Introduction
Dystrophiamyotonica (DM) is a systemic autosomal
dominant disorder and follows a progressive, chronic course
(1,2). Incidence rates of DM range from 1 in
500 to 3 per 100,000, depending on the population (3). DM affects the skeletal muscles, ocular
lens, lungs, heart and gastrointestinal tract, as well as the
endocrine and central nervous systems (1,2,4). The clinical manifestations of DM vary,
however typical symptoms include myotonia, weakness and atrophy of
the skeletal muscles (5,6). Poor sleep quality, fatigue and
excessive daytime sleepiness have an important effect on the
quality of life of DM patients. Mathieu et al (7) proposed that DM mortality rate was 7.3
times higher than that of the normal population, and the average
age of death was 53 years old in one 10-year follow-up study.
Seventy percent of cases of mortality in DM are associated with
cardiorespiratory disorders and there is solid evidence that timely
intervention and active monitoring significantly reduces morbidity
and mortality, although there is no curative therapy (8).
DM belongs to the RNA-dominant disorders of the
family and are autosomal dominant neuromuscular diseases caused by
microsatellite expansions (9). Based
on the mutations that occur within the DM protein kinase
(DMPK) gene, DM is categorized as two distinct types: Types
1 and 2 (5). DM type 1 is caused by
expansion of CTG triplet repeat in the 3′ untranslated region of
the DMPK gene on chromosome 19q13.3 (5). DM type 2 is caused by the progressive
expansion of a CCTG tetramer repeat in ZNF9. To date, only a
small number of cases of white matter lesions in patients with DM1
have been reported. The current study presents the case of a
28-year-old male diagnosed with DM type 1, who also presented with
dysarthria and associated multifocal hyperintense lesions in the
white matter. Diagnosis of DM type 1 was confirmed by genetic
analysis.
