Introduction
The H7 subtype of avian-origin influenza viruses
(AIV-H7) initially emerged in Italy in 1902 as H7N7 (1). During its period of evolution, AIV-H7
has repeatedly caused pandemics among poultry and has been
associated with huge losses to livestock. However, AIV-H7
infections in humans have rarely been reported. One of the largest
infections of humans occurred in the Netherlands in 2007 (2); the outbreak was caused by the H7N7
subtype and led to keratitis and other minor symptoms in humans,
although no mortalities were reported. However, in 2013 an AIV-H7N9
epidemic occurred in China. By May 1st, 2014, China had reported
422 confirmed cases (3), including
people suffering from pneumonia, respiratory distress syndrome,
septic shock and other life-threatening diseases (4). The exact death toll had not been
updated in 2014; however, in 2013, >57 mortalities were
reported, with a fatality rate of >33%. Therefore, H7-AIV has
aroused global concern.
Similar to other influenza viruses, AIV-H7 can be
divided into North American and Euro-Asiatic lineages (5); the 2013 outbreak in China belonged to
the Euro-Asiatic lineage (6). The
major glycoprotein of AIV is hemagglutinin (HA), which has an
important role in binding the virus to sialic acid on the membranes
of host cells, including cells in the upper respiratory tract or
erythrocytes (7). Phylogenetic tree
analyses suggested that the HA of H7N9 was derived from a
reassortment of H7N3 in Zhejiang ducks (8). HA, which is a large protein of 560
amino acids, possesses various epitopes that can be divided into
linear and conformational types (9).
The linear epitopes consist of conserved amino acids, whereas the
conformational epitopes contain the adjacent amino acids in space
that may lie far away from the linear epitopes in the primary
sequence (10). The majority of the
HA epitopes are conformational; however, linear epitopes are easier
to express and mimic (11).
Therefore, the present study aimed to predict the potential linear
epitopes of AIV-H7.
The present study was divided into two parts. First,
the linear epitopes of HA were predicted using a combination of
three epitope prediction softwares, including: Artificial Neural
Network based B-cell Epitope Prediction (ABCpred), B-cell Epitope
Prediction (BepiPred) and Linear B-cell Epitope Prediction
(LBtope). Each software constituted a specific algorithm with the
highest predictive accuracy being ≤66% (12). Second, 24 strains of Euro-Asiatic
AIV-H7 that had infected humans or had caused avian pandemics in
the past 30 years (13) were
selected, and the amino acid sequences of HA were compared in order
to identify the conserved region of HA. By using epitope prediction
softwares and comparing reference strains, the present study
effectively screened for invalid and mutant epitopes, which was
time-saving and inexpensive. In addition, preliminary
investigations on the antigenicity of HA, as well as amino acid
mutations and epitopes associated with secondary structure, were
performed and may be considered useful for future research. The
results of the present study may have a profound impact on the
diagnosis and future research of, and design of vaccines for,
AIV-H7, as well as for other virulent viruses such as the Ebola
virus.
Data and methods
Sequence availability and
comparisons
Sequences were obtained from the National Center for
Biotechnology Information database (http://ncbi.nlm.nih.gov) and the Global Initiative on
Sharing Avian Influenza Data (GISAID) database (http://platform.gisaid.org/epi3/frontend). Since
AIV-H7 has frequent variation, 24 strains of Euro-Asiatic lineage
H7 subtype AIV that had triggered serious poultry pandemics or had
