complex. Finally, inactivated C1INH can be synthesized locally in
inflamed tissue such as the I/R myocardium (13). The aforementioned data collectively
suggest that activation of the complement system serves an
important role in hypoxia/reoxygenation-induced myocardial
injury.
Apoptotic cells are able to directly activate the
classical complement pathway by binding C1 (14). Therefore, apoptosis contributes to
hypoxia/reoxygenation-induced cell injury and organ dysfunction,
and may serve as a target for the potential protective effects of
acute phase proteins (15).
Similarly, cellular apoptosis has been clearly documented in
cultured neonatal rat cardiomyocytes experiencing hypoxia (16).
C1INH, a serine protease inhibitor, which has been
used in the treatment of patients with paroxysmal nocturnal
hemoglobinuria, has been evaluated for its potential clinical
utility in studies of sepsis, I/R injury and capillary leak
(17,18). It has been considered as a major
inhibitor of complement system activation. C1INH is an acute phase
protein that has a mean plasma level of ~250 mg/l and can be
markedly upregulated up to 2.5-fold during the inflammation process
(19). Previous data indicate that
C1INH exerts an anti-apoptotic effect on vascular endothelial cell
injury induced by gram-negative lipopolysaccharide (5). Therefore, inhibition of complement
activation may attenuate CIH-induced myocardial apoptosis. In the
present study, using an established CIH rat model, it was observed
that C1INH was able to reduce myocardial apoptosis during CIH.
Similarly, C1INH has been indicated to inhibit apoptosis in an H9c2
rat myocyte cell line in vitro (5), suggesting that a cardioprotective
pathway may be initiated by C1INH. Furthermore, as C1INH is able to
attenuate the apoptosis of H9c2 rat cardiomyocytes in vitro,
it may be suggested that its anti-apoptotic effect is exerted
through a mechanism that functions independently from the mediation
of complement system activation (5).
The mechanisms underlying the potential cardioprotective and
anti-apoptotic effects of C1INH in myocardial cells have yet to be
determined, and further investigation is warranted.
Although the activation of the complement system
subsequent to hypoxia/reoxygenation injury has been attributed to
various pathways, including the mannose-binding lectin pathway and
the classical pathway for the heart, in addition to the alternative
pathway for the kidney (20,21), complement C3 has been recognized as
the most abundant complement protein in the circulation and is
vital in the complement cascade. The classical, alternative and
lectin pathways are activated via cleavage of the C3 molecule into
various fragments that possess opsonic, chemotactic, anaphylotoxic
and immunoregulatory properties. Typically, the liver produces the
highest C3 levels under normal conditions; however, heart tissue
undergoing hypoxia/reoxygenation produces substantially higher
levels of C3 component compared with those in the liver (22). Local C3 synthesis may be an important
pathological event during the complement-dependent processes,
involving mediation of hypoxia/reoxygenation injury and
immune-mediated tissue injury. Incremental increases in circulating
C3a, and to a lesser extent C5a, are attenuated by C1INH treatment
(23). The results of the present
study also demonstrate that C1INH application is able to suppress
the upregulation of cytoplasmic C3 mRNA expression levels and
protein synthesis in rat myocardium induced by CIH.
In addition, the inflammatory response in the
myocardium involves the local production of chemotactic factors,
neutrophil infiltration and activation, cytokine production and the
expression of adhesion molecules. Thus, the ability of neutrophils
to adhere to cardiac myocytes is enhanced, and the local activation
of the complement system is induced (24). The results of the present study
indicate that C1INH is required for the inhibition of neutrophil
influx into the myocardium. Such effects on inflammation are most
likely the result of direct prevention of early primary apoptosis
or complement activation. Alternatively, direct anti-inflammatory
effects of C1INH may also be involved. A reduction in neutrophil
influx may explain the observed attenuation of apoptosis in rats
treated with C1INH.
Collectively, the above results indicate that C1INH
may exert its cardioprotective effects during CIH-induced
myocardial cell injury and cardiac dysfunction through several
different mechanisms, including the inhibition of complement
activation induced by CIH, inhibition of proinflammatory events
such as neutrophil accumulation, and the inhibition of locally
synthesized C3. Thereby, application of C1INH may directly inhibit
the activation of the complement pathway in hypoxic tissue. In
addition, C1INH may also exert direct anti-apoptotic effects in
CIH-mediated myocardial cell injury. These results collectively
suggest that treatment with C1INH may provide a useful therapeutic
approach for the treatment of patients with OSA.
Future animal studies are warranted to continue to
enhance the present understanding of the pathogenesis of
OSA-associated CV diseases, perhaps through the investigation of
knock-out and genetically engineered mice or by performing
selective pharmacological interventions. This may enable
identification of the fundamental molecular pathways underlying the
association between OSA and CV diseases, and novel cardioprotective
treatment options may emerge for the relatively large proportion of
patients with OSA who are unable to tolerate continuous positive
airway pressure therapy.
Acknowledgements
This study was supported by a grant from the
National Natural Sciences Foundation in China, awarded to Dr Ke Hu
(grant no. 81370181).