Case report
A 28-year-old man presented with a 6-year history of
progressive dysarthria and 2-year history of bilateral hand
weakness. The patient was admitted to the Department of Neurology
in the First Teaching Hospital of Jilin University (Changchun,
China) in December 2014. On admission, the patient underwent
physical examination that revealed exiguous hair, dysarthria,
limited movement of the eyes, facial muscle weakness, glossal
amyotrophy, bilateral sternocleidomastoid atrophy and left
major-thenar amyotrophy. Muscle strength in the hands and muscle
tone of all four limbs was decreased and a lack of tendon reflexes
was observed in the extremities. Furthermore, bilateral mammary
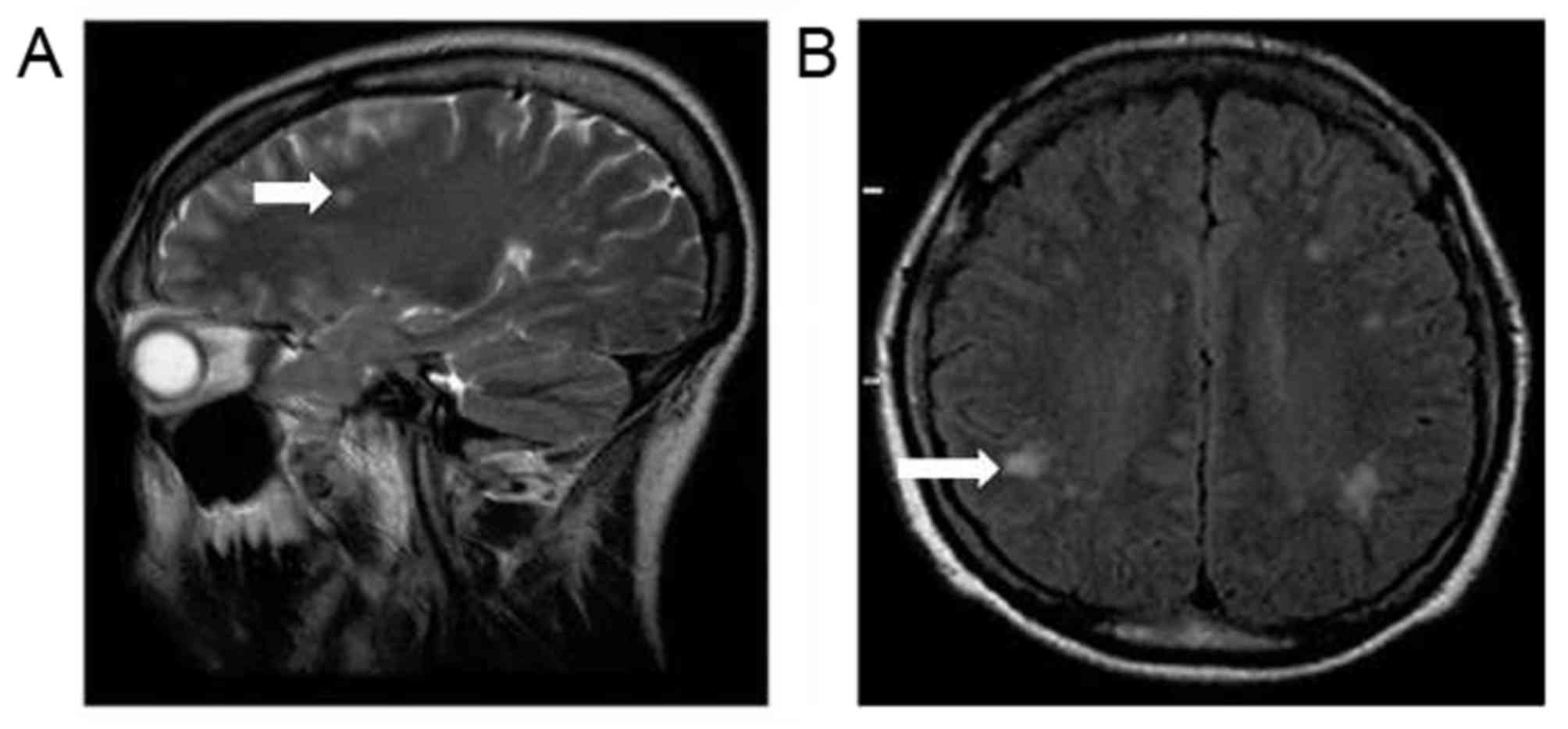
gland hyperplasia was detected. Magnetic resonance imaging (MRI)
scans of the brain identified multifocal hyperintense lesions in
the white matter of the bilateral parietal and frontal lobes, which
were suggestive of demyelination (Fig.
1). The patient complained of a 4-year history of intermittent
diarrhea. The patient denied any relevant family history of
DM-associated pathologies. Written informed consent was provided by
the patient prior to the current study.
Laboratory tests identified elevated levels of
progesterone [0.30 nmol/l (normal range, 0.138–2.671 nmol/l)] and
luteinizing hormone [9.518 mIU/ml (normal range 1.24–8.62 mIU/ml)],
as well as reduced levels of vitamin B12 [107.59 pmol/l (normal
range 133–675 pmol/l)]. Creatinase levels, blood routine
examination, conventional coagulation parameters, liver and kidney
function, pituitary endocrine examination, hypersensitive
C-reactive protein level, cortisol urine levels and glycosylated
hemoglobin levels were all normal. An electrocardiogram detected
first-degree atrioventricular block and ST segment elevation in
leads II, III and avf. Electromyography (EMG) identified myotonic
discharges in all extremities.
