Introduction
With the increase in the aging world population, the
prevalence of major neurodegenerative diseases has become a serious
public health problem worldwide, particularly Alzheimer's disease
(AD). AD is a degenerative, progressive and incurable neurological
syndrome accompanied by a gradual decline in learning, memory and
intelligence (1). A series of
hypotheses have been proposed for the mechanism of AD, and it is
now widely accepted that the deposit of amyloid β protein (Aβ) in
cerebral centers involved in cognition and memory is the most
important pathological feature of AD (2). There are two types of AD; familial and
sporadic AD (3). Familial AD is
associated with a genetic predisposition, which is associated by
the production and deposition of Aβ (4).
Notably, the pathogenesis of another type of
degenerative disease, type 2 diabetes mellitus (T2DM), has certain
similarities with AD, such as Aβ deposits in the islets of T2DM
patients. Further studies are necessary to assess whether
therapeutic methods for T2DM could antagonize the neurotoxicity of
Aβ peptides and ameliorate symptoms of AD patients. Glucagon-like
peptide-1 (GLP-1) is a T2DM novel therapeutic drug has been
suggested as a therapeutic target for AD (5). Due to the relatively short half-life of
native GLP-1, which is easily degraded by dipeptidyl peptidase-4
(DPP-4), it has been limited in its clinical application (6). Exendin-4 is a GLP-1 analogue which is
more slowly degraded, with a half-life of 9.57 h, and has been
approved by the Federal Drug Administration as a therapeutic for
T2DM (7).
Recently, studies have been published addressing the
neuroprotective effect of GLP-1 analogues on neurodegenerative
disorders (8,9). Our previous study indicated that
Val8-GLP-1, another GLP-1 analog, was able to
effectively antagonize synaptic dysfunction and intracellular
Ca2+ overload induced by Aβ1–40 (10). This neuroprotection may be associated
with the exhaustive regulation of synaptic transmission and
intracellular calcium homeostasis by Val8-GLP-1
(11). Val8-GLP-1
prevented synaptic degeneration and reversed hippocampal synaptic
plasticity in mice (12). In
addition, Exendin-4 appears to increase the expression level of
soluble polypeptide by enhancing the activity level of α-secretase
of amyloid precursor protein (APP), thus antagonizing Aβ toxicity
(13). The above findings suggest
that GLP-1 analogs such as Exendin-4 may exert neuroprotective
effects by improving synaptic function and antagonizing Aβ
toxicity. However, whether Exendin-4 is able to mitigate spatial
learning and memory damage resulting from Aβ fragment remains
unclear. The present study investigated the effects of Exendin-4 on
Aβ1-42-induced spatial learning and memory impairment, and on the
overall behavior of rats in order to evaluate any neuroprotective
effects induced by Exendin-4, and further investigated the
underlying electrophysiological and molecular mechanisms.
Materials and methods
Experimental animal and reagents
A total of 80 adult male Sprague-Dawley rats
(220–260 g) were used for the present study. All animals were
provided by the Research Animal Center of Shanxi Medical University
(Taiyuan, China). The study protocol received the approval of the
Shanxi Animal Research Ethics Committee.
Aβ1–42 [Bachem (UK) Ltd, Saint Helens, UK] was
dissolved in dimethyl sulfoxide (Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany) then diluted with normal saline to
6.25×10−4 mol/l and incubated at 37°C for 36 h to form
oligomers (14). Exendin-4
(Sigma-Aldrich) was diluted with normal saline to 2×10−4
mol/l. The administration was performed as follows:
Intra-hippocampal injection 0.625 nmol (1 µl) Aβ1–42 (Aβ1–42
group); 0.2 nmol (1 µl) Exendin-4 (Exendin-4 group); Exendin-4 +
Aβ1–42 group received Exendin-4 injection at 15 min following
Aβ1–42 oligomers; and the control group received 1 µl normal saline
injection. The intra-hippocampal injection was performed via a
microinjection pump (KD Scientific, Inc., Holliston, MA, USA), with
an injection rate of 0.1 µl/min. The experiments were then
performed 5 days after.
Morris water maze test
The spatial learning and memory test was performed
using a Morris water maze (MWM), as described previously (10). The MWM consists of a circular pool
with a diameter of 150 cm (Zhenghua Biological Instrument Equipment
Co., Ltd., Anhui, China), containing tap water at ~25°C. A
underwater platform (14 cm in diameter) was placed under ~1.0 cm
below the horizontal plane. At two weeks after drug injection, the
hidden platform test was conducted. Animals underwent consecutive
six-day trial periods, involving four trials per day using a random
set of start locations. The spatial bias of the four quadrants was
measured in the probe trial on the seventh day (10). Then the visible platform test was
performed in order to measure the time of swimming to the platform
(10).
Hippocampal CA1 region long-term
potentiation (LTP) in vivo recording
Following the MWM test, the electrophysiological
experiment was conducted as described in our previous study
(10). In the present study, LTP was
evaluated in vivo. The field excitatory postsynaptic
potential (fEPSP) of the hippocampal CA1 region was recorded.
Baseline fEPSP with test stimuli and LTP with high frequency
stimulation (HFS) for at least 60 min were recorded. The averaged
value of baseline fEPSP amplitude was taken as 100%.
Bicinchoninic acid (BCA) protein
assay
Following the LTP experiment, the rats were
sacrificed using an overdose of 25% urethane (5 ml/kg;
Sigma-Aldrich) and the hippocampal tissues were dissected and
frozen at −80°C. Then the tissues were homogenized with protease
lysate, followed by centrifugation at 16,000 × g for 10 min
at 4°C, after which the supernatant was collected. The total
protein was quantified using a BCA kit (Westang Biotech Co., Ltd.,
Shanghai, China), according to the manufacturer's instructions. The
BCA assay was conducted according to the manufacturer's
instructions. Absorbance was recorded at 562 nm using a microplate
reader (MK3; Thermo Fisher Scientific, Inc., Waltham, MA, USA). A
standard curve was drawn and the total protein concentration was
calculated.
Enzyme-linked immunosorbent assay
(ELISA)
Subsequently, an ELISA experiment was performed
using a commercial kit (cat. no. F15181; Westang Biotech Co., Ltd.)
to detect the expression of cyclic adenosine monophosphate (cAMP).
The polystyrene ELISA plate was arranged and each group was marked
clearly. Next, 100-µl standards of fold dilution were added to 8
wells of ELISA plate successively, then 100-µl samples were added
to each well, with mixing and incubating at 37°C for 40 min, then
washed 4–6 times and dried. Subsequently 50 µl distilled water and
rabbit anti-rat cAMP antibody was added to each well (except the
eighth standard well), mixed, incubated at 37°C for 20 min and
washed with PBST. Next, 100 µl horse radish peroxidase working
solution was added, mixed, incubated at 37°C for 10 min, washed and
printed. Next, 100 µl substrate working solution was added at 37°C
for 15 min, then subsequently 100 µl stop solution was added.