infected humans in the past 30 years were selected (13). Of the 24 strains, two represented the
latest H7N9 virus, and were selected from 94 of the latest complete
records of H7N9 in the GISAID database.
ClustalW (www.ebi.ac.uk/Tools/msa/clustalw2/) was used to
compare the HA amino acid sequence of 24 strains and to identify
their conserved region. The accession numbers of the HA proteins of
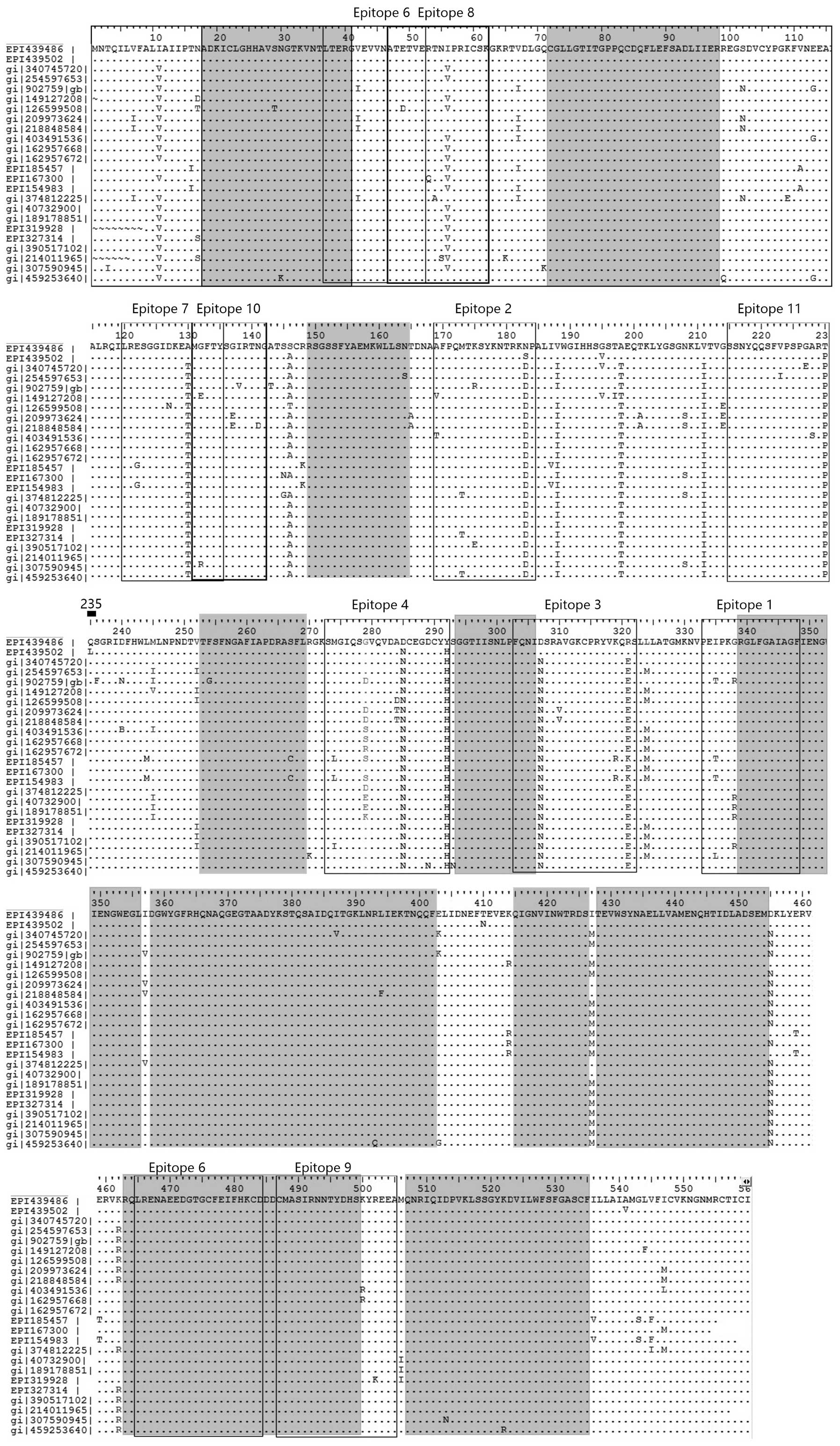
the 24 strains are listed in Fig. 1.
The HA gene with 560-amino acids from H7N9 (A/tree
sparrow/Shanghai/01/2013 (H7N9), gi|546235348| or EPI439486) was
selected as the reference sequence as it was the latest HA
gene.
Primary sequence and structure
analysis
Various properties of the 560-amino acid HA protein
from H7N9, including the theoretical isoelectric point (pI), amino
acid composition and molecular weight, were tested using the
ProtParam tool from the ExPASy Bioinformatics Resource Portal
(http://web.expasy.org/protparam/).
Subsequently, a structural search was performed using the Protein
Data Bank database (http://rcsb.org). PDB:4N5J was
identified as the three dimensional (3D) structure of AIV-H7.
Prediction of linear B-cell
epitopes
Linear B-cell epitopes were predicted using the
algorithms of ABCped, BepiPred and LBtope. The sequence of HA
protein were downloaded and analyzed by each software. ABCpred has
developed a systematic method based on a neural network (12). The amino acid length was set at 10,
12, 14, 16, 18 and 20 mer and the scoring threshold at 0.8.
BepiPred, which was developed by Larsen et al (14), employs the hidden Markov model and
apropensity scale method developed by Parker et al (15). A threshold of 0.350 was selected as
it is the point at which sensitivity (0.49)/specificity (0.75) is
maximized (14). LBtope is a new
method for predicting linear epitopes and was created by Singh
et al (16) in 2013. The
Lbtope_Confirm dataset was selected since it has previously shown
the best performance. The amino acid length was set to 15 mer and
the scoring threshold to 60%. The results from the three softwares
were assembled and the overlapping regions were considered
predicted epitopes.
Analysis of predicted epitopes
Using a combination of software prediction and
sequence comparisons, the conserved epitopes were predicted and a
domain enhanced lookup time accelerated-Basic Local Alignment
Search Tool (BLAST) analysis (http://blast.ncbi.nlm.nih.gov/Blast.cgi) was performed
to determine the specificity of the epitopes. Furthermore, the
location and structures of the epitopes were denoted in 3D
images.
Analysis of glutamine
(Gln)235-to-leucine (Leu)235 mutation
The 235th amino acid of HA from 24 AIV-H7 strains,
as well as 100 strains of human H7N9 viruses from the GISAID
database, were compared. The 235th amino acid of HA from 24 AIV-H7
strains, as well as 100 strains of human H7N9 viruses from the
GISAID database, were distinguished based on whether mutations were
present.
Results
Similarity searches and primary
sequence analyses
Primary sequence analyses revealed that the
theoretical pI of HA was pH 6.25 and the molecular weight was
62.062 kDa. The location and variation of HA from 24 AIV-H7 strains
are presented in Fig. 1. Following
the comparison, the highly conserved sequences included amino acids
18–41, 72–98, 149–164, 253–269, 294–306, 339–356, 358–402, 415–426,
428–454, 463–499 and 507–535. The majority of mutations were
located in the N-terminal 339 amino acids, which comprised the HA1
subunit of HA. The HA1 subunit forms the head of HA, which has a
vital role in interacting with the environment and host receptor.
Thus, the HA1 subunit is readily mutated in order to avoid the
attack of antibodies (17).
