Cardiovascular disease (CVD) is a major health
problem worldwide and despite significant advances in treatment
approaches that led to reduced CVD-related mortality over the last
few years, the number of individuals living with damaged heart has
increased. It is well recognized that elevated plasma level of
low-density lipoprotein-cholesterol (LDL-C) is a cardinal risk
factor for atherosclerosis and CVD (1,2).
Deposition of pro-atherogenic LDL-C, on the intima of arterial
wall, a process mediated by macrophages, contributes to the plaque
formation and atherosclerosis. This further leads to lowered blood
flow to vital organs and increased risk of atherothrombotic and
atheroembolic sequelae. Several clinical studies have shown a
direct association between LDL-C reduction and coronary heart
disease prevention (1,3). It is known that every 1 mM decrease in
plasma LDL-C lowers the risk of cardiovascular events by 20–22%
(4,5). A target reduction of LDL-C to <2.5
mM for high-risk patients and <1.8 mM for very high-risk
patients has been suggested by the European guidelines for managing
dyslipidemia (6). The identification
of statins, the inhibitors of 3-hydroxy-3-methylglutaryl-coenzyme A
(HMG-CoA) reductase, the rate-limiting enzyme of cholesterol
biosynthesis, revolutionized the treatment of dyslipidemia and due
to their overwhelming success in reducing hypercholesterolemia and
reducing LDL-C they rapidly became the choice of first line therapy
for this indication (7). However,
statin therapy was effective in reducing dyslipidemia and
preventing cardiovascular events only in 50% of the patient
population (4) and in some patients,
particularly those with familial hypercholesterolemia, these drugs
were not effective to meet the required goals of lower LDL-C, and
to reduce the CVD risk (8). In
addition, many patients even develop intolerability to statins and
resistance (9).
Thus there has been a significant impetus to develop
alternate therapeutics to meet the requirements of reducing LDL-C
and the incidence of cardiovascular events. Several such programs
to develop novel therapeutics were not successful due to safety
issues and lack of better efficacy over statins (10,11),
except ezetimide, which has been found to further improve
dyslipidemia over statin therapy (12). Even combination of lipid lowering
drugs such as fibrates and nicotinic acid derivatives failed to
improve CVD events, when used in conjunction with statins (13). Thus, clinical guidelines do not
recommend the use of non-statin drugs in combination with
high-intensity statin treatment (14). The finding of pro-protein convertase
subtilisin/kexin type 9 (PCSK9) (15) and association of mutations in this
protein with familial hypercholesterolemia (16) led to the identification of PCSK9 as a
new therapeutic target for lowering LDL-C and
dyslipidemia-associated CVD.
PCSK9 belongs to a family of intracellular
‘convertase’ or subtilase enzymes that process precursor protein,
and are generally inactive, to active mature products that are
functional. Substrates for these convertases include a wide array
of precursor proteins, such as hormones, receptors, growth factors
and other enzymes (17). However,
PCSK9 per se does not have an enzymatic function except for
the autocatalytic cleavage of its own pro-domain, to become a
mature protein. PCSK9 is encoded by a 22-kb long gene
consisting of 12 exons and located at chromosome 1p32. PCSK9
gene codes for a 692-amino acid protein of 74 kDa molecular weight,
which later undergoes autocatalytic cleavage to the mature 62 kDa
form, in endoplasmic reticulum/Golgi bodies, from where it is
secreted into circulation. The cleaved prodomain remains
non-covalently associated with the mature 62 kDa protein and is
essential for the biological function of PCSK9 (18). PCSK9 is primarily synthesized in
hepatocytes but other tissues including intestine, brain and
kidneys are also known to express this protein (19,20). The
transcription factor sterol regulatory element-binding protein 2
(SREBP-2) upregulates PCSK9 expression (21) and LDL-receptor (LDLR) as well as
enzymes involved in cholesterol biosynthesis, such as HMG-CoA
reductase. Unlike other proconvertase enzymes, PCSK9 is secreted as
a complex of mature PCSK9 (153–692 aa) and its inhibitory
pro-segment (aa 32–152) (15,22).
This complex of PCSK9 is enzymatically inactive as its active site
is blocked by the inhibitory pro-segment and thus prevents it from
binding with any other substrates (23). Thus, it appears that PCSK9 is its own
substrate and its physiological activity probably resides in its
ability to bind specific target proteins to escort them towards
intracellular degradation compartments.