Absorbance was recorded at 450 nm (within 30 min) using a
microplate reader (MK3; Thermo Fisher Scientific, Inc.) and used to
draw the standard curve. Finally, the content of cAMP was
calculated. Absorbance was recorded at 450 nm (within 30 min) using
a microplate reader and the content of cAMP was calculated
according to the cAMP standard curve that was created with SkanIt
Software (Thermo Fisher Scientific, Inc.).
Western blot analysis
The experiment was used to evaluate
phosphorylated-cAMP response element binding protein (p-CREB)
expression in rat hippocampal tissue. The bilateral hippocampus
tissue (100 mg) was lysed in RIPA buffer (Beyotime Institute of
Biotechnology, Nanjing, China). Loading sample (40 µg) was selected
for separation using 12% SDS-PAGE, then transferred to a PVDF
membrane (Whatman; GE Healthcare, Chalfont, UK). The membrane was
blocked with 5% non-fat milk powder for 2 h and washed 3 times with
1X TBST buffer for 10 min. Then monoclonal primary antibodies
against p-CREB (1:1,000; cat. no. 9198; Cell Signaling Technology,
Inc., Danvers, MA, USA) were incubated with the membrane at 4°C
overnight. Then the membrane was incubated with the corresponding
secondary antibody for 1 h (peroxidase-conjugated goat anti-rat
IgG; 1:3,000; cat. no. ZB-2307; Beijing Zhongshan Golden Bridge
Biotechnology, Beijing, China). The blot was developed using a
super enhanced chemiluminescence detection kit (cat. no. P1030;
Applygen Technologies, Inc., Beijing, China), then the membrane was
placed in a Kodak Image Station 400 (Kodak Company, Rochester, NY,
Japan) for exposure and images of membrane signal bands were
obtained. Image J software (imagej.nih.gov/ij/) was used to analyze the western
blot bands.
Statistical analysis
Experimental data are presented as the mean ±
standard deviation. SPSS 17.0 software (SPSS, Inc., Chicago, IL,
USA) was used to perform statistical analysis. One-way analysis of
variance and repeated measures analysis of variance were used. Data
were considered statistically significant when P<0.05.
Results
Intra-hippocampal injection of Aβ1–42
induced abnormal behavior
The experiment was performed using an MWM test to
characterize the changes, if any, in rat spatial learning and
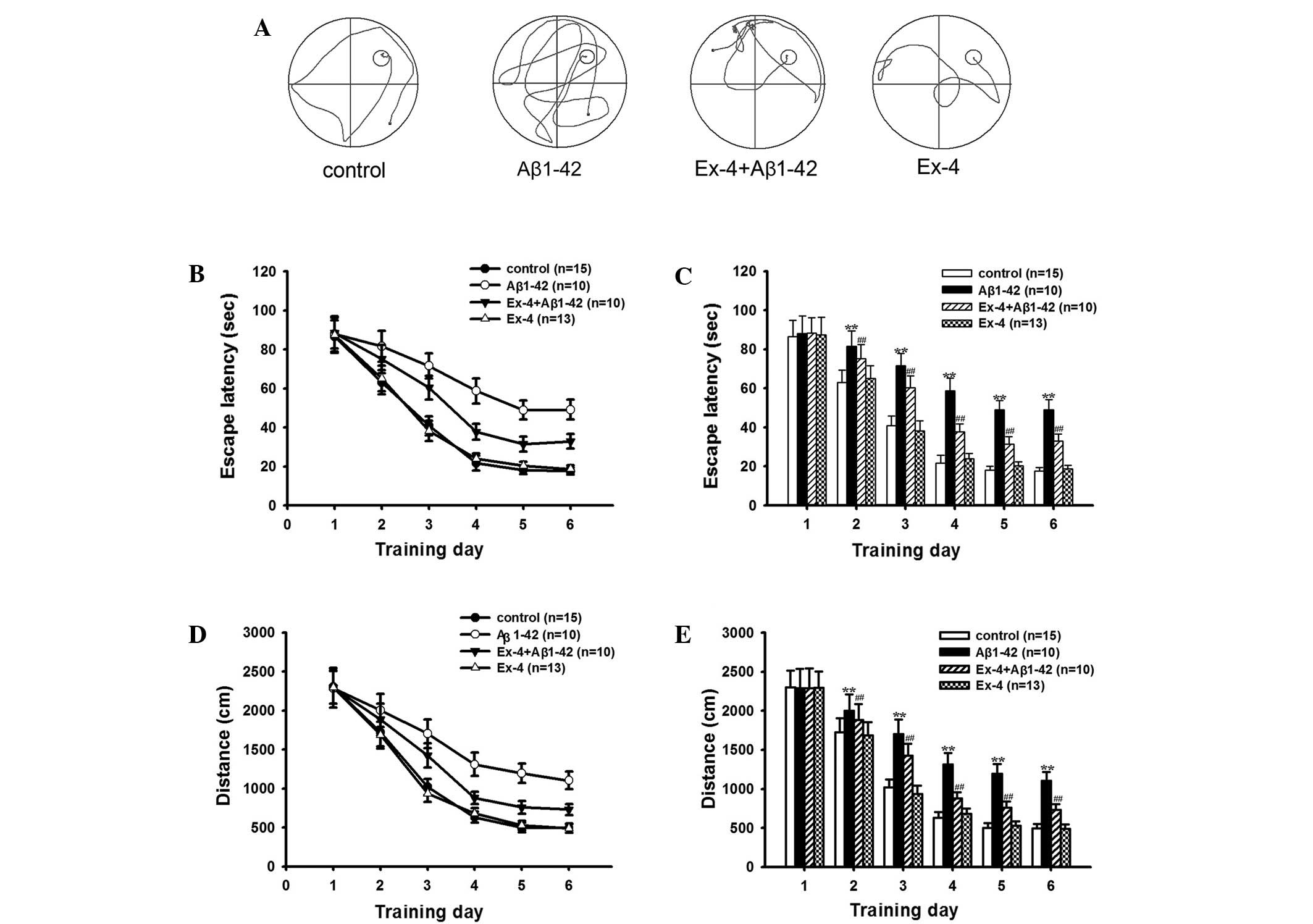
memory after intra-hippocampal injection of Aβ1-42. Fig. 1A shows Typical swim trajectories of
rats in searching for the hidden platform in all groups in the
sixth training day. Fig. 1B-E shows
the mean escape latencies and distances in the learning ability
test. As shown in Fig. 1, the
average escape latencies and distances gradually reduced with the
increase of training days, and these indexes remained relatively
stable on the fifth day in all groups. The average escape latencies
and distances significantly prolonged in the Aβ1–42 group from the
second to the sixth training day compared with the control group,
although the value of the first day maintained consistent. Fig. 1A shows typical swim trajectories of
all groups on the sixth training day.