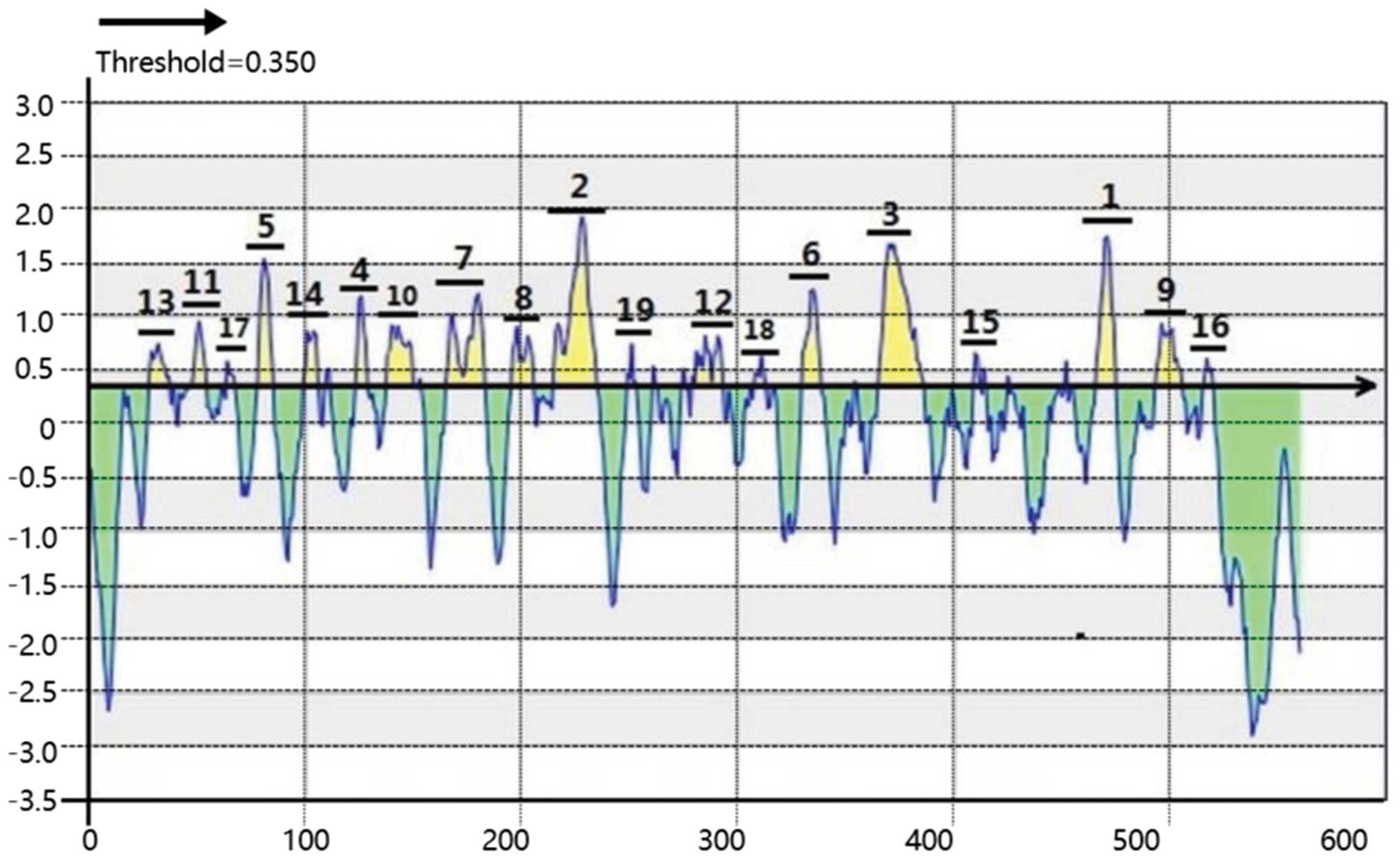
Epitope prediction by BepiPred
BepiPred software was used to produce a Linear
Epitope Prediction map (Fig. 2) and
BepiPred Prediction Details (Table
I). BepiPred analyzes each amino acid independently and assigns
it a score between −3 and 3. A higher score indicates a higher
probability for the existing epitope (14). The threshold was set at 0.35 and
scores >0.35 were regarded as positive. From the Fig. 2, beyond the axis, the higher of the
peak, the higher scores of prediction. Therefore, the 19 peaks of
the map formed by consecutive amino acids were regarded as 19
predicted linear epitopes. They were ranked by mean residue score
(mean residue score was averaged by every residue in the predicted
peptide), as presented in Table I.
The peaks 1–4 were the most likely epitopes to be predicted by
BepiPred.
 | Table I.Epitopes of hemagglutinin predicted by
BepiPred. |
Table I.
Epitopes of hemagglutinin predicted by
BepiPred.
| Rank | Sequence | Location | Mean residue
score |
|---|
| 1 | RENAEEDGTG | 466–475 | 1.130 |
| 2 |
SSNYQQSFVPSPGARPQVNG | 215–234 | 1.126 |
| 3 |
HQNAQGEGTAADYKSTQSAIDQ | 365–386 | 1.086 |
| 4 | TITGPPQCDQ | 77–86 | 0.857 |
| 5 | KNVPEIPKGR | 330–339 | 0.850 |
| 6 |
TDNAAFPQMTKSYKNTRKS | 165–183 | 0.732 |
| 7 | STAEQTKLYG | 196–205 | 0.707 |
| 8 | NNTYDHSKYREEA | 493–505 | 0.688 |
| 9 |
IRTNGATSACRRSGSS | 138–153 | 0.636 |
| 10 | VNATETVERT | 45–54 | 0.587 |
| 11 |
MGIQSGVQVDANCEGDCYHS | 274–293 | 0.539 |
| 12 | VSNGTKVNTLTE | 28–39 | 0.537 |
| 13 | REGSDVCYPGKFV | 99–111 | 0.523 |
| 14 | IDSRAVGKCPR | 306–316 | 0.378 |
| 15 | NDTVTFSFNGAFIA | 249–262 | 0.091 |
| 16 | KGKRTV | 62–67 | 0.426 |
| 17 | GGTDK | 124–128 | 0.946 |
| 18 | FNEVEK | 409–414 | 0.472 |
| 19 | KLSSG | 516–520 | 0.484 |
Epitope prediction by LBtope and
ABCpred
The output of the LBtope analysis is presented in
Fig. 3. LBtope assigns scores
between 0 and 100% to each epitope it predicts (16). A higher score indicates a higher
probability of the epitope existing (16). According to the mean residue score
(Table II), the software selected
14 consecutive amino acids that have a possibility of 61 to 100% of
being an epitope. As the threshold was set at 60%, these 14
consecutive amino acids were regarded as 14 predicted epitopes.
