Introduction
Breast cancer is the most common malignant tumor
type in females, and the second most common cancer type worldwide
(1). It is the leading cause of
cancer mortality in women, responsible for 14% of all
cancer-related fatalities (2,3). Notable
improvements in the diagnosis and treatment of breast cancer have
been made in recent decades, and the mortality rate of this disease
has decreased by more than 30% (2,3). A
better understanding of the molecular mechanisms underlying the
malignant progression of breast cancer will be beneficial for
improving the efficacy of breast cancer treatment strategies
(4).
Hormone 17-β-estradiol (E2), one of the most
prevalent endogenous estrogens, has been found to be involved in
physiological and pathological processes, including the development
and malignant progression of breast cancer (5). E2 functions through activation of
estrogen receptor (ER) α-mediated signaling pathways, which are
involved in the mediation of cell proliferation, differentiation
and migration, as well as homeostasis (6–8). Almost
70% of breast cancers are ERα positive (9), thus ERα has been developed into a
therapeutic target for breast cancer (10,11). For
instance, tamoxifen is a selective ER modulator and represents the
standard treatment for the majority of breast cancer patients
(9). However, the regulatory
mechanism of ERα expression in breast cancer is still largely
unknown.
MicroRNAs (miRs) are endogenous non-coding RNAs that
can suppress gene expression by directly binding to the
3′untranslated region (3′UTR), causing translation inhibition or
mRNA degradation (12). They have
been demonstrated to play crucial roles in various biological
processes, such as cell proliferation, differentiation, apoptosis
and migration, as well as tumorigenesis (13,14).
Moreover, the development and malignant progression of breast
cancer is tightly associated with gene mutation and deregulation,
as well as epigenetic mechanisms including miRs (15,16).
Previously, downregulation of miR-148a has been implicated in
breast cancer (17,18) and found to inhibit the migration and
invasion of breast cancer cells (19,20). For
instance, Jiang et al recently found that miR-148a
suppressed the migration and invasion of breast cancer cells by
directly targeting WNT-1 (19). Xue
et al reported that miR-148a inhibited the migration of
breast cancer cells by targeting MMP-13 (20). However, to the best of our knowledge,
the exact role of miR-148a in ERα-positive breast cancer remains
unclear. In the present study, we aimed to reveal the underlying
mechanism of miR-148a in mediating E2-induced viability and
migration of ERα-positive breast cancer cells.
Materials and methods
Cell culture and E2 treatment
A human embryonic kidney cell line, HEK293 and a
human breast cancer cell line, MCF7, were obtained from the Cell
Bank of Central South University (Changsha, China). MCF7 cells were
cultured in Dulbecco's modified Eagle's medium (DMEM; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) with 10% fetal bovine serum
(Thermo Fisher Scientific, Inc.) at 37°C with 5% CO2.
MCF7 cells were treated with E2 for 3 h, then cell viability and
migration rates were analyzed.
MTT assay
An MTT assay was used to examine cell viability.
MCF7 cells (5,000 per well) were cultured in a 96-well plate. Each
well contained 100 µl fresh serum-free DMEM with 0.5 g/l MTT.
Following an incubation at 37°C for 0, 12, 24, 48 or 72 h, the
medium was removed by aspiration and 50 µl dimethyl sulfoxide (5
mg/ml; Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) was added to
each well. After incubation at room temperature for 10 min,
formazan production was detected by measuring the optical density
at 570 nm using an AIA 600II Automated Enzyme Immunoassay Analyzer
(Tosoh Corporation, Tokyo, Japan).
Wound healing assay
A wound healing assay was used to examine cell
migration. MCF7 cells were cultured at 37°C with 5% CO2
to full confluence in 6-well plates. Wounds of ~1 mm width were
created with a plastic scriber. After that, cells were washed with
phosphate-buffered saline (PBS) and incubated at 37°C with 5%
CO2 for 48 h. Then, the wounds were observed and
photographed under an Eclipse Ti-E inverted microscope (Nikon
Corporation, Tokyo, Japan).
Cell transfection
MCF7 cells were transfected with a scrambled miR
mimic (Yearthbio, Changsha, China) as a negative control (miR-NC),
an miR-148a mimic (Yearthbio), a negative control inhibitor
(Yearthbio), an miR-148a inhibitor (Yearthbio), ERα siRNA
(Yearthbio) or pcDNA3.1-ERα ORF plasmid (Yearthbio) using
Lipofectamine® 2000 (Thermo Fisher Scientific, Inc.), in
accordance with the manufacturer's protocols. Transfection
efficiency was measured using reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) and western blotting as
follows.
RT-qPCR assay
Total RNA from MCF7 cells was isolated using Trizol
reagent (Thermo Fisher Scientific, Inc.), according to the
manufacturer's instructions. Total RNA was reverse transcribed
using the RevertAid Reverse Transcription kit (Thermo Fisher
Scientific, Inc.) according to the manufacturer's instructions.
Then, qPCR was conducted using the SYBR-Green miScript PCR kit
(Qiagen, Inc., Valencia, CA, USA) and a Roche LightCycler 480 PCR
machine (Roche Diagnostics, Basel, Switzerland). All primers were
purchased from Sangon Biotech Co., Ltd. (Shanghai, China). The
primers for ERα were: Forward, 5′-CCCACTCAACAGCGTGTCTC-3′ and
reverse, 5′-CGTCGATTATCTGAATTTGGCCT-3′. The primers for GAPDH were:
Forward, 5′-ACAACTTTGGTATCGTGGAAGG-3′ and reverse,
5′-GCCATCACGCCACAGTTTC-3′. The reaction conditions were: 95°C for
10 min, followed by 45 cycles of 95°C for 15 sec and 60°C for 30
sec. The relative mRNA expression of ERα was normalized against
that of GAPDH. The relative expression of miR-148a was normalized
against that of U6. The relative expression was analyzed by the
2−ΔΔCq method (21).