The first and probably the most studied and
important target of PCSK9 is LDLR on the hepatocyte surface in
liver (22). It has been shown in a
mouse model that PCSK9 inactivation leads to a significant
reduction in cholesteryl esters and atherosclerosis, whereas the
overexpression of PCSK9 led to opposite changes and excessive
atherosclerosis and all these effects of PCSK9 expression were
absent in the LDLR-KO mouse (24).
PCSK9 is shown to associate with the epidermal growth factor-A
(EGF-A) domain of LDLR and to other similar receptors such as the
VLDL receptor (25). Previous
studies have indicated that PCSK9 is involved in the regulation of
plasma triglyceride rich protein and thus it was observed that a
deficiency of PCSK9 is associated with significantly lowered plasma
triglycerides both in the clinical setting (26) and in genetically altered mice
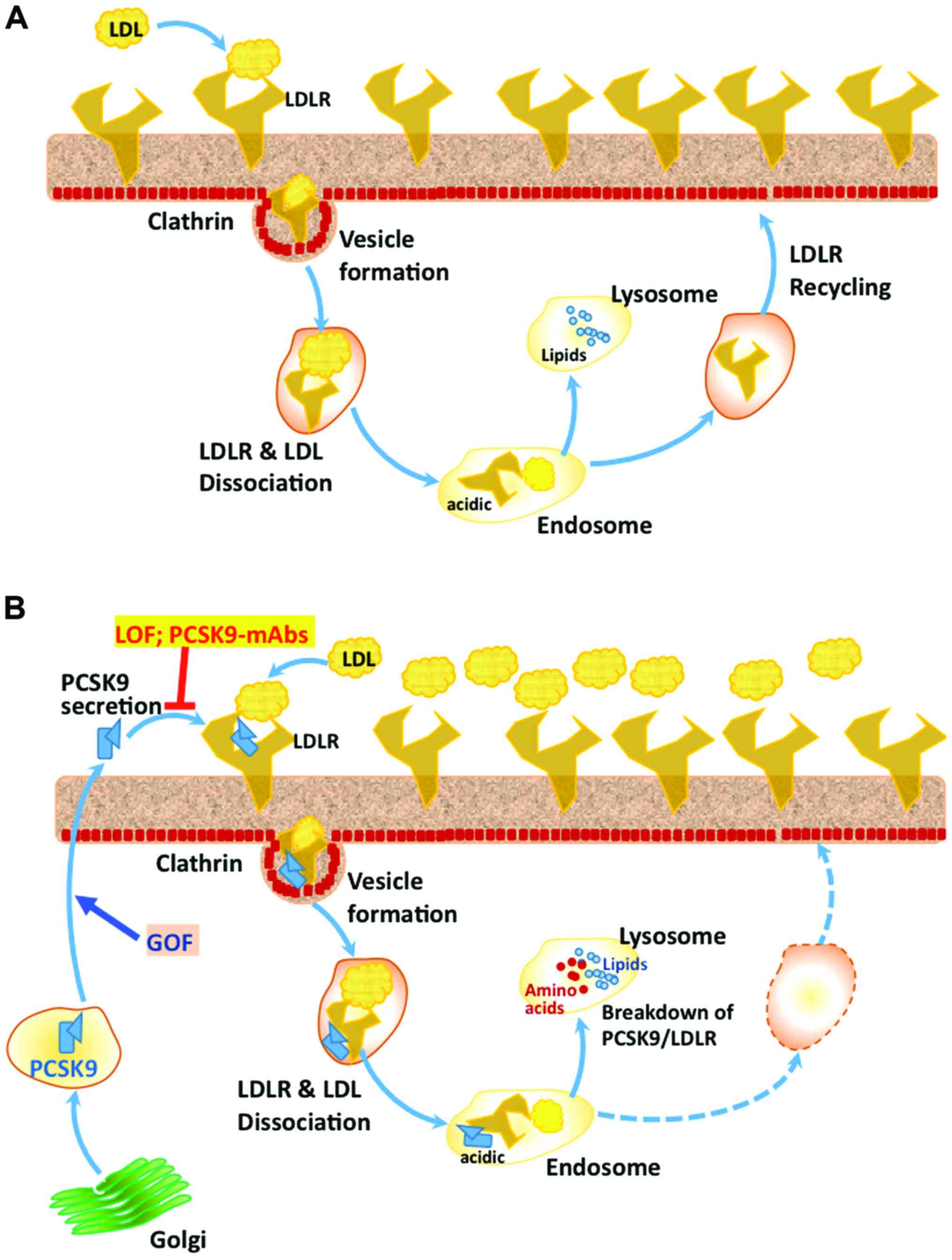
(27). The complex of LDL-C/LDLR
normally enters cells through clathrin heavy chain-coated vesicles,
followed by its dissociation in the acidic environment of endosomes
to LDLR and and LDL-C. LDLR is usually recycled back to the cell
surface, whereas the LDL-C is degraded in lysosomes, where the
recovered cholesterol is reutilized in the cell (Fig. 1) (28).