They were ranked by mean residue score, as shown in Table II. The sequences 1–2 were the most
likely linear epitopes as their scores were >80%.
| Table II.Epitopes of hemagglutinin predicted
by LBtope. |
Table II.
Epitopes of hemagglutinin predicted
by LBtope.
| Rank | Sequence | Location | Mean residue
score |
|---|
| 1 |
RNNTYDHSKYREEAM | 492–506 | 86.80 |
| 2 |
LYGSGNKLVTVGSSNYQQSFVPSPGARP | 203–230 | 82.37 |
| 3 | IDSRAVGKCPRY | 306–317 | 78.94 |
| 4 | QITGKLNRLIEK | 386–397 | 77.98 |
| 5 | IIERREGSDVCYPG | 95–108 | 76.45 |
| 6 |
TNIPRICSKGKRTVDL | 54–69 | 75.26 |
| 7 | SYKNTRKSPAL | 176–186 | 75.10 |
| 8 | DGWYGFRHQN | 358–367 | 72.99 |
| 9 |
NTLTERGVEVVNATET | 35–50 | 72.11 |
| 10 | LRENAEEDGTGCF | 465–477 | 72.04 |
| 11 |
LRGKSMGIQSGVQVDANCEG | 269–288 | 71.31 |
| 12 |
DKEAMGFTYSGIRTNGATSA | 127–146 | 70.84 |
| 13 |
SLLLATGMKNVPEIPK | 322–337 | 69.75 |
| 14 | SITEVWSYNA | 426–435 | 67.15 |
ABCpred is able to predict antigens that vary in
length from 10 to 20 residues and assigns a score between 0 and 1
to each epitope it predicts. A score that is closer to 1 indicates
a higher probability of the epitope existing and a score closer to
0 suggests that the amino acid sequence is unlikely to be an
epitope (12). In order to avoid
omissions, the amino acid length was set to 10, 12, 14, 16, 18 and
20 mer and the scoring threshold to 0.8. In total, 67 sequences met
the requirements.
Prediction of potential epitopes
The overlapping epitopes predicted by the three
softwares were considered as potential epitopes. Following a
comparison and analysis, 11 epitopes met the requirements. Each of
the potential epitopes were above the thresholds set for the 3
software packages, had a suitable length and were highly antigenic.
Therefore, they adequately represented the linear epitopes of HA.
The 11 epitopes are ranked by ABCpred scores in Table III.
| Table III.Potential epitopes, as ranked by
ABCpred scores. |
Table III.
Potential epitopes, as ranked by
ABCpred scores.
| Number | Sequence | Start position | End position | ABCpred score |
|---|
| 1 |
PEIPKGRGLFGAIAGF | 333 | 348 | 0.91 |
| 2 |
AFPQMTKSYKNTRKSP | 169 | 184 | 0.91 |
| 3 |
FQNIDSRAVGKCPRYVKQRS | 303 | 322 | 0.88 |
| 4 |
SMGIQSGVQVDANCEGDCYH | 273 | 292 | 0.88 |
| 5 |
ATETVERTNIPRICSK | 47 | 62 | 0.85 |
| 6 |
LRENAEEDGTGCFEIFHKCD | 465 | 484 | 0.85 |
| 7 |
LRESGGIDKEAMGFTY | 120 | 135 | 0.84 |
| 8 |
LTERGVEVVNATETVE | 37 | 52 | 0.83 |
| 9 |
CMASIRNNTYDHSKYREEA | 487 | 505 | 0.82 |
| 10 | MGFTYSGIRTNG | 131 | 142 | 0.82 |
| 11 |
SSNYQQSFVPSPGARPQVNG | 215 | 234 | 0.80 |
HA antigenicity and the conservation
of predicted epitopes
The locations of the 11 potential epitopes were
determined. The entire HA protein can be divided into three parts:
Amino acids 1–18 form the signal peptide, amino acids 19–339 form
the HA1 subunit and amino acids 340–560 form the HA2 subunit
(17). Eight of the 11 epitopes were
in the HA1 subunit, two were in the HA2 subunit and one was at the
junction of the two subunits. These results suggest that the HA1
subunit is the immunodominant antigen and the HA2 subunit is more
conserved.
Five epitopes showing minimal variation were
selected from the 11 epitopes by observation and comparison
(Fig. 1). Two of the five epitopes
were from the HA2 subunit and three were from the HA1 subunit.
These five epitopes showed high antigenicity and were highly
conserved; thus they were called the conserved predicted epitopes.
Three of the five epitopes were 20 amino acids in length, one was
16 amino acids and the other was 12 amino acids. The exact position
and composition of amino acids are shown in Table IV.
| Table IV.Five potential conserved
epitopes. |
Table IV.