In order to further investigate the changes in rat
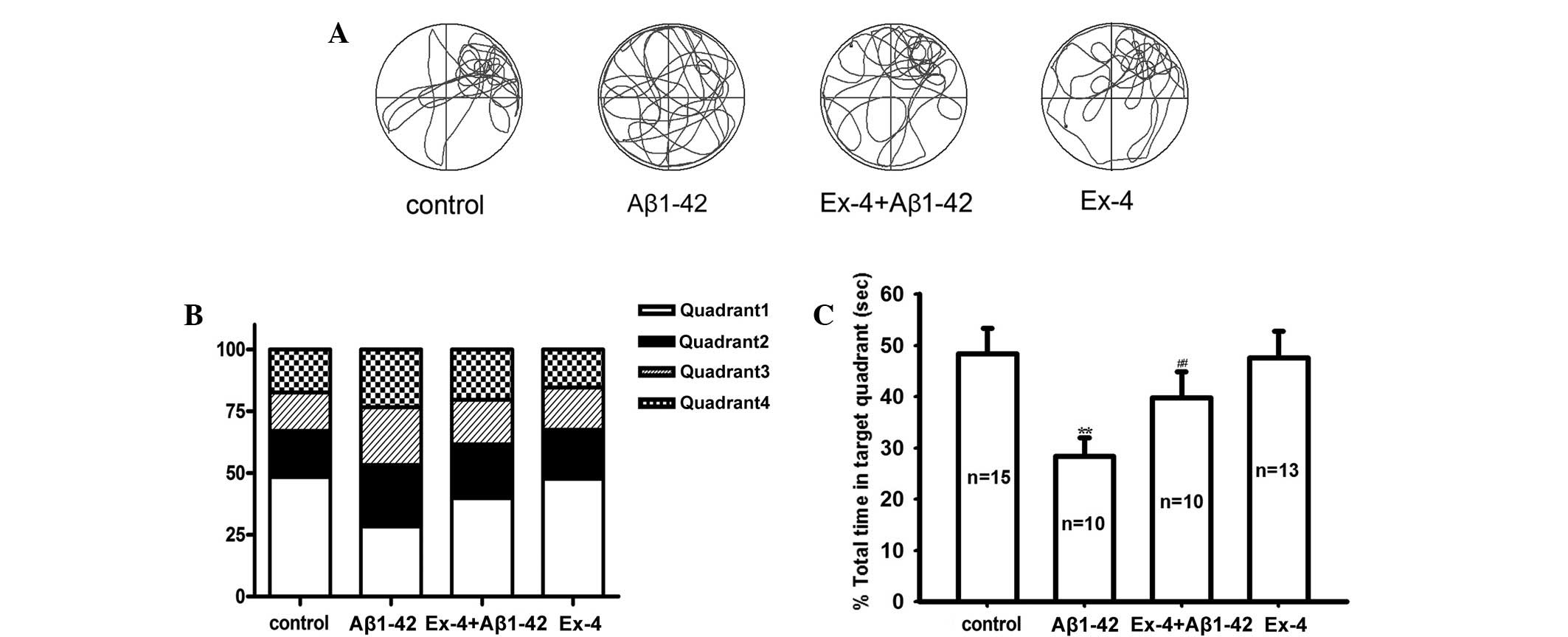
spatial memory ability, a probe trial was conducted. Fig. 2A showed typical swim trajectories in
all groups in the probe trial. Approximately the same time in the
target quadrant and the other quadrants was spent in the Aβ1–42
group; however, the rats in the control group spent more time in
the target quadrant. and the time was significantly longer than the
other quadrants (Fig. 2B and C).
Exendin-4 mitigated the behavior
ability caused by Aβ1–42
The subsequent results showed that there was no
significant change of the typical swim trajectories between
Exendin-4 and control group in the hidden platform test (Fig. 1A). Firstly, the application of
Exendin-4 alone did not change the mean escape latencies and
distances on any learning training day (Fig. 1B-E), the time percentage in the
target quadrant maintained consistent with control group in probe
trial (Fig. 2A-C).
Furthermore, the neuroprotective impact of Exendin-4
on the behavioral ability was investigated. Mean escape latency and
distance were decreased after pretreatment with Exendin-4 compared
to the Aβ1–42 group in the learning ability test (Fig. 1). In the probe trial, the time in the
target quadrant obviously prolonged compared with the Aβ1–42 group
(Fig. 2C).
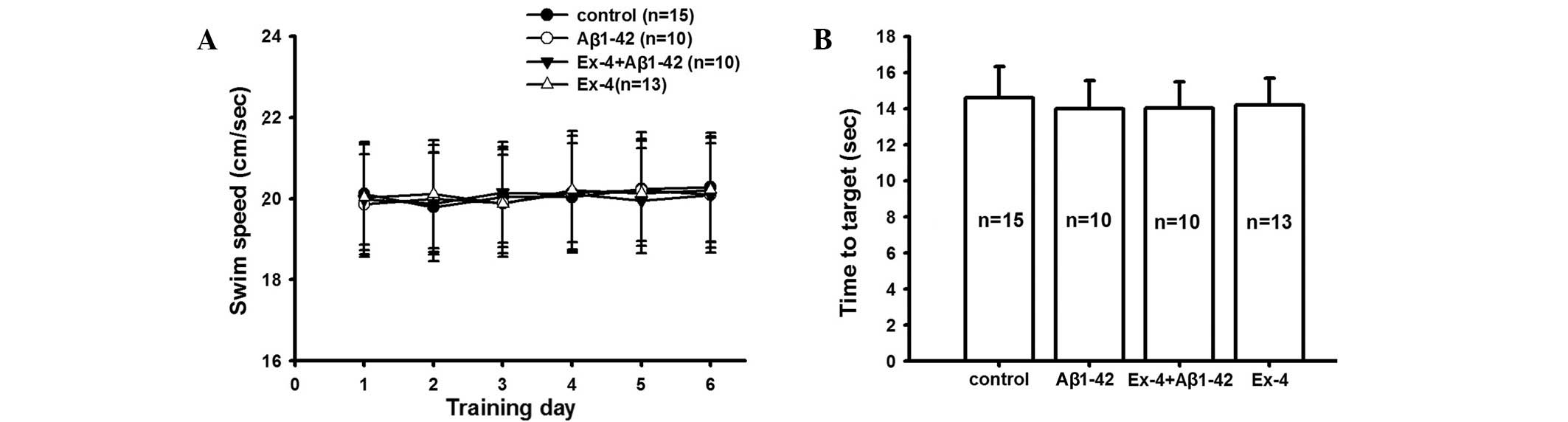
The latency and swimming speed were consistent among
all groups during successive six days in the hidden platform and
visible platform tests (Fig. 3).
These data confirmed that the visual and motion ability of all rats
was consistent, and the aforementioned behavioral experimental
results indicate the effect of Exendin-4 and Aβ1-42.