However, the complex of LDLR-PCSK9, which also
enters the cells through the clathrin-coated vesicles, does not
dissociate and is degraded in lysosomes, via some unknown mechanism
(29). PCSK9 is shown to direct LDLR
for degradation through intracellular pathways 48 and also after
its secretion extracellularly by binding to the LDLR (30). While there is ample evidence for
these two pathways in hepatocytes, it is not clear if the two
pathways are operational in other cells where PCSK9 is expressed.
The Cys/His-rich domain present at the C-terminus of PCSK9 appears
to be essential for the intracellular and the extracellular LDLR
degradation pathways mediated by PCSK9 (31). It has been suggested that the acidic
environment of endosomes causes His residues of CHRD domain to be
positively charged, making its interaction with the negatively
charged ligand-binding domain of LDLR stronger and facilitating its
targeting to lysosomes for degradation (Fig. 1) (32). Even though it has been suggested that
the amyloid precursor-like protein 2 (APLP-2) participates in the
targeting of PCSK9-LDLR complex to lysosomes, through its
interaction with CHRD at the surface (33), knockdown of APLP-2 had no effect on
PCSK9-mediated LDLR degradation, either via the intracellular or
extracellular pathways (18).
Interaction of plasma LDL, in particular, the ApoB component of
LDL, can bind to circulating PCSK9 and compromise its ability to
bind with cell surface LDLR. Thus several players influence the
PCSK9-mediated targeting and degradation of LDLR, including protein
X, ApoB, and sec24a, although the mechanistic details of these
regulations are not entirely clear. Thus, there is a positive
correlation between circulating PCSK9 and LDL-C, as increased
levels of PCSK9 lead to LDLR degradation, rendering the cells
unable to import LDL-C from circulation. By contrast, binding to
LDLR followed by intracellular clearance seems to be the major
route for the removal of PCSK9 from circulation, as demonstrated in
LDLR-KO mice (34) and in homozygous
familial hypercholesterolemia patients (35).
Besides the gain-of-function mutations, mutations
that led to its loss of function were also identified and were
found to be beneficial. Thus, it was observed that carriers of two
nonsense PCSK9 variants, Y142X and C679X, decreased plasma LDL-C by
~1.0 mM and the incidence of CVD in these subjects was reduced by
~88%. Similarly, another PCSK9 variant, R46L, is known to be
associated with an LDL-C reduction of 0.5 mM, and a 47% reduction
in CVD risk (39). Protein stability
of R46L mutant PCSK9 is reported to be decreased with increased
degradation, as well as its affinity for LDLR (40). There is also certain
ethnicity-dependent occurrence of a given mutation. Thus, Y142X and
C679X mutations are seen in 2% of African-Americans whereas R46L is
present in 2–4% of Caucasian populations (41). R46L mutation is also reported to
protect against myocardial infarction (42). Other losses of function PCSK9
mutations are the truncating mutations W428X and L82X (43). In an interesting case of compound
heterozygous woman, Y142X mutation of PCSK9 was inherited from the
mother and c290-292 delGCC mutation was from the father. The woman
had no detectable PCSK9 in circulation with only 0.36 mM LDL-C and
lived a healthy normal life, indicating that PCSK9 is not necessary
for normal life (44). A specific
French-Canadian mutation Q152H, which causes loss of function of
PCSK9, has been shown to prevent the autocatalytic processing of
proPCSK9, leading to the formation of a dominant negative form of
PCSK9, which reduces the circulating levels of PCSK9 and also LDL-C
by ~50% (45).