Five potential conserved
epitopes.
| Number | Sequence | Start position | End position | ABCpred score |
|---|
| 6 |
LRENAEEDGTGCFEIFHKCD | 465 | 484 | 0.85 |
| 8 |
LTERGVEVVNATETVE | 37 | 52 | 0.83 |
| 9 |
CMASIRNNTYDHSKYREEA | 487 | 505 | 0.82 |
| 10 | MGFTYSGIRTNG | 131 | 142 | 0.82 |
| 11 |
SSNYQQSFVPSPGARPQVNG | 215 | 234 | 0.80 |
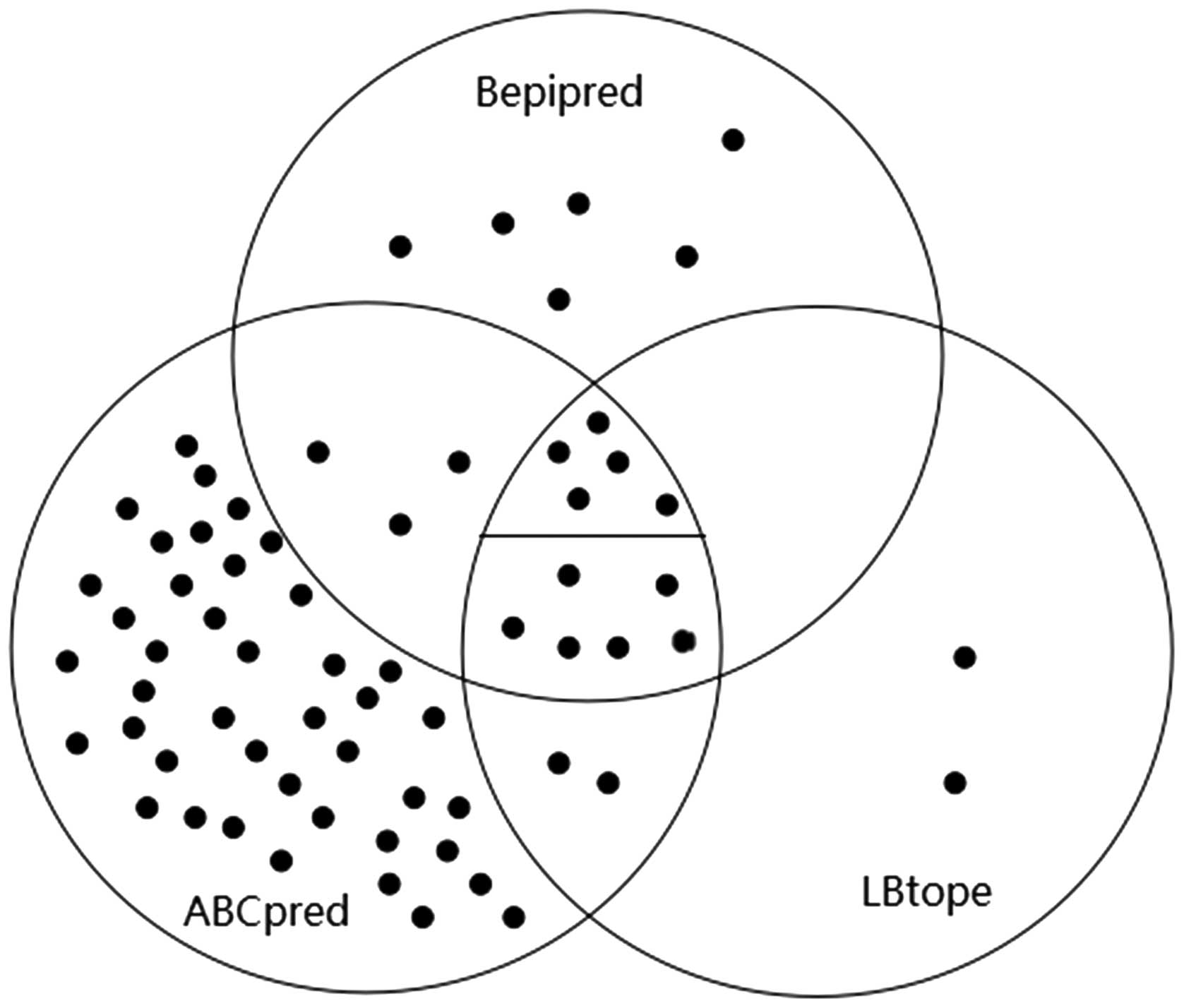
A Venn diagram was used to show the analysis process
and overall results (Fig. 4). The 11
epitopes that were detected by all three algorithms were in the
overlapping part of the three circles. The middle overlapping part
was divided into two parts by a short line. The five dots laying
above the line represented the five conserved predicted epitopes
among the 11 potential epitopes.