Western blot analysis
Cells were lysed using RIPA buffer (Beyotime
Institute of Biotechnology, Shanghai, China). The concentration of
protein was quantified using the Pierce BCA Protein Assay kit
(Thermo Fisher Scientific, Inc.). After that, proteins (50 µg) were
separated using 10% SDS-PAGE gels (Beyotime Institute of
Biotechnology) and blotted onto a polyvinylidene difluoride (PVDF)
membrane (Life Technologies, Grand Island, NY, USA), which was then
blocked with 5% non-fat dried milk (Mengniu, Beijing, China) in PBS
with Tween (PBST; Beyotime Institute of Biotechnology) overnight at
4°C. The PVDF membrane was then incubated with rabbit monoclonal
anti-human ERα antibody (1:100; ab32063; Abcam, Cambridge, MA,
USA), or rabbit monoclonal anti-human GAPDH antibody (1:100;
ab9485; Abcam) antibody at room temperature for 3 h. After being
washed with PBST three times, the PVDF membrane was incubated with
goat anti-rabbit horseradish peroxidase-conjugated secondary
antibody (1:5,000; ab7090; Abcam) for 40 min at room temperature.
Chemiluminescent detection was performed with an ECL kit (Thermo
Fisher Scientific, Inc.). The relative protein expression was
determined using Image-Pro plus software version 6.0 (Media
Cybernetics, Inc., Rockville, MD, USA), represented as the density
ratio vs. GAPDH.
Bioinformatics analysis and luciferase
reporter assay
TargetScan 3.1 online software (Whitehead Institute
for Biomedical Research, Cambridge, MA, USA) was used to analyze
the putative target genes of miR-148a (22).
A wild type (WT)-or mutant type (MT)-ERα 3′UTR was
inserted downstream of the luciferase reporter gene in a
pMIR-REPORT vector (Yearthbio), generating a WT-ERα-3′UTR reporter
vector or MT-ERα-3′UTR reporter vector, respectively. HEK293 cells
were co-transfected with miR-148a mimic (Yearthbio) or miR-NC
(Yearthbio), a WT-ERα-3′UTR or MT-ERα-3′UTR reporter vector, and
pRL-SV40 (Promega Corporation, Madison, WI, USA) expressing Renilla
luciferase using Lipofectamine® 2000 according to the
manufacturer's protocol. Following incubation at 37°C with 5%
CO2 for 48 h, the luciferase activities were measured
using the Dual-Luciferase Reporter Assay System (Promega
Corporation), according to the manufacturer's instructions.
Statistical analysis
Data are expressed as the mean ± standard deviation
of three independent experiments. The Student's t-test was used to
compare differences between two groups. One-way analysis of
variance was used to analyze differences among more than two
groups. Statistical analysis was conducted using SPSS 17.0 software
(SPSS, Inc., Chicago, IL, USA) and P<0.05 was considered to
indicate a statistically significant difference.
Results
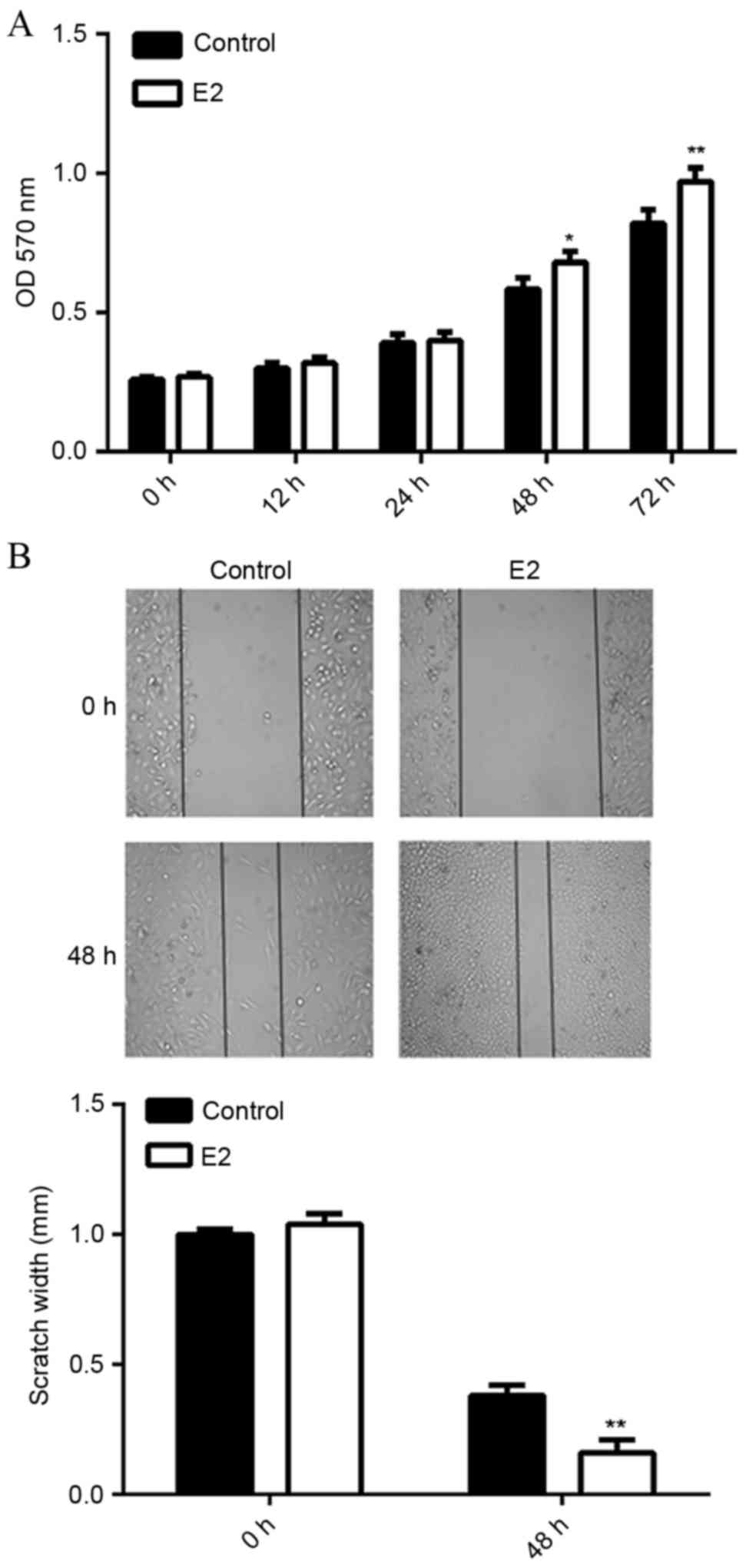
E2 treatment promotes MCF7 cell
viability and migration
It has been well established that MCF7 is an
ERα-positive breast cancer cell line (23). In the current study, MCF7 were
treated with E2 (1 mM) for 3 h, followed by analyses of cell
viability and migration. MTT assay data indicated that MCF7 cell
viability was significantly increased following treatment with E2
for 48 h (P<0.05) or 72 h (P<0.01), as compared with the
control group (Fig. 1A).