Several strategies have been examined to develop
PCSK9 antagonists because of the promise they hold as therapeutic
agents against dyslipidemia and associated CVD. Attempts to develop
small molecule inhibitors, peptidomimetics, were not successful
because of specificity and accessibility issues. To date it appears
that monoclonal antibodies against PCSK9 are the best approach to
block PCSK9 function (Fig. 1), in a
therapeutic manner (18,46). Many pharmaceutical companies such as
Sanofi (Gentilly, France) and Amgen (Thousand Oaks, CA, USA) have
developed different therapeutic monoclonal antibodies against PCSK9
(evolocumab, alirocumab and bococizumab), which have been
clinically tested in several trials and found to be safe and
efficacious in reducing LDL-C and also triglycerides.
Evolocumab (AMG145), developed by Amgen, is a fully
human monoclonal antibody inhibitor of PCSK9 and several
meta-analyses recently have shown that this mAb is well tolerated
and more efficacious than statins in patients with familial
hypercholesterolemia (47,48). Evolocumab was also found to reduce
cardiovascular outcomes in statin treated patients (49). Alirocumab (SAR236553/REGN727), also a
fully humanized monoclonal antibody was developed by Sanofi and is
found to be almost equipotent as evolocumab in controlling LDL-C,
triglycerides and also cardiovascular outcomes (49).
Clinical experience with the anti-PCSK9-monoclonal
antibodies indicates that these therapeutic agents are safe and
well tolerated, without any major safety issues and serious
drug-related adverse events (50).
The most common adverse events were nasopharyngitis, upper
respiratory tract infections, influenza-like symptoms and back pain
(51). Even though treatment related
adverse effects were not noted in patients receiving PCSK9 mAbs,
caution needs to be exercised for possible hypocholesterolemia
associated adverse effects such as cognitive impairment as well as
hemorrhagic stroke (49). More
clinical data are needed to ascertain if these are potential
problems or of not much concern.
Plasma level of LDL-C is a major risk factor for
atherosclerosis and CVD. The most commonly used statin therapy is
effective in reducing dyslipidemia and preventing cardiovascular
events only in about half of the patient population and these drugs
are not effective in patients with familial hypercholesterolemia,
to meet the required goals of lower LDL-C, to reduce the CVD risk.
The finding of PCSK9, which promotes LDLR breakdown and association
of PCSK9 gain of mutations with familial hypercholesterolemia and
loss of mutations with reduced levels of LDL-C, led to the
identification of PCSK9 as a new therapeutic target for lowering
LDL-C and dyslipidemia associated CVD. Monoclonal antibodies
against PCSK9 are currently being tested in clinical trials and
have been found to be safe without any major adverse effects and
efficacious in countering the activity of PCSK9 and thus control
the plasma LDL-C and triglycerides even in statin-non responsive
patients and protect against dyslipidemia-related CVD. Further
research is needed to clearly establish their protective effect
against CVD and related mortality.
|
1
|
Law MR, Wald NJ and Rudnicka AR:
Quantifying effect of statins on low density lipoprotein
cholesterol, ischaemic heart disease, and stroke: systematic review
and meta-analysis. BMJ. 326:14232003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mendis S, Puska P and Norrving B: Global
atlas on cardiovascular disease prevention and control. World
Health Organization; Geneva: 2011
|
|
3
|
Brown MS and Goldstein JL: Biomedicine.
Lowering LDL - not only how low, but how long? Science.
311:1721–1723. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mihaylova B, Emberson J, Blackwell L,
Keech A, Simes J, Barnes EH, Voysey M, Gray A, Collins R and
Baigent C: Cholesterol Treatment Trialists' (CTT) Collaborators:
the effects of lowering LDL cholesterol with statin therapy in
people at low risk of vascular disease: meta-analysis of individual
data from 27 randomised trials. Lancet. 380:581–590. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Baigent C, Blackwell L, Emberson J,
Holland LE, Reith C, Bhala N, Peto R, Barnes EH, Keech A, Simes J,
et al: Cholesterol Treatment Trialists' (CTT) Collaboration:
Efficacy and safety of more intensive lowering of LDL cholesterol:
a meta-analysis of data from 170,000 participants in 26 randomised
trials. Lancet. 376:1670–1681. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Reiner Z, Catapano AL, De Backer G, Graham
I, Taskinen MR, Wiklund O, Agewall S, Alegria E, Chapman MJ,
Durrington P, et al: European Association for Cardiovascular
Prevention & Rehabilitation; ESC Committee for Practice
Guidelines (CPG) 2008–2010 and 2010–2012 Committees: ESC/EAS
Guidelines for the management of dyslipidaemias: The Task Force for
the management of dyslipidaemias of the European Society of
Cardiology (ESC) and the European Atherosclerosis Society (EAS).