Secondary structure and specificity of
the five conserved predicted epitopes
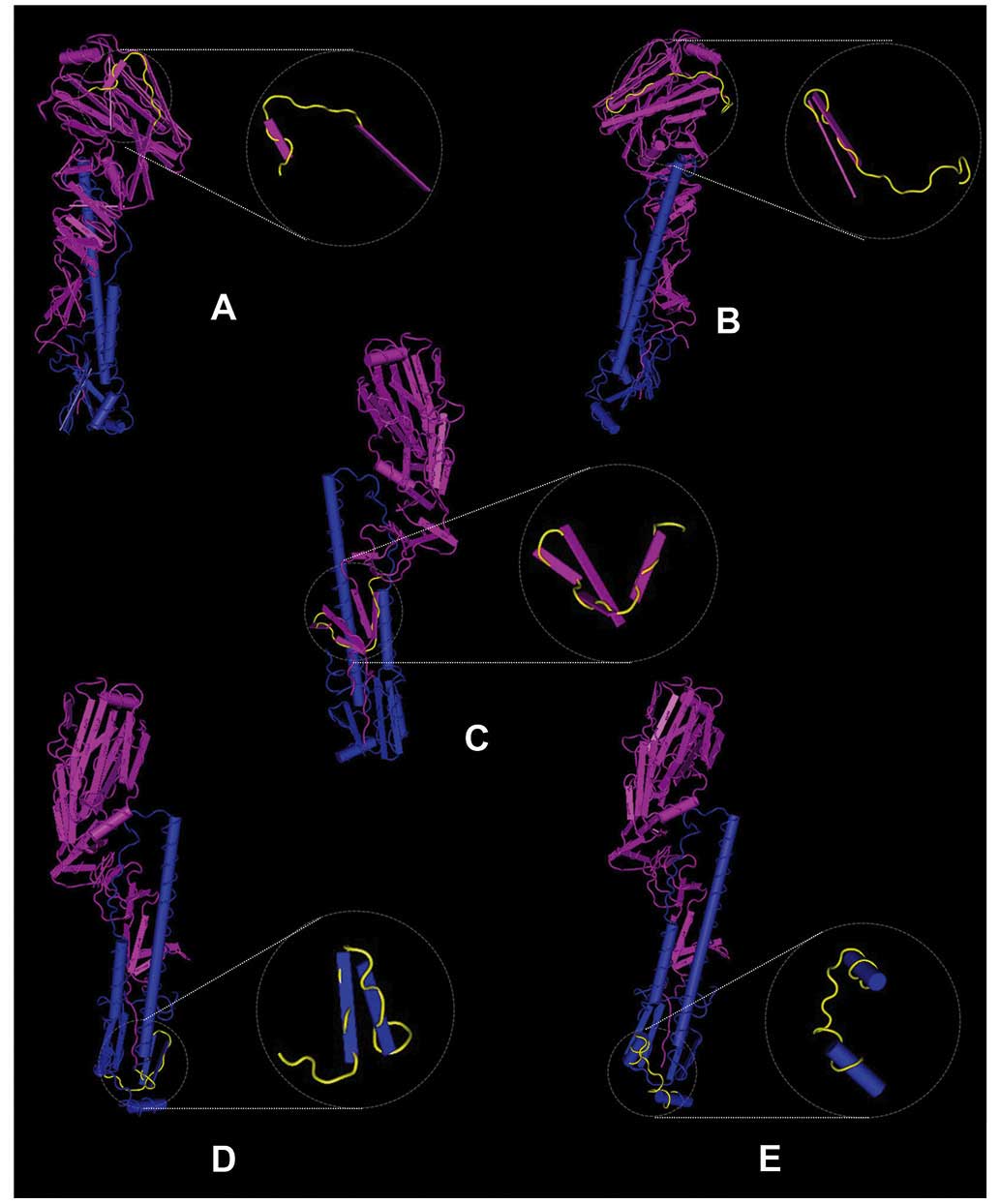
The secondary structures of the five conserved
predicted epitopes were analyzed and are presented in Fig. 5. Epitope 6 contains two β-strands and
one turn, epitope 8 contains three β-strands and one β-bridge,
epitope 9 consists of one turn and two parts of an α-helix, epitope
10 contains one β-strand and one β-bridge and epitope 11
constitutes one β-strand, one complete β-bridge, one partial
β-bridge and one turn. All of the five epitopes are on the surface
of HA protein, which exposes them to the environment and makes them
more likely to be antigenic (18).
Previous studies have demonstrated that β-bridges and turns are
more likely to form epitopes (18,19) and,
in the present study, at least one β-bridge or turn was identified
in every conserved predicted epitope. The exact locations of the
epitopes are displayed in Fig.
5.
Epitopes 8, 9 and 11 had no significant similarity
with other organisms, as demonstrated by a BLAST analysis. Epitope
6 showed 70% similarity with H5N1 and H2N2. Epitope 9 showed 74%
similarity with H3N1 and H3N2, 68% with H5N1, H5N2 and H1N1, and
63% with H12N4. Since changing one amino acid in an epitope can
markedly decrease the antigen-antibody interaction involved in
antibody recognition, it was concluded that all five epitopes were
specific to AIV-H7 (9).
Results of Gln235
to-Leu235 mutation
The present study demonstrated that the 235th amino
acid of HA (Gln) was a highly conserved residue for the majority of
H7-AIV, including those associated with previous human outbreaks
(Fig. 1). However, in the 2013 H7N9
AIV, the 235th residue had mutated to Leu; in the 100 strains of
human H7N9 viruses collected from across China, only 16 carried
Gln235, with the remaining (84%) all carrying
Leu235. These results suggest that the
Gln235-to-Leu235 mutation exists in the novel
H7N9 viruses.
Discussion
Bioinformatics is a promising and standard approach
for the identification of specific and immunogenic epitopes and it
has important applications in vaccine design, epitope mapping and
antibody research (20,21). Previous studies have demonstrated
that the efficiency of discovering novel epitopes is improved 10–20
times by immunoinformatics; the experimental work was reduced by
95% in one study (22). In 2008,
Frikha-Gargouri et al (23)
predicted a specific and immunogenic antigen of the OmcB protein
for the serodiagnosis of Chlamydia trachomatis infections.
Their results indicated that the use of sequence alignment tools
may be useful for identifying specific regions of an immunodominant
antigen (23). Furthermore, Jones
and Carter (24) used bioinformatics
tools to predict the B-cell epitopes of Listeria
monocytogenes and develop immunity to the bacterium in 2013.