Glossary
Abbreviations
Abbreviations:
|
C1INH
|
C1 inhibitor
|
|
CIH
|
chronic intermittent hypoxia
|
References
|
1
|
Baguet JP, Barone-Rochette G, Tamisier R,
Levy P and Pépin JL: Mechanisms of cardiac dysfunction in
obstructive sleep apnea. Nat Rev Cardiol. 9:679–688. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Beaudin AE, Pun M, Yang C, Nicholl DD,
Steinback CD, Slater DM, Edwards KE Wynne, Hanly PJ, Ahmed SB and
Poulin MJ: Cyclooxygenases 1 and 2 differentially regulate blood
pressure and cerebrovascular responses to acute and chronic
intermittent hypoxia: Implications for sleep apnea. J Am Heart
Assoc. 3:e0008752014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chen L, Zhang J, Gan TX, Chen-Izu Y,
Hasday JD, Karmazyn M, Balke CW and Scharf SM: Left ventricular
dysfunction and associated cellular injury in rats exposed to
chronic intermittent hypoxia. J Appl Physiol (1985). 104:218–223.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yeung HM, Hung MW, Lau CF and Fung ML:
Cardioprotective effects of melatonin against myocardial injuries
induced by chronic intermittent hypoxia in rats. J Pineal Res.
58:12–25. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Fu J, Lin G, Wu Z, Ceng B, Wu Y, Liang G,
Qin G, Li J, Chiu I and Liu D: Anti-apoptotic role for C1 inhibitor
in ischemia/reperfusion-induced myocardial cell injury. Biochem
Biophys Res Commun. 349:504–12. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Fu J, Lin G, Zeng B, Wu Z, Wu Y, Chu H,
Qin G, Liang G, Li J, Gan X, et al: Anti-ischemia/reperfusion of C1
inhibitor in myocardial cell injury via regulation of local
myocardial C3 activity. Biochem Biophys Res Commun. 350:162–168.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chen L, Zhang J, Hu X, Philipson KD and
Scharf SM: The Na+/Ca2+ exchanger-1 mediates
left ventricular dysfunction in mice with chronic intermittent
hypoxia. J Appl Physiol (1985). 109:1675–1685. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Liu D, Zhang D, Scafidi J, Wu X, Cramer CC
and Davis AE: 3rd: C1 inhibitor prevents Gram-negative bacterial
lipopolysaccharide-induced vascular permeability. Blood.
105:2350–2355. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Buerke M..Schwertz H..Seitz W..Meyer
J..Darius H.: Novel small molecule inhibitor of C1s exerts
cardioprotective effects in ischemia–reperfusion injury in rabbits.
J Immunol. 167:5375–5380. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Konings J, Govers-Riemslag JW, Spronk HM,
Waltenberger JL and ten Cate H: Activation of the contact system in
patients with a first acute myocardial infarction. Thromb Res.
132:138–142. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Onat A, Can G, Rezvani R and Cianflone K:
Complement C3 and cleavage products in cardiometabolic risk. Clin
Chim Acta. 412:1171–1179. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Uyar IS, Onal S, Akpinar MB, Gonen I,
Sahin V, Uguz AC and Burma O: Alpha lipoic acid attenuates
inflammatory response during extracorporeal circulation. Cardiovasc
J Afr. 24:322–326. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lu F, Fernandes SM and Davis AE III: The
effect of C1 inhibitor on myocardial ischemia and reperfusion
injury. Cardiovasc Pathol. 22:75–80. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shi H, Williams JA, Guo L, Stampoulis D,
Cordeiro M Francesca and Moss SE: Exposure to the complement C5b-9
complex sensitizes 661W photoreceptor cells to both apoptosis and
necroptosis. Apoptosis. 20:433–443. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ding W and Zhang X, Huang H, Ding N, Zhang
S, Hutchinson SZ and Zhang X: Adiponectin protects rat myocardium
against chronic intermittent hypoxia-induced injury via inhibition
of endoplasmic reticulum stress. PLoS One. 9:e945452014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang M, Lu L, Liu Y, Gu G and Tao R:
FTY720 attenuates hypoxia-reoxygenation-induced apoptosis in
cardiomyocytes. Exp Mol Pathol. 97:218–224. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Caliezi C, Zeerleder S, Redondo M, Regli
B, Rothen HU, Zürcher-Zenklusen R, Rieben R, Devay J, Hack CE,
Lämmle B and Wuillemin WA: C1-inhibitor in patients with severe
sepsis and septic shock: Beneficial effect on renal dysfunction.
Crit Care Med. 30:1722–1728. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Emlen W, Li W and Kirschfink M:
Therapeutic complement inhibition: New developments. Semin Thromb
Hemost. 36:660–668. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bäck J, Lood C, Bengtsson AA, Ekdahl KN
and Nilsson B: Contact activation products are new potential
biomarkers to evaluate the risk of thrombotic events in systemic
lupus erythematosus. Arthritis Res Ther. 15:R2062013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Marcheix B, Carrier M, Martel C, Cossette
M, Pellerin M, Bouchard D and Perrault LP: Effect of pericardial
blood processing on postoperative inflammation and the complement
pathways. Ann Thorac Surg. 85:530–535. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Collard CD, Väkevä A, Büküsoglu C, Zünd G,
Sperati CJ, Colgan SP and Stahl GL: Reoxygenation of hypoxic human
umbilical vein endothelial cells activates the classic complement
pathway. Circulation. 96:326–333. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Devlin LA, Nguyen MD, Figueroa E, Gordon
LE, Feldhoff PW and Lassiter HA: Effects of endotoxin
administration and cerebral hypoxia-ischemia on complement activity
and local transcriptional regulation in neonatal rats. Neurosci
Lett. 390:109–113. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bengtson A, Millocco I, Heideman M and
Berggren H: Altered concentrations of terminal complement
complexes, anaphylatoxins, and leukotrienes in the coronary sinus
during cardiopulmonary bypass. J Cardiothorac Anesth. 3:305–310.
1989. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gluszek J: Ischaemic heart disease and
hypertension in patients with chronic obstructive pulmonary disease
and obstructive sleep apnoea. Pneumonol Alergol Pol. 81:567–574.
2013.(In Polish). PubMed/NCBI
|