Furthermore, wound healing assay data showed that E2 treatment also
upregulated the migration of MCF7 cells at 48 h, when compared to
the control group (P<0.01; Fig.
1B). These results suggest that E2 activates ERα-mediated
viability- and migration-related signaling pathways in MCF7
cells.
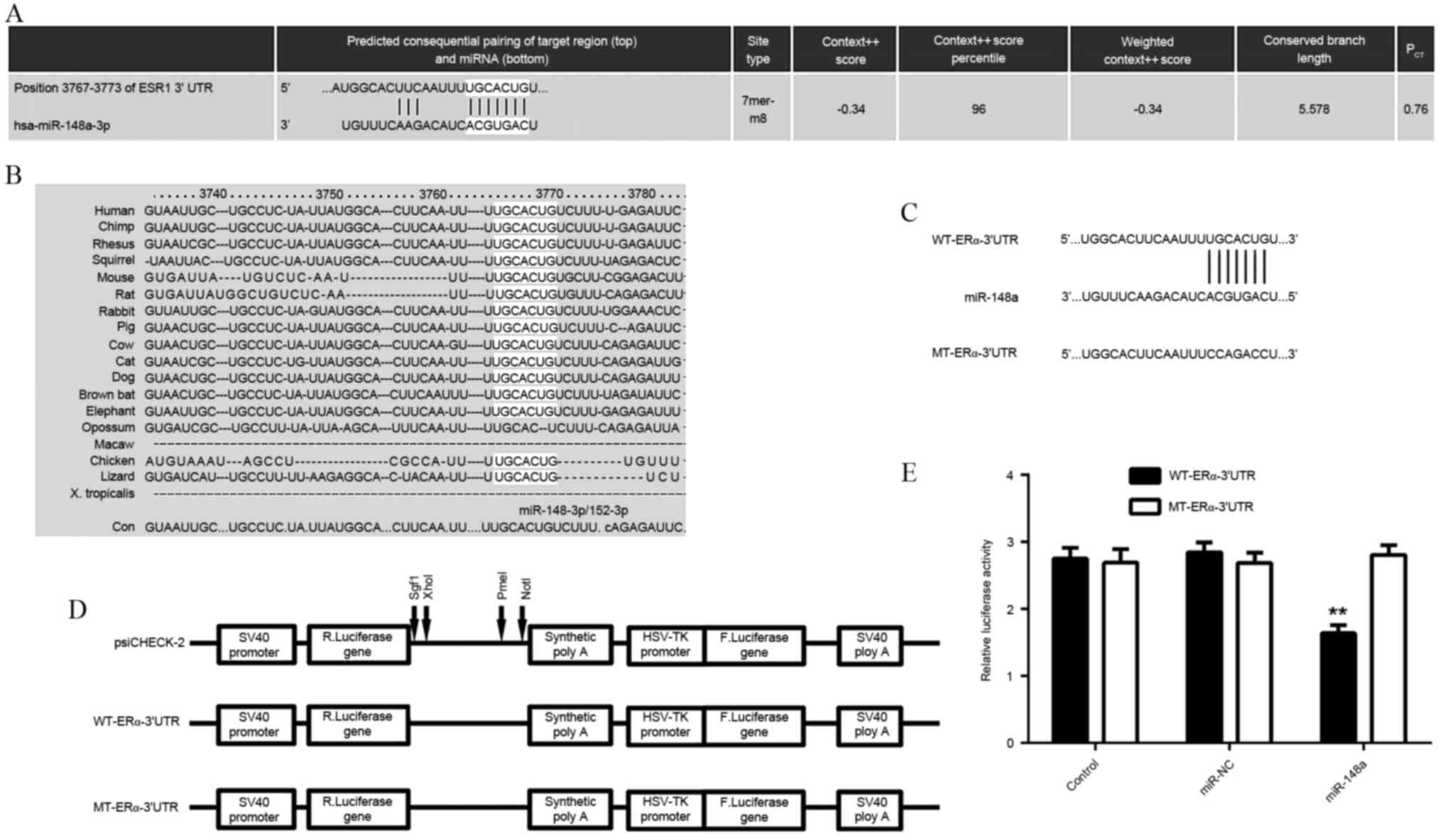
ERα is a direct target of
miR-148a
As the function of E2 is through activation of
ERα-mediated downstream signaling, a bioinformatics search was
conducted for putative miRs that directly target ERα. As indicated
in Fig. 2A and B, ERα was predicted
to be a direct target of miR-148a, and the targeting relationship
was evolutionarily conserved. In order to examine this targeting
relationship, the WT- or MT-ERα-3′UTR was inserted downstream of
the luciferase reporter gene in a pMIR-REPORT vector (Fig. 2C and D). A luciferase reporter assay
demonstrated that transfection with the miR-148a mimic
significantly reduced the luciferase activity in HEK293 cells
transfected with WT-ERα-3′UTR reporter vector, but not in cells
transfected with MT-ERα-3′UTR reporter vector, when compared to the
control group (P<0.01; Fig. 2E).
These findings indicated that ERα was a direct target of
miR-148a.