Their results may be used to investigate the pathogenesis of L.
monocytogenes infections, as well as to develop an inexpensive
assay. Maksimov et al (25)
used in silico-predicted epitopes for the serological
diagnosis of Toxoplasma gondii infection in humans, and
established a peptide-based microarray assay to assess the
diagnostic performance of the selected peptides. The present study
used bioinformatics to predict the linear B-cell epitopes of HA of
H7-AIV.
ABCpred, which is an algorithm that was created by
Saha and Raghava (12) in 2006, is
able to predict epitopes with 66% accuracy, 67% sensitivity and 65%
specificity. Initially, the recurrent neural network was used, and
it was trained with a dataset of 700 experimentally detected B-cell
epitopes from the BciPep database (26) and 700 random peptides from the
Swiss-Prot database, for which no antibody binding is reported as a
negative dataset. The ABCpred algorithm was shown to have a better
predictive performance, as compared with various physicochemical
properties, including hydrophilicity (15), flexibility (27) and accessibility (28). In addition, Costa et al
(29) demonstrated that AAPPred and
ABCpred obtained the best results in terms of epitope prediction,
as compared with other programs, although AAPPred is no longer
available. BepiPred is a traditional algorithm developed by Larsen
et al (14) in 2006 using 14
epitope-annotated proteins and a human immunodeficiency virus
dataset. BepiPred analyzes each amino acid independently and does
not require a minimum or maximum number of amino acids to predict
an epitope. While we can distinguish the epitopes by the scores,
Reimer (30) demonstrated that the
predictions made by BepiPred were better than a random guess for
8/11 proteins. LBtope is a novel tool for the prediction of
epitopes that was developed by Singh et al (16) who exploited the availability of
several thousands of experimentally verified epitopes and
non-epitopes. Singh et al (16) derived five datasets from the Immune
Epitope Database called Lbtope_Fixed, Lbtope_Fixed_non_redundant,
Lbtope_Variable, Lbtope_Confirm and Lbtope_Variable_non_redundant
dataset (13). The greatest
advantage of LBtope is the ability to rule out nonepitopes that are
neglected by other algorithms, and it has been shown to compensate
for the inadequacies of the other two methods (16). However, users are advised to predict
linear epitopes using all existing methods and then identify the
target predicted by the majority of the methods (12). Therefore, the present study combined
the advantages of three algorithms and regarded the overlapped
results as the potential epitopes. Due to limitations of epitope
prediction methods, the prediction was unable to reach an accuracy
of 100% and further studies are required.
In conclusion, the present study identified 11
potential epitopes of the HA protein, and demonstrated that the HA1
subunit was the immunodominant antigen, whereas HA2 was more
conserved. Five potential conserved epitopes were selected and were
analyzed for secondary structure, software prediction and sequence
comparison; they all showed a high antigenicity and low variation.
Previous studies demonstrated that the
Gln235-to-Leu235 mutation was associated with
an improved affinity for human receptors, in particular when sialic
acid is 2-6-linked to galactose in novel H7N9 viruses (31,32).
This mutation was detected in the present study, thus suggesting
that it may have an important role in the increased virulence of
novel H7N9 viruses.
In the 2013–2014 period, H7N9 caused an epidemic in
humans that was associated with severe morbidity and mortality.
Investigation into the epitopes of H7-AIV may accelerate the
diagnosis of epidemic disease, permit the prediction of epidemics
and allow viral mutation, pathogenesis and recombination mechanisms
to be monitored. Future studies should extend to the Euro-Asiatic
H5, H1 and other subtypes of AIV as well as to other virulent
viruses, such as the Ebola virus.
Acknowledgements
The present study was supported by a grant from the
National Natural Science Foundation of China (grant no.
81371765).
References
|
1
|
Klimov A, Prösch S, Schäfer J and Bucher
D: Subtype H7 influenza viruses: Comparative antigenic and
molecular analysis of the HA-, M-, and NS-genes. Arch Virol.
122:143–161. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fouchier RA, Schneeberger PM, Rozendaal
FW, Broekman JM, Kemink SA, Munster V, Kuiken T, Rimmelzwaan GF,
Schutten M, Van Doornum GJ, et al: Avian influenza A virus (H7N7)
associated with human conjunctivitis and a fatal case of acute
respiratory distress syndrome. Proc Natl Acad Sci USA.
101:1356–1361. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
World Health Organization, . Human
infection with avian influenza A (H7N9) virus - update. http://www.who.int/csr/don/2014_01_20/en/May
1–2014
|
|
4
|
Sun Y, Shen Y and Lu H: Discovery process,
clinical characteristics, and treatment of patients infected with
avian influenza virus (H7N9) in Shanghai. Chin Med J (Engl).
127:185–186. 2014.PubMed/NCBI
|
|
5
|
Li YJ, Jiao XA, Pan ZM, Sun L, Wang CB,
Zhang SH, Sun QY and Liu XF: Development and characterization of
monoclonal antibodies against H7 hemagglutinin of avian influenza
virus. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi. 23:953–955. 2007.(In
Chinese). PubMed/NCBI
|
|
6
|
Zhao B, Zhang X, Zhu W, Teng Z, Yu X, Gao
Y, Wu D, Pei E, Yuan Z, Yang L, et al: Novel avian influenza
A(H7N9) virus in tree sparrow, Shanghai, China, 2013. Emerg Infect
Dis. 20:850–853. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Russell RJ, Kerry PS, Stevens DJ,
Steinhauer DA, Martin SR, Gamblin SJ and Skehel JJ: Structure of
influenza hemagglutinin in complex with an inhibitor of membrane
fusion. Proc Natl Acad Sci USA. 105:17736–17741. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wang Y, Dai Z, Cheng H, Liu Z, Pan Z, Deng
W, Gao T, Li X, Yao Y, Ren J and Xue Y: Towards a better
understanding of the novel avian-origin H7N9 influenza A virus in
China. Sci Rep. 3:23182013.PubMed/NCBI
|
|
9
|
Barlow DJ, Edwards MS and Thornton JM:
Continuous and discontinuous protein antigenic determinants.