Biopsy of the right bicep muscle was performed
according to routine surgical aseptic operation under local
infiltration anesthetic. A dose of 5–20 ml of 1% lidocaine
(Shanghai Zhaohui Pharmaceutical Co., Ltd.) was used (10). Fresh muscle specimens were placed in
tragacanth on a piece of cork and inverted into liquid nitrogen
cooled isopentane, shaken gently and removed after 20–30 sec. The
muscle was then placed in a cryostat and cut into 6-µm sections at
−25°C. In general, based on the differential expression of isoforms
of myosin heavy chains, skeletal muscle consists of four types of
muscle fiber: Types I, IIA, IIB and IIX (11). Type I is a slow aerobic metabolism
muscle fiber, which contains relatively more mitochondria and
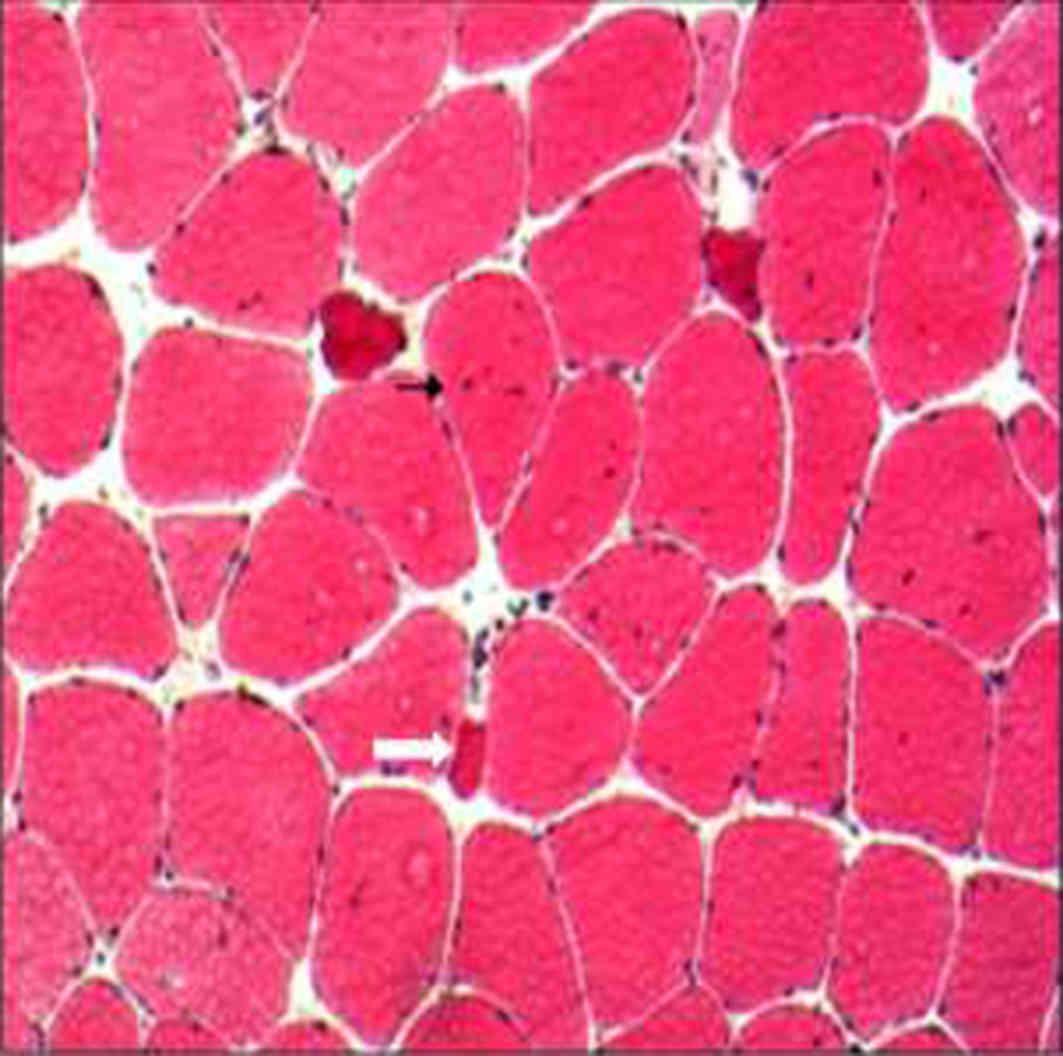
cytochromes and has low glycogen content (11). Subsequent hematoxylin stainig at 60°C
for 30–60 sec and eosin staining at room temperature for 1–3 min
identified muscle fibers of variable sizes (primarily type I) and
some atrophic fibers with an angular shape, observed by electron
microscopy (Fig. 2). In addition,
necrotic and regenerated fibers were identified, however, there was