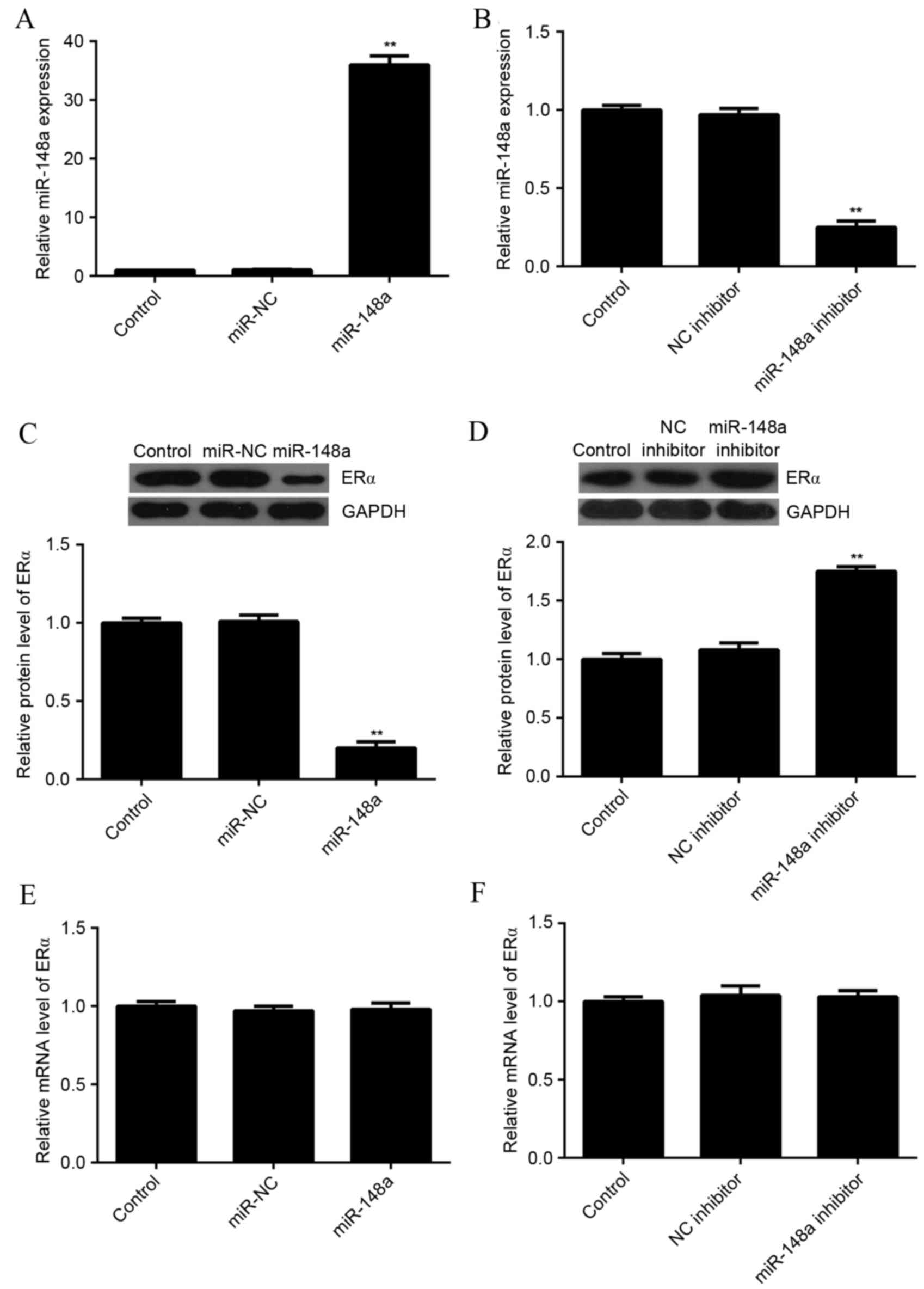
miR-148a negatively regulated the
protein expression of ERα in MCF7 cells
As miRs most often inhibit the expression of their
target genes at the post-transcriptional level, the effects of
miR-148a upregulation and downregulation on the protein level of
ERα in MCF7 cells were evaluated. MCF7 cells were transfected with
miR-NC, miR-148a mimic, NC inhibitor, or miR-148a inhibitor. After
transfection, qPCR was used to evaluate the level of miR-148a
expression in each group. As indicated in Fig. 3A and B, transfection with an miR-148a
mimic significantly increased the level of miR-148a expression,
while transfection with an miR-148a inhibitor significantly
decreased the level of miR-148a expression in MCF7 cells as
compared with the control group (P<0.01 for both).
Next, a western blot analysis was conducted to
determine the protein level of ERα. Overexpression of miR-148a
significantly reduced the level of ERα protein (Fig. 3C), while knockdown of miR-148a
significantly increased the level of ERα protein in MCF7 cells, as
compared with the control (P<0.01 for both; Fig. 3D). In addition, neither miR-148a
upregulation nor downregulation affected the mRNA expression of ERα
in MCF7 cells (Fig. 3E and F). These
results suggested that miR-148a negatively regulated the expression
of ERα at the post-transcriptional level in MCF7 cells.