Eur Heart J. 32:1769–1818. 2011.PubMed/NCBI
|
|
7
|
Koo BK: Statin for the primary prevention
of cardiovascular disease in patients with diabetes mellitus.
Diabetes Metab J. 38:32–34. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Joy TR: Novel therapeutic agents for
lowering low density lipoprotein cholesterol. Pharmacol Ther.
135:31–43. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Pijlman AH, Huijgen R, Verhagen SN, Imholz
BP, Liem AH, Kastelein JJ, Abbink EJ, Stalenhoef AF and Visseren
FL: Evaluation of cholesterol lowering treatment of patients with
familial hypercholesterolemia: a large cross-sectional study in The
Netherlands. Atherosclerosis. 209:189–194. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Barter PJ, Caulfield M, Eriksson M, Grundy
SM, Kastelein JJ, Komajda M, Lopez-Sendon J, Mosca L, Tardif JC,
Waters DD, et al: ILLUMINATE Investigators: Effects of torcetrapib
in patients at high risk for coronary events. N Engl J Med.
357:2109–2122. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Landray MJ, Haynes R, Hopewell JC, Parish
S, Aung T, Tomson J, Wallendszus K, Craig M, Jiang L, Collins R, et
al: HPS2-THRIVE Collaborative Group: Effects of extended-release
niacin with laropiprant in high-risk patients. N Engl J Med.
371:203–212. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cannon CP, Blazing MA, Giugliano RP,
McCagg A, White JA, Theroux P, Darius H, Lewis BS, Ophuis TO,
Jukema JW, et al: IMPROVE-IT Investigators: Ezetimibe added to
statin therapy after acute coronary syndromes. N Engl J Med.
372:2387–2397. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ginsberg HN, Elam MB, Lovato LC, Crouse JR
III, Leiter LA, Linz P, Friedewald WT, Buse JB, Gerstein HC,
Probstfield J, et al: ACCORD Study Group: Effects of combination
lipid therapy in type 2 diabetes mellitus. N Engl J Med.
362:1563–1574. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Stone NJ, Robinson JG, Lichtenstein AH,
Merz CN Bairey, Blum CB, Eckel RH, Goldberg AC, Gordon D, Levy D,
Lloyd-Jones DM, et al: American College of Cardiology/American
Heart Association Task Force on Practice Guidelines: 2013 ACC/AHA
guideline on the treatment of blood cholesterol to reduce
atherosclerotic cardiovascular risk in adults: a report of the
American College of Cardiology/American Heart Association Task
Force on Practice Guidelines. J Am Coll Cardiol. 63:2889–2934.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Seidah NG, Benjannet S, Wickham L,
Marcinkiewicz J, Jasmin SB, Stifani S, Basak A, Prat A and Chretien
M: The secretory proprotein convertase neural apoptosis-regulated
convertase 1 (NARC-1): liver regeneration and neuronal
differentiation. Proc Natl Acad Sci USA. 100:928–933. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Abifadel M, Varret M, Rabès JP, Allard D,
Ouguerram K, Devillers M, Cruaud C, Benjannet S, Wickham L, Erlich
D, et al: Mutations in PCSK9 cause autosomal dominant
hypercholesterolemia. Nat Genet. 34:154–156. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Seidah NG, Sadr MS, Chrétien M and Mbikay
M: The multifaceted proprotein convertases: their unique,
redundant, complementary, and opposite functions. J Biol Chem.
288:21473–21481. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Seidah NG, Awan Z, Chrétien M and Mbikay
M: PCSK9: a key modulator of cardiovascular health. Circ Res.