no evidence of sarcoplasmic reticulum or inflammatory cell
infiltration. Enzymatic activity of nicotinamide adenine
dinucleotide (NADH) and succinic dehydrogenase (SDH) was assayed by
placing the slides in NADH and SDH incubating solution, containing
NADH or SDH as a substrate and nitroblue tetrazolium for
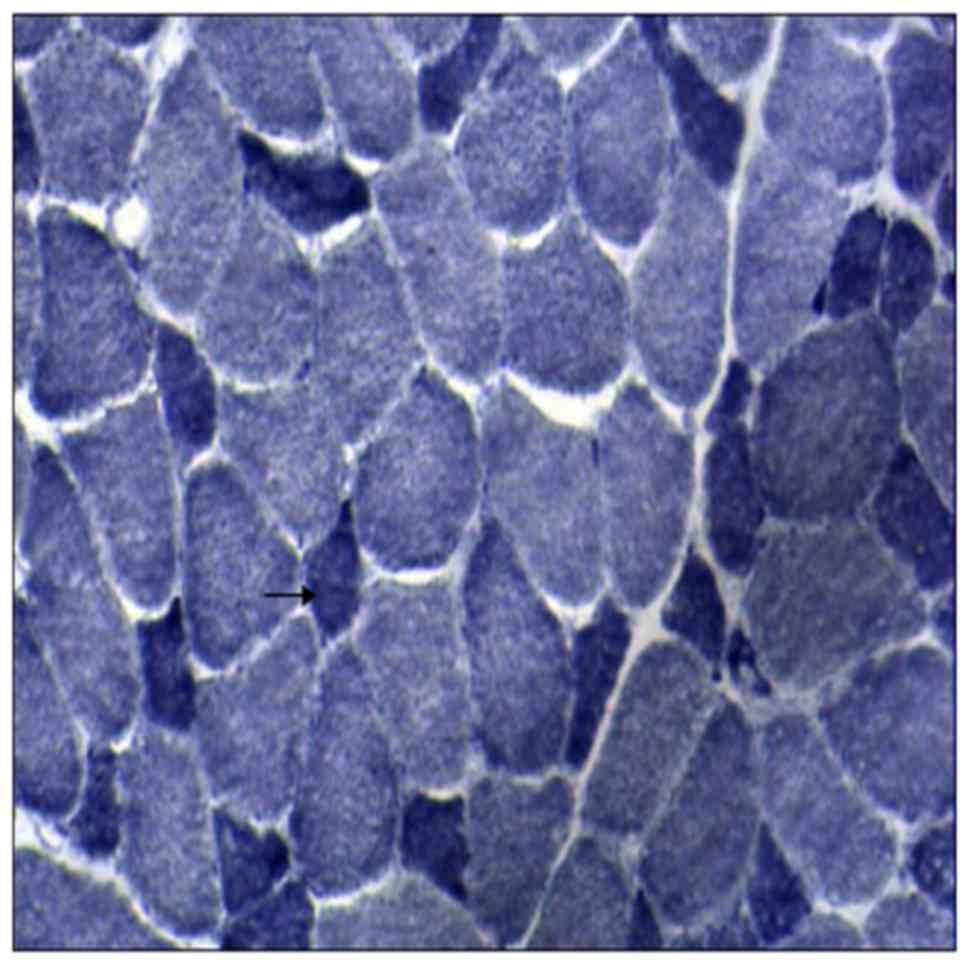
visualization of reaction for 1 h at 37°C. NADH staining exhibited
small type I fibers and reduced activity of NADH, observed by
electron microscopy (Fig. 3).
Glutamyltranspeptidase staining was also performed.
Fresh muscle specimens were placed in liquid nitrogen cooled
isopentane to be frozen. Muscle tissue specimens were then placed
at room temperature (25°C), relative humidity 70%. The dried frozen
sections (6 µm) were stained with hematoxylin for 10–20 min, then
rinsed with tap water for 5 min and dried with filter paper. The
sections were then placed into a vat dye of Gomori trichrome stain
for 10 min at room temperature (25°C). The sections were
differentiated using 0.2% acetic acid for 2 min at room temperature
(25°C). Sections were dehydrated in ascending alcohol solutions.