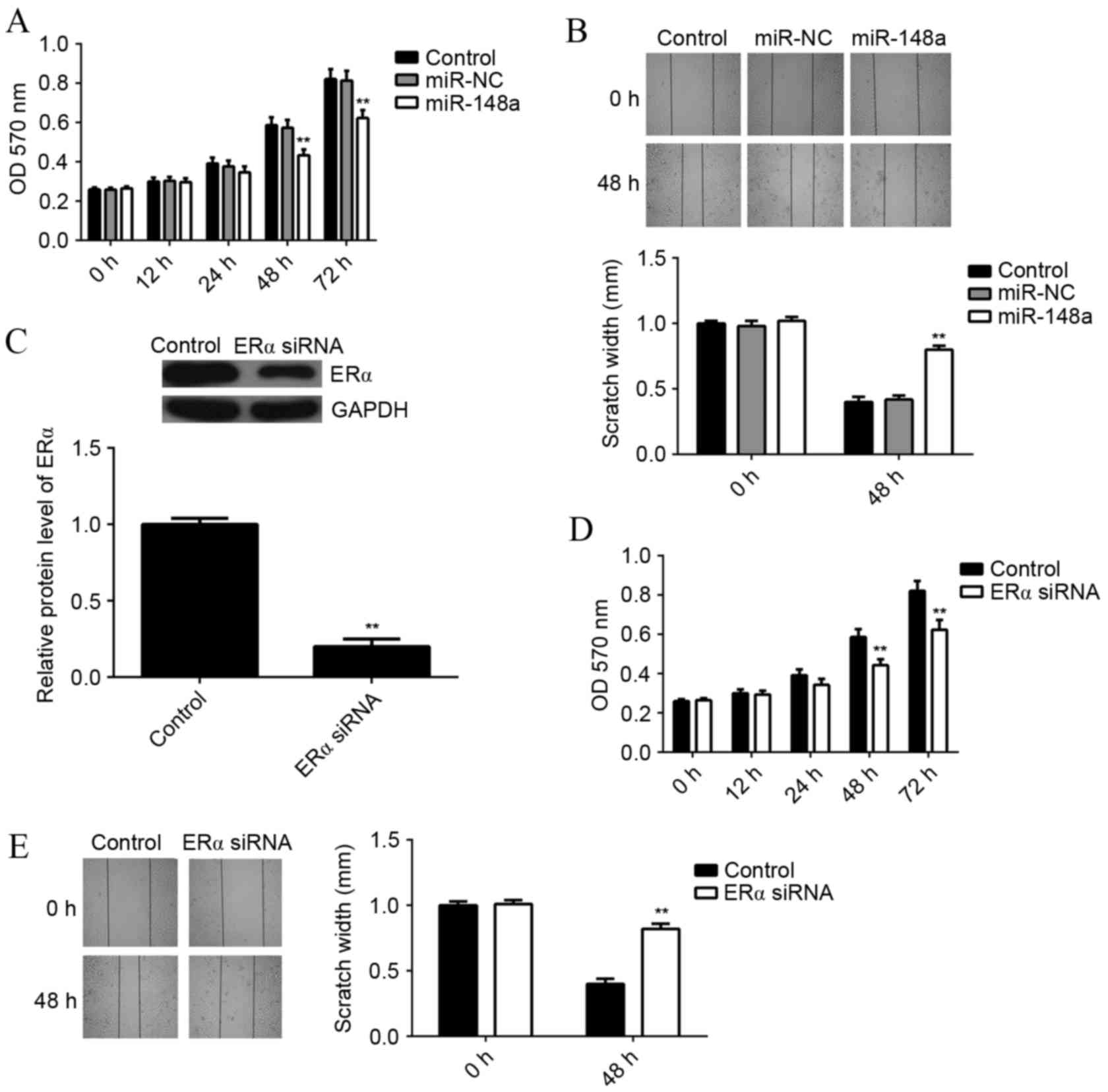
miR-148a overexpression or ERα
knockdown inhibits E2-induced MCF7 cell viability and
migration
As E2 functions through activation of ERα-mediated
signaling, it was examined whether miR-148a could affect E2-induced
MCF7 cell viability and migration. MCF7 cells with or without
miR-148a overexpression were treated with E2 for 3 h. An MTT assay
was then used to examine the cell viability in each group. It was
found that the viability rate of miR-148a-overexpressing cells was
significantly reduced at 48 and 72 h, as compared with the control
group (P<0.01; Fig. 4A). A wound
healing assay showed that the scratch width for
miR-148a-overexpressing cells was significantly larger at 48 h
compared with the control group, indicating that the migration rate
of miR-148a-overexpressing cells was significantly reduced
(P<0.01; Fig. 4B).
In order to investigate the underlying mechanism of
miR-148a further, MCF7 cells were transfected with ERα-specific
small interfering RNA (siRNA), which significantly decreased the
protein level of ERα as compared with the control (P<0.01;
Fig. 4C). MCF7 cells with or without
ERα knockdown were treated with E2 for 3 h. An MTT assay showed
that the viability of MCF7 cells was significantly decreased after
knockdown of ERα for 48 or 72 h, when compared to the control group
(P<0.01; Fig. 4D). A wound
healing assay showed that the scratch width for MCF7 ERα-knockdown
cells was significantly larger at 48 h compared with the control
group, indicating that the migration rate of MCF7 cells was
significantly decreased after knockdown of ERα (P<0.01; Fig. 4E). These results suggest that both
miR-148a overexpression and ERα knockdown result in suppressed
E2-induced viability and migration of MCF7 cells.
miR-148a inhibits E2-induced MCF7 cell
viability and migration via inhibition of ERα expression
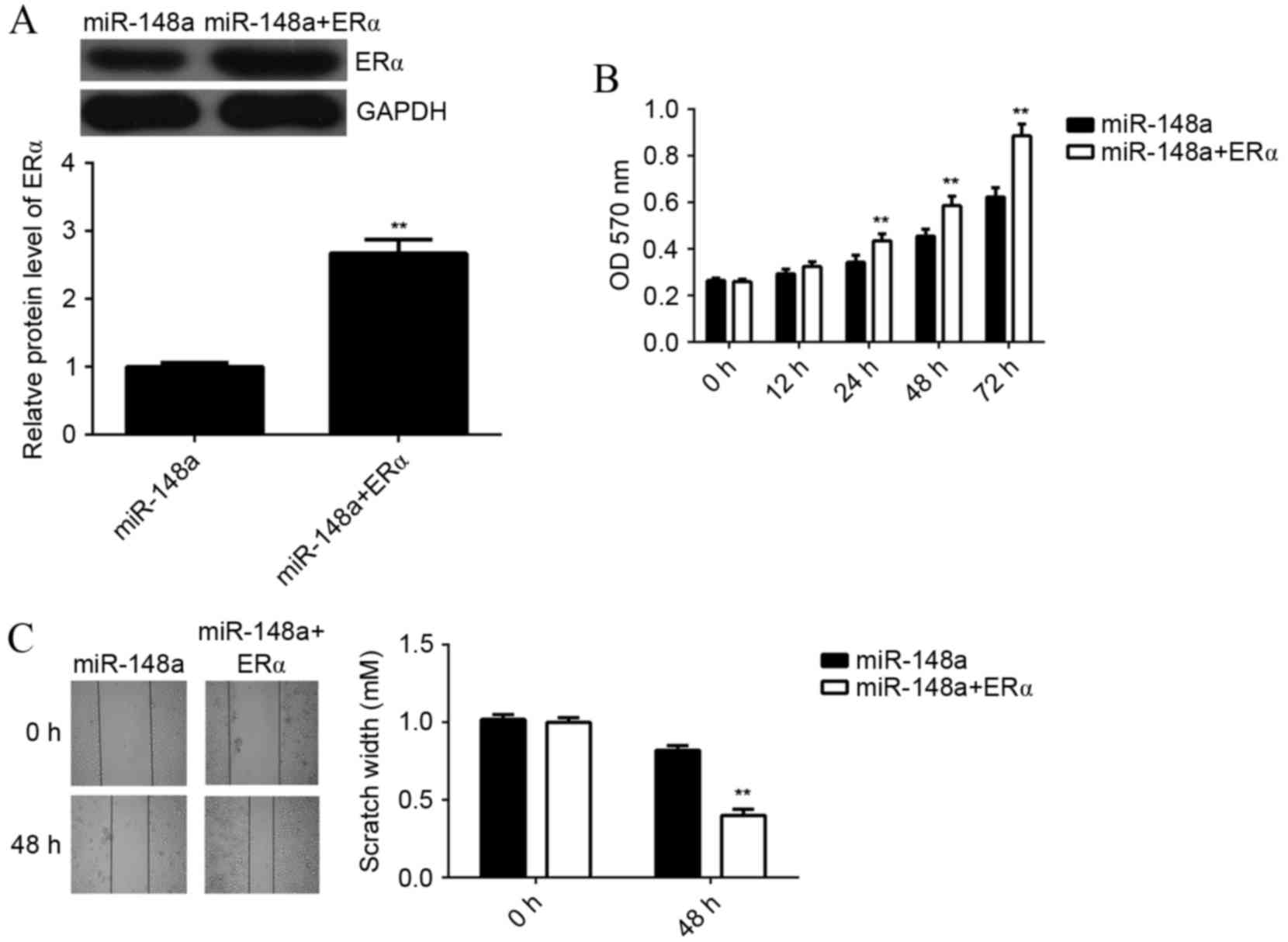
In order to investigate whether the effects of
miR-148a on E2-induced MCF7 cell viability and migration were