LTP was evidently reduced in the
Aβ1–42 group
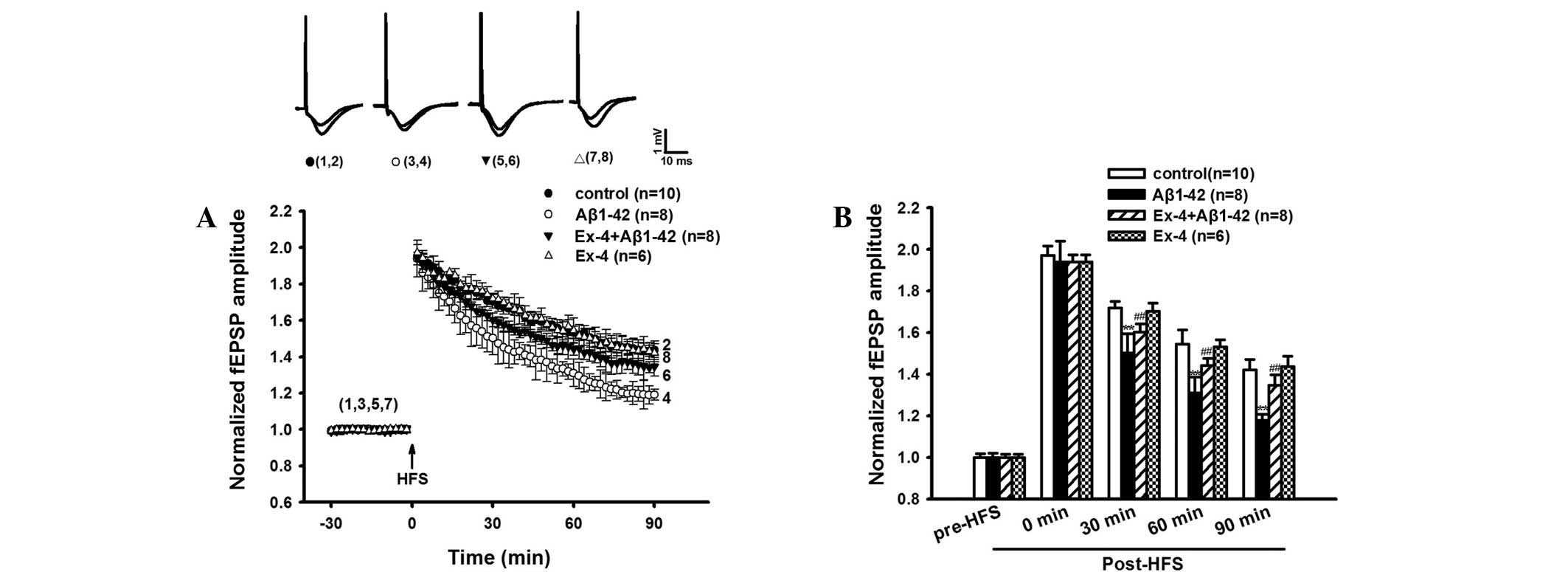
The LTP experiment was conducted following the MWM
test. Aβ1–42 significantly suppressed LTP in the rat hippocampus
in vivo compared with control group, the average fEPSP
amplitudes at 30, 60 and 90 min after HFS showed a significant
difference compared to the control group (Fig. 4A and B).
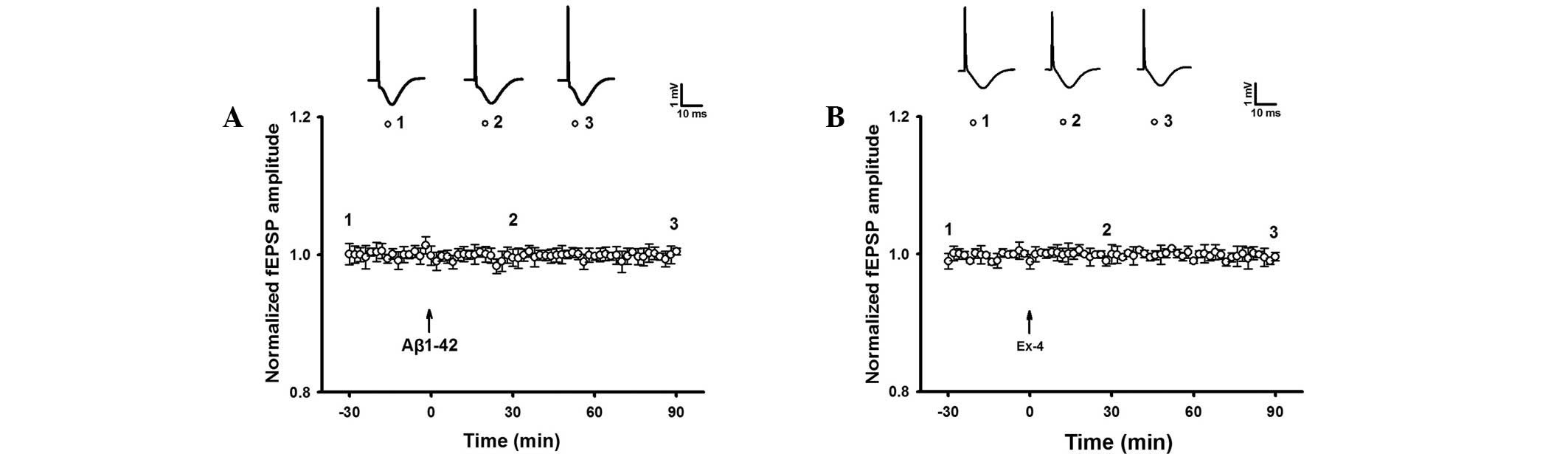
The baseline fEPSP of intra-hippocampal injection of
Aβ1–42 was observed. The result indicated that fEPSP without HFS
was not significantly different during at least 90 min of recording
in the Aβ1–42 group (Fig. 5). The
results suggested that Aβ1–42 induced hippocampal LTP damage.
Exendin-4 prevented Aβ1-42-induced
impairment of LTP
Similarly, Exendin-4 alone did not impact the
baseline fEPSP (Fig. 5). However,
Exendin-4 effectively prevented Aβ1-42-induced deficits of LTP. As
shown in Fig. 4A and B, Aβ1–42
induced LTP inhibition was significantly antagonized by Exendin-4.
The average fEPSP amplitudes at 30, 60 and 90 min post-HFS were
significantly higher compared with the Aβ1–42 group.