Nature. 322:747–748. 1986. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Evans MC: Recent advances in
immunoinformatics: Application of in silico tools to drug
development. Curr Opin Drug Discov Devel. 11:233–241.
2008.PubMed/NCBI
|
|
11
|
Van Regenmortel MH: Synthetic peptides
versus natural antigens in immunoassays. Ann Biol Clin (Paris).
51:39–41. 1993.PubMed/NCBI
|
|
12
|
Saha S and Raghava GP: Prediction of
continuous B-cell epitopes in an antigen using recurrent neural
network. Proteins. 65:40–48. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhu WF, Gao RB, Wang DY, Yang L, Zhu Y and
Shu YL: A review of H7 subtype avain influenza virus. Bing Du Xue
Bao. 29:245–249. 2013.(In Chinese). PubMed/NCBI
|
|
14
|
Larsen JE, Lund O and Nielsen M: Improved
method for predicting linear B-cell epitopes. Immunome Res.
2:22006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Parker JM, Guo D and Hodges RS: New
hydrophilicity scale derived from high-performance liquid
chromatography peptide retention data: Correlation of predicted
surface residues with antigenicity and X-ray-derived accessible
sites. Biochemistry. 25:5425–5432. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Singh H, Ansari HR and Raghava GP:
Improved method for linear B-cell epitope prediction using
antigen's primary sequence. PLoS One. 8:e622162013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Xu R, de Vries RP, Zhu X, Nycholat CM,
McBride R, Yu W, Paulson JC and Wilson IA: Preferential recognition
of avian-like receptors in human influenza A H7N9 viruses. Science.
342:1230–1235. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Krchnák V, Mach O and Malý A: Computer
prediction of B-cell determinants from protein amino acid sequences
based on incidence of beta turns. Methods Enzymol. 178:586–611.
1989. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Pellequer JL, Westhof E and Van
Regenmortel MH: Correlation between the location of antigenic sites
and the prediction of turns in proteins. Immunol Lett. 36:83–99.
1993. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Dudek NL, Perlmutter P, Aguilar MI, Croft
NP and Purcell AW: Epitope discovery and their use in peptide based
vaccines. Curr Pharm Des. 16:3149–3157. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bryson CJ, Jones TD and Baker MP:
Prediction of immunogenicity of therapeutic proteins: Validity of
computational tools. BioDrugs. 24:1–8. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
De Groot AS, Sbai H, Aubin CS, McMurry J
and Martin W: Immuno-informatics: Mining genomes for vaccine
components. Immunol Cell Biol. 80:255–269. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Frikha-Gargouri O, Gdoura R, Znazen A,
Gargouri B, Gargouri J, Rebai A and Hammami A: Evaluation of an in
silico predicted specific and immunogenic antigen from the OmcB
protein for the serodiagnosis of Chlamydia trachomatis infections.
BMC Microbiol. 8:2172008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Jones MS and Carter JM: Prediction of
B-cell epitopes in listeriolysin O, a cholesterol dependent
cytolysin secreted by listeria monocytogenes. Adv Bioinformatics.
2014:8716762014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Maksimov P, Zerweck J, Maksimov A, Hotop
A, Gross U, Pleyer U, Spekker K, Däubener W, Werdermann S,
Niederstrasser O, et al: Peptide microarray analysis of in
silico-predicted epitopes for serological diagnosis of Toxoplasma
gondii infection in humans. Clin Vaccine Immunol. 19:865–874. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Saha S, Bhasin M and Raghava GP: Bcipep: A
database of B-cell epitopes. BMC Genomics. 6:792005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Karplus PA and Schulz GE: Prediction of
chain flexibility in proteins. Naturwissenschaften. 72:212–213.
1985. View Article : Google Scholar
|
|
28
|
Emini EA, Hughes JV, Perlow DS and Boger
J: Induction of hepatitis A virus-neutralizing antibody by a
virus-specific synthetic peptide. J Virol. 55:836–839.
1985.PubMed/NCBI
|
|
29
|
Costa JG, Faccendini PL, Sferco SJ, Lagier
CM and Marcipar IS: Evaluation and comparison of the ability of
online available prediction programs to predict true linear B-cell
epitopes. Protein Pept Lett. 20:724–730. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Reimer U: Prediction of linear B-cell
epitopes. Methods Mol Biol. 524:335–344. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kageyama T, Fujisaki S, Takashita E, Xu H,
Yamada S, Uchida Y, Neumann G, Saito T, Kawaoka Y and Tashiro M:
Genetic analysis of novel avian A (H7N9) influenza viruses isolated
from patients in China, February to April 2013. Euro Surveill.
18:204532013.PubMed/NCBI
|
|
32
|
Liu D, Shi W, Shi Y, Wang D, Xiao H, Li W,
Bi Y, Wu Y, Li X, Yan J, et al: Origin and diversity of novel avian
influenza A H7N9 viruses causing human infection: Phylogenetic,
structural, and coalescent analyses. Lancet. 381:1926–1932. 2013.
View Article : Google Scholar : PubMed/NCBI
|