through directly targeting ERα, MCF7 cells were co-transfected with
miR-148a mimic and ERα ORF plasmid. A western blot analysis showed
that the protein level of ERα was significantly higher in cells
co-transfected with miR-148a mimic and ERα ORF plasmid, when
compared with cells transfected with miR-148a mimic alone
(P<0.01; Fig. 5A). This indicated
that transfection with ERα ORF plasmid rescued the suppressive
effect of miR-148 on the protein expression of ERα in MCF7 cells.
Next, MCF7 cells in each group were treated with E2 for 3 h, and
MTT and wound healing assays were performed. The viability of MCF7
cells was significantly increased in cells at 24, 48 and 72 h after
co-transfection with miR-148a mimic and ERα ORF plasmid, as
compared with miR-148a-overexpressing cells without plasmid
(P<0.01; Fig. 5B). In a wound
healing assay, the scratch width of MCF7 cells at 48 h after
co-transfection with miR-148a and ERα ORF plasmid was significantly
smaller compared with miR-148a-overexpressing cells without
plasmid, indicating that the migration rate of was significantly
increased (P<0.01; Fig. 5C).
These results suggest that miR-148a inhibits E2-induced MCF7 cell
viability and migration via inhibition of ERα expression.
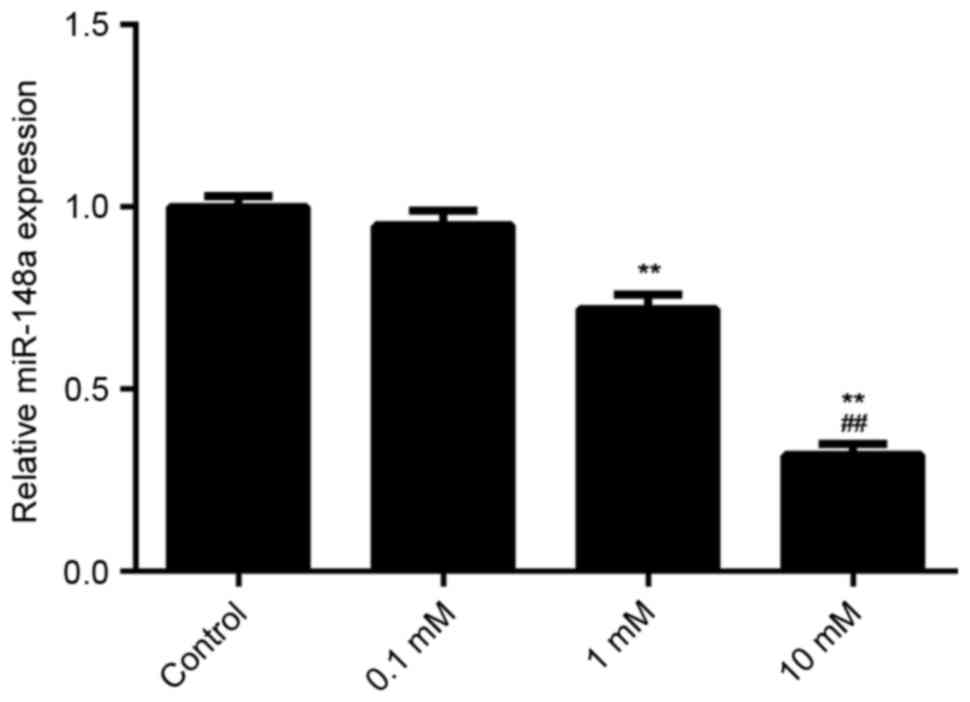
E2 treatment decreases the expression
of miR-148a in MCF7 cells
The effect of E2 treatment on the expression of
miR-148a in MCF7 cells was examined using qPCR. It was found that
E2 treatment decreased the miR-148a levels in MCF7 cells in a
dose-dependent manner (Fig. 6). A
dosage of 1 mM significantly reduced miR-148a expression as
compared with the control (P<0.01). Furthermore, a dosage of 10
mM significantly reduced miR-158a expression as compared with a
dosage of 1 mM. These results suggest that E2 treatment decreases
miR-148a expression, which increases the ERα protein level in MCF7
cells. Through activation of ERα-mediated downstream signaling, E2
treatment induces the viability and migration of breast cancer
cells.