Exendin-4 antagonized Aβ1–42 induced
decrease of cAMP and p-CREB in rat hippocampus tissue
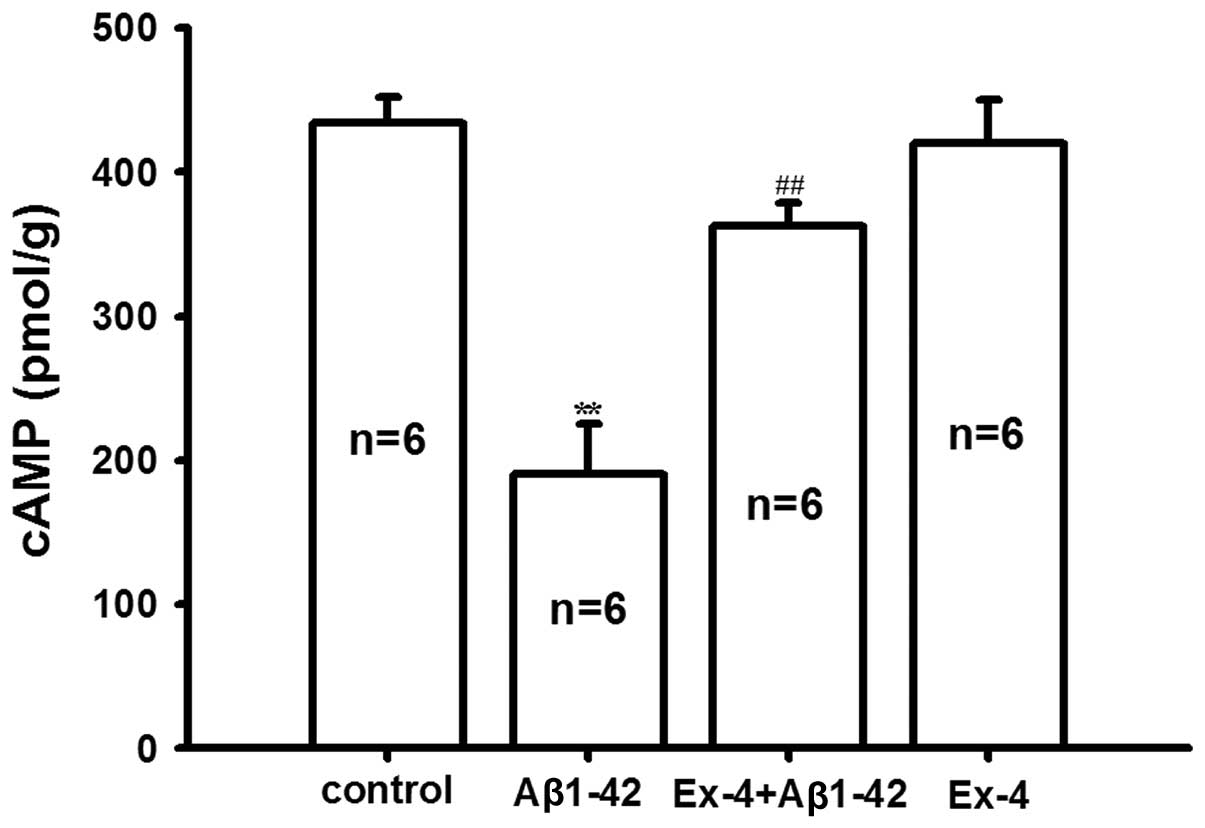
In order to investigate the molecular mechanism of
antagonism of Exendin-4 for Aβ1-42, the levels of cAMP and p-CREB
in the rat hippocampus were determined. We found that
Aβ1-42-induced the cAMP level was significantly reduced compared
with the control group, pretreatment of Exendin-4 antagonized the
decrease of cAMP induced by Aβ1-42; however, the level of cAMP was
not affected by Exendin-4 application alone (Fig. 6).
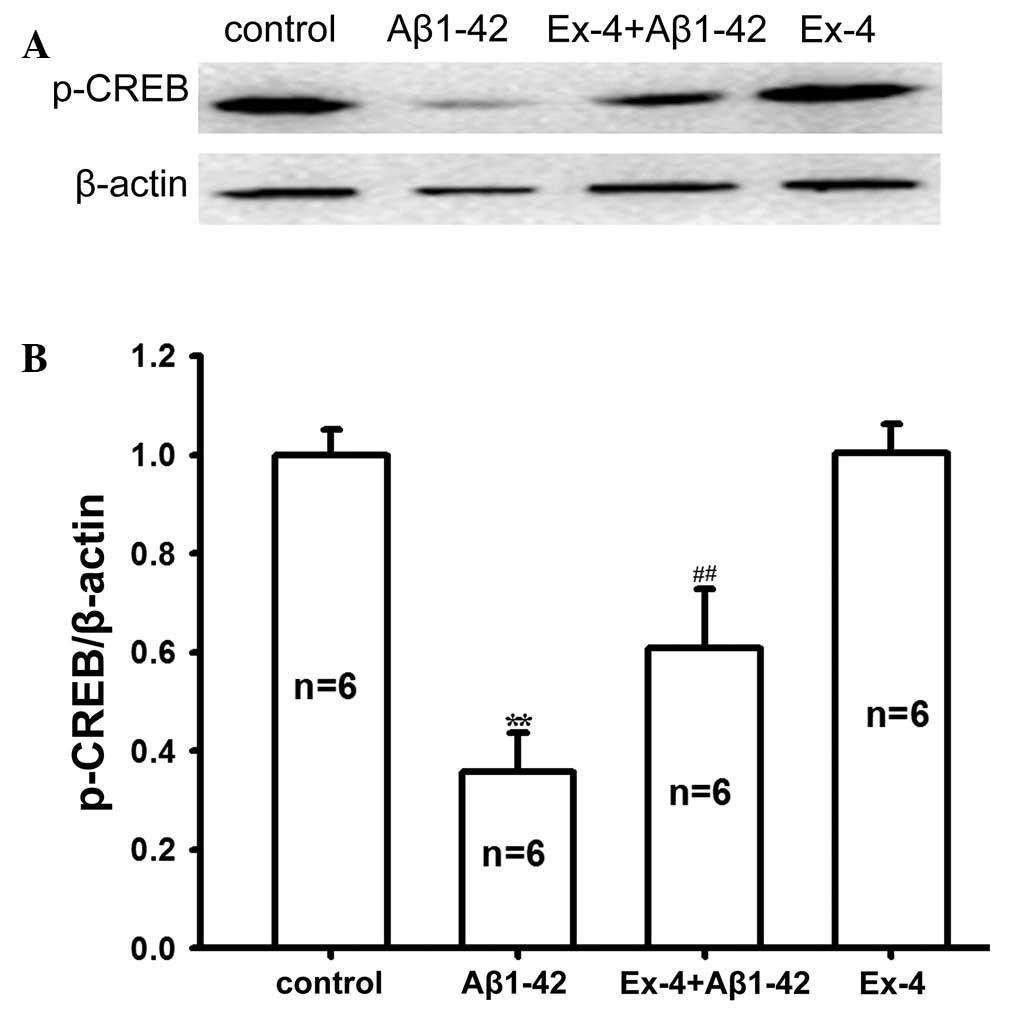
Subsequently, p-CREB expression in rat hippocampus
tissue was determined in all groups. As shown in Fig. 7, Aβ1–42 evidently reduced the protein
expression level of p-CREB. p-CREB protein expression markedly
improved after pretreatment with Exendin-4 compared with the
application of Aβ1–42 alone. These results suggested that
pretreatment with Exendin-4 may reverse the decreased p-CREB
protein level induced by Aβ1-42. Furthermore, the protein
expression level of p-CREB was not affected by Exendin-4
application alone. Thus, the present results imply that Exendin-4
may antagonize Aβ1–42 toxicity by mediating the cAMP/PKA/CREB
pathway.
Discussion
In the present study, intra-hippocampal injection of
Aβ1–42 was adopted to generate rat models, then behavioral,
electrophysiological and molecular experiments were performed. The
results imply that the impairment of learning and memory induced by
Aβ1–42 was significantly reversed by Exendin-4 treatment.
Furthermore, the electrophysiology and molecular mechanisms
underlying the neuroprotective effect of Exendin-4 were
investigated.