114:1022–1036. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Artenstein AW and Opal SM: Proprotein
convertases in health and disease. N Engl J Med. 365:2507–2518.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Seidah NG and Prat A: The biology and
therapeutic targeting of the proprotein convertases. Nat Rev Drug
Discov. 11:367–383. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Jeong HJ, Lee HS, Kim KS, Kim YK, Yoon D
and Park SW: Sterol-dependent regulation of proprotein convertase
subtilisin/kexin type 9 expression by sterol-regulatory element
binding protein-2. J Lipid Res. 49:399–409. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Benjannet S, Rhainds D, Essalmani R, Mayne
J, Wickham L, Jin W, Asselin MC, Hamelin J, Varret M, Allard D, et
al: NARC-1/PCSK9 and its natural mutants: Zymogen cleavage and
effects on the low density lipoprotein (LDL) receptor and LDL
cholesterol. J Biol Chem. 279:48865–48875. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cunningham D, Danley DE, Geoghegan KF,
Griffor MC, Hawkins JL, Subashi TA, Varghese AH, Ammirati MJ, Culp
JS, Hoth LR, et al: Structural and biophysical studies of PCSK9 and
its mutants linked to familial hypercholesterolemia. Nat Struct Mol
Biol. 14:413–419. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Denis M, Marcinkiewicz J, Zaid A, Gauthier
D, Poirier S, Lazure C, Seidah NG and Prat A: Gene inactivation of
proprotein convertase subtilisin/kexin type 9 reduces
atherosclerosis in mice. Circulation. 125:894–901. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lo Surdo P, Bottomley MJ, Calzetta A,
Settembre EC, Cirillo A, Pandit S, Ni YG, Hubbard B, Sitlani A and
Carfí A: Mechanistic implications for LDL receptor degradation from
the PCSK9/LDLR structure at neutral pH. EMBO Rep. 12:1300–1305.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Akram ON, Bernier A, Petrides F, Wong G
and Lambert G: Beyond LDL cholesterol, a new role for PCSK9.
Arterioscler Thromb Vasc Biol. 30:1279–1281. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Le May C, Kourimate S, Langhi C,
Chétiveaux M, Jarry A, Comera C, Collet X, Kuipers F, Krempf M,
Cariou B, et al: Proprotein convertase subtilisin kexin type 9 null
mice are protected from postprandial triglyceridemia. Arterioscler
Thromb Vasc Biol. 29:684–690. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Huang S, Henry L, Ho YK, Pownall HJ and
Rudenko G: Mechanism of LDL binding and release probed by
structure-based mutagenesis of the LDL receptor. J Lipid Res.
51:297–308. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Nassoury N, Blasiole DA, Oler A Tebon,
Benjannet S, Hamelin J, Poupon V, McPherson PS, Attie AD, Prat A
and Seidah NG: The cellular trafficking of the secretory proprotein
convertase PCSK9 and its dependence on the LDLR. Traffic.
8:718–732. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Cameron J, Holla OL, Ranheim T, Kulseth
MA, Berge KE and Leren TP: Effect of mutations in the PCSK9 gene on
the cell surface LDL receptors. Hum Mol Genet. 15:1551–1558. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Saavedra YG, Day R and Seidah NG: The M2
module of the Cys-His-rich domain (CHRD) of PCSK9 protein is needed
for the extracellular low-density lipoprotein receptor (LDLR)
degradation pathway. J Biol Chem. 287:43492–43501. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Tveten K, Holla OL, Cameron J, Strøm TB,
Berge KE, Laerdahl JK and Leren TP: Interaction between the
ligand-binding domain of the LDL receptor and the C-terminal domain
of PCSK9 is required for PCSK9 to remain bound to the LDL receptor
during endosomal acidification. Hum Mol Genet. 21:1402–1409. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
DeVay RM, Shelton DL and Liang H:
Characterization of proprotein convertase subtilisin/kexin type 9
(PCSK9) trafficking reveals a novel lysosomal targeting mechanism
via amyloid precursor-like protein 2 (APLP2). J Biol Chem.
288:10805–10818. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Tavori H, Fan D, Blakemore JL, Yancey PG,
Ding L, Linton MF and Fazio S: Serum proprotein convertase
subtilisin/kexin type 9 and cell surface low-density lipoprotein
receptor: Evidence for a reciprocal regulation. Circulation.
127:2403–2413. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Raal F, Scott R, Somaratne R, Bridges I,
Li G, Wasserman SM and Stein EA: Low-density lipoprotein
cholesterol-lowering effects of AMG 145, a monoclonal antibody to
proprotein convertase subtilisin/kexin type 9 serine protease in
patients with heterozygous familial hypercholesterolemia: The
Reduction of LDL-C with PCSK9 Inhibition in Heterozygous Familial
Hypercholesterolemia Disorder (RUTHERFORD) randomized trial.
Circulation. 126:2408–2417. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Leren TP: Mutations in the PCSK9 gene in
Norwegian subjects with autosomal dominant hypercholesterolemia.