Discussion
The present study investigated the function and
underlying mechanism of miR-148a in mediating the E2-induced
viability and migration of ERα-positive breast cancer MCF7 cells.
The results showed that treatment with E2 significantly enhanced
the viability and migration of MCF7 cells. miR-148a was found to
negatively regulate the protein expression of ERα in MCF7 cells
through directly binding to the 3′UTR of ERα mRNA and thus causing
translation inhibition. Ectopic expression of miR-148a
significantly suppressed the E2-induced viability and migration of
MCF7 cells, similar to the effect of ERα knockdown. Furthermore,
overexpression of ERα rescued the suppressive effect of miR-148a on
E2-induced viability and migration of MCF7 cells. Finally,
treatment with E2 was found to suppress miR-148a expression in MCF7
cells in a dose-dependent manner.
ERα, coded by gene ESR1, is a ligand-activated
transcription factor composed of several domains important for
hormone binding, DNA binding and activation of transcription
(24). ERα localizes to the nucleus
where it may form a homodimer or a heterodimer with ERβ (25). It has previously been demonstrated
that E2 and ERα are essential for sexual development and
reproductive function, and are involved in some human cancers such
as breast cancer and endometrial cancer (26–28). In
the present study, treatment with E2 promoted the viability and
migration of ERα-positive breast cancer MCF7 cells. Therefore, the
E2/ERα-mediated signaling pathway is a promising target in the
treatment of ERα-positive breast cancer. Shang et al
demonstrated that baicalein could inhibit E2-induced migration,
adhesion and invasion of breast cancer cells via the
G-protein-coupled receptor 30 signaling pathway (29).
Furthermore, treatment with E2 has previously been
found to cause deregulation of many miRs during the mammary
carcinogenesis process (30),
suggesting that miRs may play a role in breast cancer. For
instance, miR-125b was found to be significantly upregulated in
breast cancer, and its upregulation was associated with poor
prognosis and aromatase inhibitor resistance (31). miR-200b was reported to be
downregulated in breast cancer, and its low expression was
correlated with late Tumor, Node, Metastasis stages, negative ER
and positive HER-2 statuses, and poor prognosis (32).
miR-148a has been demonstrated to serve a
suppressive function in multiple cancer types (33,34). For
instance, miR-148a was downregulated in gastric cancer due to
hypermethylation in its promoter region (35). Furthermore, miR-148a was found to
regulate numerous target genes and pathways involving tumor
proliferation, invasion and metastasis (35). It is also significantly downregulated
in non-small cell lung cancer (NSCLC) compared to adjacent
non-cancerous lung tissues, and its low level is significantly
associated with lymph-node metastasis (36). In addition, it can suppress
epithelial-to-mesenchymal transition by targeting ROCK1 in NSCLC
cells (36). In the present study,
ERα was found to be a direct target gene of miR-148a, and its
expression was negatively regulated by miR-148a at the
post-transcriptional level in MCF7 cells. To further reveal the
role of miR-148a in ERα-positive breast cancer, MCF7 cells were
transfected with miR-148a mimic to increase its expression level.
Overexpression of miR-148a suppressed the E2-induced viability and
migration of MCF7 cells. Moreover, siRNA-induced ERα downregulation
also inhibited the E2-induced MCF7 cell viability and migration,
similar to the effect of miR-148a upregulation. These results
suggested that miR-148a affected the E2-induced MCF7 cell viability
and migration through inhibiting the protein expression of ERα. To
verify this, ERα ORF plasmid was transfected into
miR-148a-overexpressing MCF7 cells. The resulting overexpression of
ERα reversed the suppressive effect of miR-148a effect on
E2-induced MCF7 cell viability and migration. Therefore, ERα has
been demonstrated to be involved in the miR-148a-mediated viability
and migration of MCF7 cells treated with E2.
In addition, it was found that treatment with E2
decreased the miR-148a levels in MCF7 cells in a dose-dependent
manner. This indicated that E2 treatment not only activates the
ERα-mediated viability and migration-related signaling in MCF7
cells, but also increases the ERα protein level via inhibition of
miR-148a expression.
In conclusion, the present study demonstrates that
miR-148a inhibits the E2-induced viability and migration of
ERα-positive breast cancer cells, at least partly via inhibition of
ERα protein expression. These findings expand the understanding of
miR function in breast cancer, and suggest that miR-148a is a
promising candidate for the treatment of ERα-positive breast
cancer.
Acknowledgements
This study was supported by the National Basic
Research Program of China (grant no. +2013CB835100 to Dr Lingjiang
Li), the Projects of the Science and Technology Department of Hunan
Province (grant no. 2011FJ6043) and the Health Department of Hunan
Province (grant no. B2011-017).
References
|
1
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2013. CA Cancer J Clin. 63:11–30. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Segovia-Mendoza M, González-Gonzalez ME,
Barrera D, Díaz L and García-Becerra R: Efficacy and mechanism of
action of the tyrosine kinase inhibitors gefitinib, lapatinib and
neratinib in the treatment of HER2-positive breast cancer:
Preclinical and clinical evidence. Am J Cancer Res. 5:2531–2561.
2015.PubMed/NCBI
|
|
5
|
Das Gupta S, Sae-Tan S, Wahler J, So JY,
Bak MJ, Cheng LC, Lee MJ, Lin Y, Shih WJ, Shull JD, Safe S, et al:
Dietary γ-Tocopherol rich mixture inhibits estrogen-induced mammary
tumorigenesis by modulating estrogen metabolism, antioxidant
response and PPARγ. Cancer Prev Res (Phila). 8:807–816. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Thomas C and Gustafsson JA: Estrogen
receptor mutations and functional consequences for breast cancer.
Trends Endocrinol Metab. 26:467–476. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Xu B, Lovre D and Mauvais-Jarvis F: Effect
of selective estrogen receptor modulators on metabolic homeostasis.