AD is a degenerative, progressive and incurable
neurological disorder which is characterized by the gradual
deterioration of cognitive function, neuropsychiatric symptoms and
behavioral disturbances, including nutritional disorders and
circadian rhythm disruption (15,16).
There are three major pathological features associated with AD;
neuronal loss, senile plaques formed by the deposition of amyloid β
protein and neurofibrillary tangles (17,18).
However, the exact pathogenesis of AD remains unclear, and thus a
series of hypotheses have been proposed to account for the
pathogenesis of AD, such as the Aβ hypothesis, cholinergic
hypothesis, tau protein hypothesis and oxidative stress hypothesis,
among which the Aβ hypothesis plays the most important role
(19,20). The Aβ hypothesis suggests that the
pathogenesis of various biological malfunctioning is associated
with Aβ deposition, for example, Aβ oligomers extracted from human
AD brain can inhibit LTP, reduce dendritic spine density and
disrupt memory and learning in vivo when directly injected
into a mouse hippocampus (21). Aβ
is derived from the precursor protein APP, which generates various
polypeptides via sequential cleaving by α-secretase and β-secretase
(22). Aβ is a insoluble polypeptide
which is generated by the β-secretase pathway, and can induce
oxidative stress and Ca2+ uptake. Oxidative stress can
in turn active apoptosis and initiate glial cells to produce large
quantities of inflammatory mediators and other toxic substances,
resulting in irreversible neuronal damage (23). Moreover, Ca2+ may be
releases from presynaptic neurotransmitters and influx into the
postsynaptic membrane, which is closely associated with learning
and memory (24).
By contrast, Aβ peptide includes numerous fragments,
including Aβ1-40, Aβ1-42, Aβ31–35 and Aβ25–35 (25,26).
Among these, Aβ1–42 exhibits the strongest toxicity owing to its
hydrophobicity and aggregation (27). Therefore, Aβ1–42 was adopted for the
present experiments. Formerly, intra-cerebroventricular injection
was widely used to investigated the toxicity of Aβ1–42 in numerous
studies (28,29); however, this delivery method has
numerous disadvantages, including drug diffusion, inaccurate
positioning and relatively high cost (10,30).
Intra-hippocampal administration effectively avoided these
shortcomings and shortened delivery time significantly in the
present study.
Following intra-hippocampal injection, rats
underwent an MWM experiment. Over the course of six consecutive
days of spatial learning ability testing, the escape latencies and
distances of Aβ1–42 group increased significantly compared with the
control group. Approximately equal time was spent in the target
quadrant compared with any other quadrant in the Aβ1–42 group in
the probe trial. It is thus indicated that the spatial learning and
memory impairment were induced by Aβ1-42. Consistent with this,
inhibitory avoidance analysis (a fear conditioning-based task) in a
previous study suggested that Aβ1–42 caused the damage to learning
and memory ability (31). Therefore,
protection against neurotoxicity of Aβ is hypothesized to be a
therapeutic target for the treatment of AD patients. Several
studies have indicated approaches to decreasing Aβ neurotoxicity,
such as reducing Aβ generation, increasing Aβ degradation and
restraining Aβ toxicity (32,33);
however, there is at present no efficacious drug directed against
Aβ neurotoxicity. The purpose of the present study was to
investigate whether Exendin-4 exhibits neuroprotective effects
against Aβ peptide neurotoxicity.
A previous study suggested an association between AD
and T2DM, which is another chronic degenerative disease (34). An investigation revealed that 85% of
AD patients were diagnosed with T2DM or accompanied by elevated
fasting blood glucose (35).
Furthermore, the risk of patients with T2DM suffering from AD is
twice that of the normal population (36). Notably, Aβ deposition, the typical
pathological change in AD patients, is similar to the
characteristic accumulation of Aβ in the islet cells of patients
with T2DM (37). Therefore,
researchers have suggested that AD associated with high blood
glucose be considered type 3 diabetes mellitus (38). Thus, pharmacological therapy intended
for the treatment of T2DM may additionally help to prevent and
improve the symptoms of AD. Insulin, the primary therapy for T2DM,
is not applicable as an AD treatment, as insulin-degrading enzyme
can recognize and combine the same domain which exists in insulin
and Aβ (39). Moreover, insulin
injection can reduce the combination between Aβ and IDE, facilitate
Aβ deposition in brain, and ultimately exacerbate the neurotoxicity
and induce clinical symptoms (40).
Furthermore, AD patients with normal blood glucose may sustain
glucopenia due to the fact that insulin can lower euglycemia
(41). However, GLP-1 and its
analogs may promote insulin synthesis and secretion, and do not
affect normal blood glucose (42).
The polypeptide chain of GLP-1 containing 30 amino acids can be
decomposed by DPP-4, therefore the plasma half-life of native GLP-1
is <2 min (6,43). GLP-1 analogs are similar to native
GLP-1 as they exhibit similar chemical structure and biological
activity (44). Fortunately, certain
GLP-1 analogs are not easily biodegraded by DPP-4, which is
attributed to the fatty acid side chains, so its plasma half-life
is longer than that of native GLP-1 and may cross the blood-brain
barrier (45). Exendin-4, derived
from American Gila monster's (Heloderma suspectum) saliva,
is the main GLP-1 analogue approved for clinical application
(46). Approximately 53% homology
exists between Exendin-4 and mammalian GLP-1, ad Exendin-4 has high
affinity to GLP-1 receptor, whose plasma half-life is 9.57 h
(47). Studies have found that
Exendin-4 protected the primary human SH-SY5Y cells and rat
hippocampal neurons against glutamate excitotoxicity (48), as well as abating the cognitive
impairment induced by traumatic brain injury (48). However, whether Exendin-4 can
mitigate the damage of spatial learning and memory derived
associated with Aβ remains unknown. The results of the present
study demonstrated that Exendin-4 was associated with a marked
reduction in Aβ1-42-induced learning and memory damage in a rat
model. In the behavioral experiment, pretreatment of Exendin-4
decreased mean escape latency and distance travelled by the rats,
compared with the Aβ1–42 group in the learning test, and the time
in the target quadrant obviously prolonged compared with the Aβ1–42
group in the memory trial. In order to investigate the mechanism
underlying the neuroprotective effect of Exendin-4, we conducted
further electrophysiological and molecular biological
experiments.
As an important electrophysiological mechanism of
learning and memory, LTP is a long-lasting increase in synaptic
efficacy which follows high-frequency stimulation of afferent
fibers (49). Our previous study
showed that Aβ1-40-induced damage of late phase LTP, learning and
memory were antagonized by Val8-GLP-1 (10). In the present study, we performed
electrophysiological experiments and proved that Aβ1-42-induced a
reduction of LTP could be reversed by Exendin-4. Baseline fEPSP in
the CA1 area of hippocampus was unchanged by Aβ1–42 or Exendin-4
alone, which indicates that Exendin-4 regulates learning and memory
impairment of AD model mediated by modifying impaired LTP.
Furthermore, synaptic plasticity, neurogenesis,
learning and memory are closely associated with cAMP/PKA/CREB
signaling pathway in the central nervous system (50,51).