Clin Genet. 65:419–422. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Stefanutti C, Morozzi C and Di Giacomo S:
New clinical perspectives of hypolipidemic drug therapy in severe
hypercholesterolemia. Curr Med Chem. 19:4861–4868. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Allard D, Amsellem S, Abifadel M, Trillard
M, Devillers M, Luc G, Krempf M, Reznik Y, Girardet JP, Fredenrich
A, et al: Novel mutations of the PCSK9 gene cause variable
phenotype of autosomal dominant hypercholesterolemia. Hum Mutat.
26:4972005. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Cohen JC, Boerwinkle E, Mosley TH Jr and
Hobbs HH: Sequence variations in PCSK9, low LDL, and protection
against coronary heart disease. N Engl J Med. 354:1264–1272. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Dewpura T, Raymond A, Hamelin J, Seidah
NG, Mbikay M, Chrétien M and Mayne J: PCSK9 is phosphorylated by a
Golgi casein kinase-like kinase ex vivo and circulates as a
phosphoprotein in humans. FEBS J. 275:3480–3493. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Benn M, Nordestgaard BG, Grande P, Schnohr
P and Tybjaerg-Hansen A: PCSK9 R46L, low-density lipoprotein
cholesterol levels, and risk of ischemic heart disease: 3
independent studies and meta-analyses. J Am Coll Cardiol.
55:2833–2842. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kathiresan S: Myocardial Infarction
Genetics Consortium: A PCSK9 missense variant associated with a
reduced risk of early-onset myocardial infarction. N Engl J Med.
358:2299–2300. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Marais AD, Kim JB, Wasserman SM and
Lambert G: PCSK9 inhibition in LDL cholesterol reduction: Genetics
and therapeutic implications of very low plasma lipoprotein levels.
Pharmacol Ther. 145:58–66. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhao Z, Tuakli-Wosornu Y, Lagace TA, Kinch
L, Grishin NV, Horton JD, Cohen JC and Hobbs HH: Molecular
characterization of loss-of-function mutations in PCSK9 and
identification of a compound heterozygote. Am J Hum Genet.
79:514–523. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
45
|
Mayne J, Dewpura T, Raymond A, Bernier L,
Cousins M, Ooi TC, Davignon J, Seidah NG, Mbikay M and Chrétien M:
Novel loss-of-function PCSK9 variant is associated with low plasma
LDL cholesterol in a French-Canadian family and with impaired
processing and secretion in cell culture. Clin Chem. 57:1415–1423.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Druce I, Abujrad H and Ooi TC: PCSK9 and
triglyceride-rich lipoprotein metabolism. J Biomed Res.
29:292015.
|
|
47
|
Giugliano RP, Desai NR, Kohli P, Rogers
WJ, Somaratne R, Huang F, Liu T, Mohanavelu S, Hoffman EB, McDonald
ST, et al: LAPLACE-TIMI 57 Investigators: Efficacy, safety, and
tolerability of a monoclonal antibody to proprotein convertase
subtilisin/kexin type 9 in combination with a statin in patients
with hypercholesterolaemia (LAPLACE-TIMI 57): a randomised,
placebo-controlled, dose-ranging, phase 2 study. Lancet.
380:2007–2017. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Raal FJ, Stein EA, Dufour R, Turner T,
Civeira F, Burgess L, Langslet G, Scott R, Olsson AG, Sullivan D,
et al: RUTHERFORD-2 Investigators: PCSK9 inhibition with evolocumab
(AMG 145) in heterozygous familial hypercholesterolaemia
(RUTHERFORD-2): a randomised, double-blind, placebo-controlled
trial. Lancet. 385:331–340. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Latimer J, Batty JA, Neely RD and Kunadian
V: PCSK9 inhibitors in the prevention of cardiovascular disease. J
Thromb Thrombolysis. Apr 19–2016.(Epub ahead of print). View Article : Google Scholar
|
|
50
|
Cicero AF, Tartagni E and Ertek S: Safety
and tolerability of injectable lipid-lowering drugs: a review of
available clinical data. Expert Opin Drug Saf. 13:1023–1030. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Roth EM, Taskinen MR, Ginsberg HN,
Kastelein JJ, Colhoun HM, Robinson JG, Merlet L, Pordy R and
Baccara-Dinet MT: Monotherapy with the PCSK9 inhibitor alirocumab
versus ezetimibe in patients with hypercholesterolemia: results of
a 24 week, double-blind, randomized phase 3 trial. Int J Cardiol.
176:55–61. 2014. View Article : Google Scholar : PubMed/NCBI
|