Biochimie. 124:92–97. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Misawa A and Inoue S: Estrogen-related
receptors in breast cancer and prostate cancer. Front Endocrinol
(Lausanne). 6:832015.PubMed/NCBI
|
|
9
|
Xiong R, Patel HK, Gutgesell LM, Zhao J,
Delgado-Rivera L, Pham TN, Zhao H, Carlson K, Martin T,
Katzenellenbogen JA, et al: Selective human estrogen receptor
partial agonists (ShERPAs) for tamoxifen-resistant breast cancer. J
Med Chem. 59:219–237. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kaaks R, Lukanova A and Kurzer MS:
Obesity, endogenous hormones, and endometrial cancer risk: A
synthetic review. Cancer Epidemiol Biomarkers Prev. 11:1531–1543.
2002.PubMed/NCBI
|
|
11
|
Fan P, Maximov PY, Curpan RF, Abderrahman
B and Jordan VC: The molecular, cellular and clinical consequences
of targeting the estrogen receptor following estrogen deprivation
therapy. Mol Cell Endocrinol. 418:245–263. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ambros V: The functions of animal
microRNAs. Nature. 431:350–355. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
John B, Enright AJ, Aravin A, Tuschl T,
Sander C and Marks DS: Human MicroRNA targets. PLoS Biol.
2:e3632004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Calin GA and Croce CM: MicroRNA signatures
in human cancers. Nat Rev Cancer. 6:857–866. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Boukerroucha M, Josse C, ElGuendi S,
Boujemla B, Frères P, Marée R, Wenric S, Segers K, Collignon J,
Jerusalem G and Bours V: Evaluation of BRCA1-related molecular
features and microRNAs as prognostic factors for triple negative
breast cancers. BMC Cancer. 15:7552015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kehl KL, Shen C, Litton JK, Arun B and
Giordano SH: Rates of BRCA1/2 mutation testing among young
survivors of breast cancer. Breast Cancer Res Treat. 155:165–173.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yu J, Li Q, Xu Q, Liu L and Jiang B:
MiR-148a inhibits angiogenesis by targeting ERBB3. J Biomed Res.
25:170–177. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Aydogdu E, Katchy A, Tsouko E, Lin CY,
Haldosén LA, Helguero L and Williams C: MicroRNA-regulated gene
networks during mammary cell differentiation are associated with
breast cancer. Carcinogenesis. 33:1502–1511. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jiang Q, He M, Ma MT, Wu HZ, Yu ZJ, Guan
S, Jiang LY, Wang Y, Zheng DD, Jin F and Wei MJ: MicroRNA-148a
inhibits breast cancer migration and invasion by directly targeting
WNT-1. Oncol Rep. 35:1425–1432. 2016.PubMed/NCBI
|
|
20
|
Xue J, Chen Z, Gu X, Zhang Y and Zhang W:
MicroRNA-148a inhibits migration of breast cancer cells by
targeting MMP-13. Tumour Biol. 37:1581–1590. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Soltani S, Mokarian F and Panjehpour M:
The expression of CK-19 gene in circulating tumor cells of blood
samples of metastatic breast cancer women. Res Pharm Sci.
10:485–496. 2015.PubMed/NCBI
|
|
22
|
Agarwal V, Bell GW, Nam JW and Bartel DP:
Predicting effective microRNA target sites in mammalian mRNAs.
Elife. 4:e050052015. View Article : Google Scholar
|
|
23
|
Schröder L, Koch J, Mahner S, Kost BP,
Hofmann S, Jeschke U, Haumann J, Schmedt J and Richter DU: The
effects of petroselinum crispum on estrogen receptor-positive
benign and malignant mammary cells (MCF12A/MCF7). Anticancer Res.
37:95–102. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Murphy E: Estrogen signaling and
cardiovascular disease. Circ Res. 109:687–696. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Matthews J and Gustafsson JA: Estrogen
signaling: A subtle balance between ER alpha and ER beta. Mol
Interv. 3:281–292. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Anbalagan M and Rowan BG: Estrogen
receptor alpha phosphorylation and its functional impact in human
breast cancer. Mol Cell Endocrinol. 418:264–272. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bondesson M, Hao R, Lin CY, Williams C and
Gustafsson JÅ: Estrogen receptor signaling during vertebrate
development. Biochim Biophys Acta. 1849:142–151. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chan S: A review of selective estrogen
receptor modulators in the treatment of breast and endometrial
cancer. Semin Oncol. 29(3 Suppl 11): S129–S133. 2002. View Article : Google Scholar
|
|
29
|
Shang D, Li Z, Zhu Z, Chen H, Zhao L, Wang
X and Chen Y: Baicalein suppresses 17-β-estradiol-induced
migration, adhesion and invasion of breast cancer cells via the G
protein-coupled receptor 30 signaling pathway. Oncol Rep.
33:2077–2085. 2015.PubMed/NCBI
|
|
30
|
Munagala R, Aqil F, Vadhanam MV and Gupta
RC: MicroRNA ‘signature’ during estrogen-mediated mammary
carcinogenesis and its reversal by ellagic acid intervention.
Cancer Lett. 339:175–184. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Vilquin P, Donini CF, Villedieu M, Grisard
E, Corbo L, Bachelot T, Vendrell JA and Cohen PA: MicroRNA-125b
upregulation confers aromatase inhibitor resistance and is a novel
marker of poor prognosis in breast cancer. Breast Cancer Res.
17:132015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yao Y, Hu J, Shen Z, Yao R, Liu S, Li Y,
Cong H, Wang X, Qiu W and Yue L: MiR-200b expression in breast
cancer: A prognostic marker and act on cell proliferation and
apoptosis by targeting Sp1. J Cell Mol Med. 19:760–769. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chen Y, Song YX and Wang ZN: The
microRNA-148/152 family: Multi-faceted players. Mol Cancer.
12:432013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Takahashi M, Cuatrecasas M, Balaguer F,
Hur K, Toiyama Y, Castells A, Boland CR and Goel A: The clinical
significance of MiR-148a as a predictive biomarker in patients with
advanced colorectal cancer. PLoS One. 7:e466842012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Xia J, Guo X, Yan J and Deng K: The role
of miR-148a in gastric cancer. J Cancer Res Clin Oncol.
140:1451–1456. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Li J, Song Y, Wang Y, Luo J and Yu W:
MicroRNA-148a suppresses epithelial-to-mesenchymal transition by
targeting ROCK1 in non-small cell lung cancer cells. Mol Cell
Biochem. 380:277–282. 2013. View Article : Google Scholar : PubMed/NCBI
|