CREB is an important nucleoprotein belonging to activating
transcription factor and CREB proteins are activated by
phosphorylation at Ser133 (p-CREB) from various kinases that are
activated by the increased intracellular cAMP (52). A previous study showed that
cAMP/PKA/CREB signaling pathway had a significant effect on memory
formation, particularly hippocampal-dependent LTP (53). Increased cAMP, activated MAPK and
p-CREB during rapid eye movement sleep ultimately contributed to
memory consolidation (54). In
addition, a previous study has confirmed that Aβ25-35-treated rats
displayed decreased levels of p-CREB (55). The results of the present study
indicate that Exendin-4 could reverse Aβ1-42-induced decline of
cAMP and p-CREB, which suggested that Exendin-4 ameliorates
Aβ1-42-induced damage to learning and memory, potentially by
modifying the cAMP/PKA/CREB signaling pathway.
In conclusion, the study demonstrated that Exendin-4
may exert a neuroprotective effect in the experimental rats, and
further revealed possible underlying electrophysiological and
molecular mechanisms. These findings provide theoretical support
for further investigation into the use of Exendin-4 as a drug for
the prophylaxis and treatment of AD.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (grant no. 81471343) and Science and
Technology Innovation Fund of Shanxi Medical University (grant no.
01201307).
References
|
1
|
Cummings JL: Alzheimer's disease. N Engl J
Med. 351:56–67. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hardy J and Allsop D: Amyloid deposition
as the central event in the aetiology of Alzheimer's disease.
Trends Pharmacol Sci. 12:383–388. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hoenicka J: Genes in Alzheimer's disease.
Rev Neurol. 42:302–305. 2006.(In Spanish). PubMed/NCBI
|
|
4
|
Bohm C, Chen F, Sevalle J, Qamar S, Dodd
R, Li Y, Schmitt-Ulms G, Fraser PE and St George-Hyslop PH: Current
and future implications of basic and translational research on
amyloid-β peptide production and removal pathways. Mol Cell
Neurosci. 66:3–11. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ohtake N, Saito M, Eto M and Seki K:
Exendin-4 promotes the membrane trafficking of the AMPA receptor
GluR1 subunit and ADAM10 in the mouse neocortex. Regul Pept
190–191. 1–11. 2014. View Article : Google Scholar
|
|
6
|
Vilsbøll T, Agersø H, Krarup T and Holst
JJ: Similar elimination rates of glucagon-like peptide-1 in obese
type 2 diabetic patients and healthy subjects. J Clin Endocrinol
Metab. 88:220–224. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lupi R, Mancarella R, Del Guerra S,
Bugliani M, Del Prato S, Boggi U, Mosca F, Filipponi F and
Marchetti P: Effects of exendin-4 on islets from type 2 diabetes
patients. Diabetes Obes Metab. 10:515–519. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bassil F, Fernagut PO, Bezard E and
Meissner WG: Insulin, IGF-1 and GLP-1 signaling in
neurodegenerative disorders: targets for disease modification? Prog
Neurobiol. 118:1–18. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Calsolaro V and Edison P: Novel GLP-1
(glucagon-like peptide-1) analogues and insulin in the treatment
for Alzheimer's disease and other neurodegenerative diseases. CNS
Drugs. 29:1023–1039. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wang XH, Li L, Hölscher C, Pan YF, Chen XR
and Qi JS: Val8-glucagon-like peptide-1 protects against
Aβ1-40-induced impairment of hippocampal late-phase long-term
potentiation and spatial learning in rats. Neuroscience.
170:1239–1248. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang XH, Yang W, Hölscher C, Wang ZJ, Cai
HY, Li QS and Qi JS: Val8-GLP-1 remodels synaptic
activity and intracellular calcium homeostasis impaired by amyloid
β peptide in rats. J Neurosci Res. 91:568–577. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gengler S, McClean PL, McCurtin R, Gault
VA and Hölscher C: Val (8)GLP-1 rescues synaptic plasticity and
reduces dense core plaques in APP/PS1 mice. Neurobiol Aging.
33:265–276. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Xie Z and Xu Z: General anesthetics and
β-amyloid protein. Prog Neuropsychopharmacol Biol Psychiatry.
47:140–146. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
O'Hare E, Weldon DT, Mantyh PW, Ghilardi
JR, Finke MP, Kuskowski MA, Maggio JE, Shephard RA and Cleary J:
Delayed behavioral effects following intrahippocampal injection of
aggregated A beta (1–42). Brain Res. 815:1–10. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Querfurth HW and LaFerla FM: Alzheimer's
disease. N Engl J Med. 362:329–344. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chiu MJ, Chen TF, Yip PK, Hua MS and Tang
LY: Behavioral and psychologic symptoms in different types of
dementia. J Formos Med Assoc. 105:556–562. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sorrentino G and Bonavita V:
Neurodegeneration and Alzheimer's disease: The lesson from
tauopathies. Neurol Sci. 28:63–71. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Blennow K, de Leon MJ and Zetterberg H:
Alzheimer's disease. Lancet. 368:387–403. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Nitta A, Fukuta T, Hasegawa T and
Nabeshima T: Continuous infusion of beta-amyloid protein into the
rat cerebral ventricle induces learning impairment and neuronal and
morphological degeneration. Jpn J Pharmacol. 73:51–57. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Robbins TW, McAlonan G, Muir JL and
Everitt BJ: Cognitive enhancers in theory and practice: Studies of
the cholinergic hypothesis of cognitive deficits in Alzheimer's
disease. Behav Brain Res. 83:15–23. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Shankar GM, Li S, Mehta TH, Garcia-Munoz
A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere
CA, et al: Amyloid-beta protein dimers isolated directly from
Alzheimer's brains impair synaptic plasticity and memory. Nat Med.
14:837–842. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Glenner GG and Wong CW: Alzheimer's
disease: Initial report of the purification and characterization of
a novel cerebrovascular amyloid protein. Biochem Biophys Res
Commun. 120:885–890. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Guan ZZ: Cross-talk between oxidative
stress and modifications of cholinergic and glutaminergic receptors
in the pathogenesis of Alzheimer's disease. Acta Pharmacol Sin.
29:773–780. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chakroborty S, Kim J, Schneider C,
Jacobson C, Molgó J and Stutzmann GE: Early presynaptic and
postsynaptic calcium signaling abnormalities mask underlying
synaptic depression in presymptomatic Alzheimer's disease mice. J
Neurosci. 32:8341–8353. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Clementi ME, Marini S, Coletta M, Orsini
F, Giardina B and Misiti F: Abeta(31–35) and Abeta(25–35) fragments
of amyloid beta-protein induce cellular death through apoptotic
signals: Role of the redox state of methionine-35. FEBS Lett.
579:2913–2918. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kanai M, Matsubara E, Isoe K, Urakami K,
Nakashima K, Arai H, Sasaki H, Abe K, Iwatsubo T, Kosaka T, et al:
Longitudinal study of cerebrospinal fluid levels of tau, A
beta1-40, and A beta1-42(43) in Alzheimer's disease: a study in
Japan. Ann Neurol. 44:17–26. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lambert MP, Barlow AK, Chromy BA, Edwards
C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL,
et al: Diffusible, nonfibrillar ligands derived from Abeta1-42 are
potent central nervous system neurotoxins. Proc Natl Acad Sci USA.
95:6448–6453. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bernardi A, Frozza RL, Meneghetti A, Hoppe
JB, Battastini AM, Pohlmann AR, Guterres SS and Salbego CG:
Indomethacin-loaded lipid-core nanocapsules reduce the damage
triggered by Aβ1-42 in Alzheimer's disease models. Int J
Nanomedicine. 7:4927–4942. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yamada K, Tanaka T, Han D, Senzaki K,
Kameyama T and Nabeshima T: Protective effects of idebenone and
alpha-tocopherol on beta-amyloid-(1–42)-induced learning and memory
deficits in rats: implication of oxidative stress in
beta-amyloid-induced neurotoxicity in vivo. Eur J Neurosci.
11:83–90. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kheirbakhsh R, Chinisaz M, Khodayari S,
Amanpour S, Dehpour AR, Muhammadnejad A, Larijani B and
Ebrahim-Habibi A: Injection of insulin amyloid fibrils in the
hippocampus of male Wistar rats: Report on memory impairment and
formation of amyloid plaques. Neurol Sci. 36:1411–1416. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Garcia-Osta A and Alberini CM: Amyloid
beta mediates memory formation. Learn Mem. 16:267–272. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Nalivaeva NN, Beckett C, Belyaev ND and
Turner AJ: Are amyloid-degrading enzymes viable therapeutic targets
in Alzheimer's disease? J Neurochem. 120:(Suppl 1). S167–S185.
2012. View Article : Google Scholar
|
|
33
|
Soininen H and Hiltunen M: Targeting
ApoE4/ApoE receptor LRP1 in Alzheimer's disease. Expert Opin Ther
Targets. 17:781–794. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Jones A, Kulozik P, Ostertag A and Herzig
S: Common pathological processes and transcriptional pathways in
Alzheimer's disease and type 2 diabetes. J Alzheimers Dis.
16:787–808. 2009.PubMed/NCBI
|
|
35
|
Leibson CL, Rocca WA, Hanson VA, Cha R,
Kokmen E, O'Brien PC and Palumbo PJ: Risk of dementia among persons
with diabetes mellitus: A population-based cohort study. Am J
Epidemiol. 145:301–308. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Janson J, Laedtke T, Parisi JE, O'Brien P,
Petersen RC and Butler PC: Increased risk of type 2 diabetes in
Alzheimer disease. Diabetes. 53:474–481. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Alafuzoff I, Aho L, Helisalmi S, Mannermaa
A and Soininen H: Beta-amyloid deposition in brains of subjects
with diabetes. Neuropathol Appl Neurobiol. 35:60–68. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Accardi G, Caruso C, Colonna-Romano G,
Camarda C, Monastero R and Candore G: Can Alzheimer disease be a
form of type 3 diabetes? Rejuvenation Res. 15:217–221. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Farris W, Mansourian S, Chang Y, Lindsley
L, Eckman EA, Frosch MP, Eckman CB, Tanzi RE, Selkoe DJ and
Guenette S: Insulin-degrading enzyme regulates the levels of
insulin, amyloid beta-protein, and the beta-amyloid precursor
protein intracellular domain in vivo. Proc Natl Acad Sci.
100:4162–4167. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Frank-Cannon TC, Alto LT, McAlpine FE and
Tansey MG: Does neuroinflammation fan the flame in
neurodegenerative diseases? Mol Neurodegener. 4:472009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kahn SE, Hull RL and Utzschneider KM:
Mechanisms linking obesity to insulin resistance and type 2
diabetes. Nature. 444:840–846. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Nadkarni P, Chepurny OG and Holz GG:
Regulation of glucose homeostasis by GLP-1. Prog Mol Biol Transl
Sci. 121:23–65. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Baggio LL and Drucker DJ: Biology of
incretins: GLP-1 and GIP. Gastroenterology. 132:2131–2157. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Manandhar B and Ahn JM: Glucagon-like
peptide-1 (GLP-1) analogs: Recent advances, new possibilities, and
therapeutic implications. J Med Chem. 58:1020–1037. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Hunter K and Hölscher C: Drugs developed
to treat diabetes, liraglutide and lixisenatide, cross the blood
brain barrier and enhance neurogenesis. BMC Neurosci. 13:332012.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Shirazi RH, Dickson SL and Skibicka KP:
Gut peptide GLP-1 and its analogue, Exendin-4, decrease alcohol
intake and reward. PLoS One. 8:e619652013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Kiesewetter DO, Gao H, Ma Y, Niu G, Quan
Q, Guo N and Chen X: 18F-radiolabeled analogs of exendin-4 for PET
imaging of GLP-1 in insulinoma. Eur J Nucl Med Mol Imaging.
39:463–473. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Eakin K, Li Y, Chiang YH, Hoffer BJ,
Rosenheim H, Greig NH and Miller JP: Exendin-4 ameliorates
traumatic brain injury-induced cognitive impairment in rats. PLoS
One. 8:e820162013. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Baudry M, Zhu G, Liu Y, Wang Y, Briz V and
Bi X: Multiple cellular cascades participate in long-term
potentiation and in hippocampus-dependent learning. Brain Res.
1621:73–81. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Li YF, Cheng YF, Huang Y, Conti M, Wilson
SP, O'Donnell JM and Zhang HT: Phosphodiesterase-4D knock-out and
RNA interference-mediated knock-down enhance memory and increase
hippocampal neurogenesis via increased cAMP signaling. J Neurosci.
31:172–183. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Brightwell JJ, Smith CA, Neve RL and
Colombo PJ: Long-term memory for place learning is facilitated by
expression of cAMP response element-binding protein in the dorsal
hippocampus. Learn Mem. 14:195–199. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Gonzalez GA and Montminy MR: Cyclic AMP
stimulates somatostatin gene transcription by phosphorylation of
CREB at serine 133. Cell. 59:675–680. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Xiong WX, Zhou GX, Wang B, Xue ZG, Wang L,
Sun HC and Ge SJ: Impaired spatial learning and memory after
sevoflurane-nitrous oxide anesthesia in aged rats is associated
with down-regulated cAMP/CREB signaling. PLoS One. 8:e794082013.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Luo J, Phan TX, Yang Y, Garelick MG and
Storm DR: Increases in cAMP, MAPK activity and CREB phosphorylation
during REM sleep: Implications for REM sleep and memory
consolidation. J Neurosci. 33:6460–6468. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Wang C, Yang XM, Zhuo YY, Zhou H, Lin HB,
Cheng YF, Xu JP and Zhang HT: The phosphodiesterase-4 inhibitor
rolipram reverses Aβ-induced cognitive impairment and
neuroinflammatory and apoptotic responses in rats. Int J
Neuropsychopharmacol. 15:749–766. 2012. View Article : Google Scholar : PubMed/NCBI
|