The sections were cleared with xylene and mounted. The results did
not reveal ragged red fibers by electron microscopy. In addition,
no abnormalities were observed following staining with cytochrome
oxidase, Periodic acid-Schiff or Oil red O (staining protocol as
described for Gomori trichome stain).
The patient's genetic testing was performed using
the fluorescent dye primer polymerase chain reaction (PCR)
technique combined with the triple repeat primed PCR (TP-PCR)
technique (12). Normal chromosome
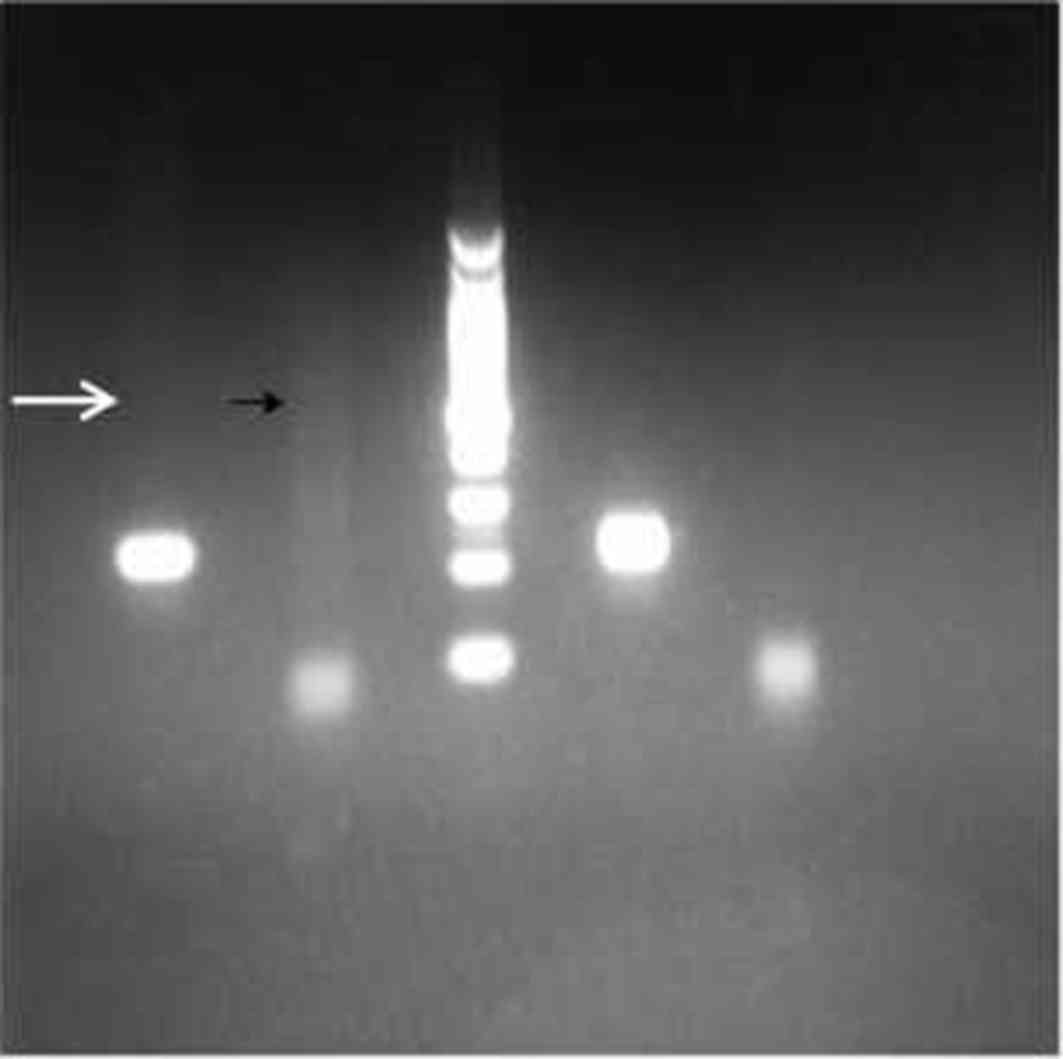
19q13 was amplified by routine PCR. PCR products were detected by
capillary electrophoresis. Abnormal chromosome 19q13 containing a
mutation in the DMPK gene was amplified into a series of
bands on the gel by TP-PCR, due to presence of the PCR primer
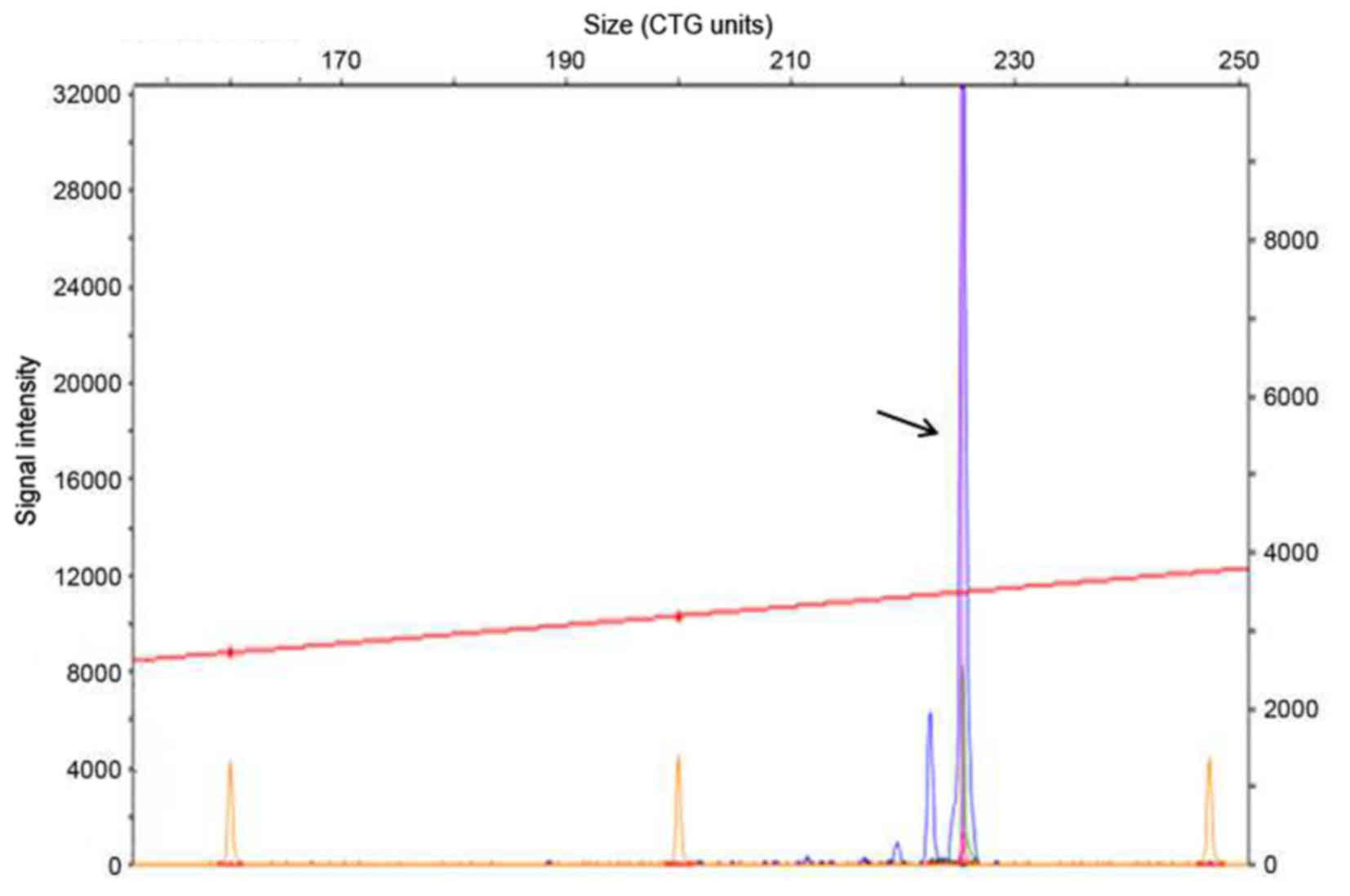
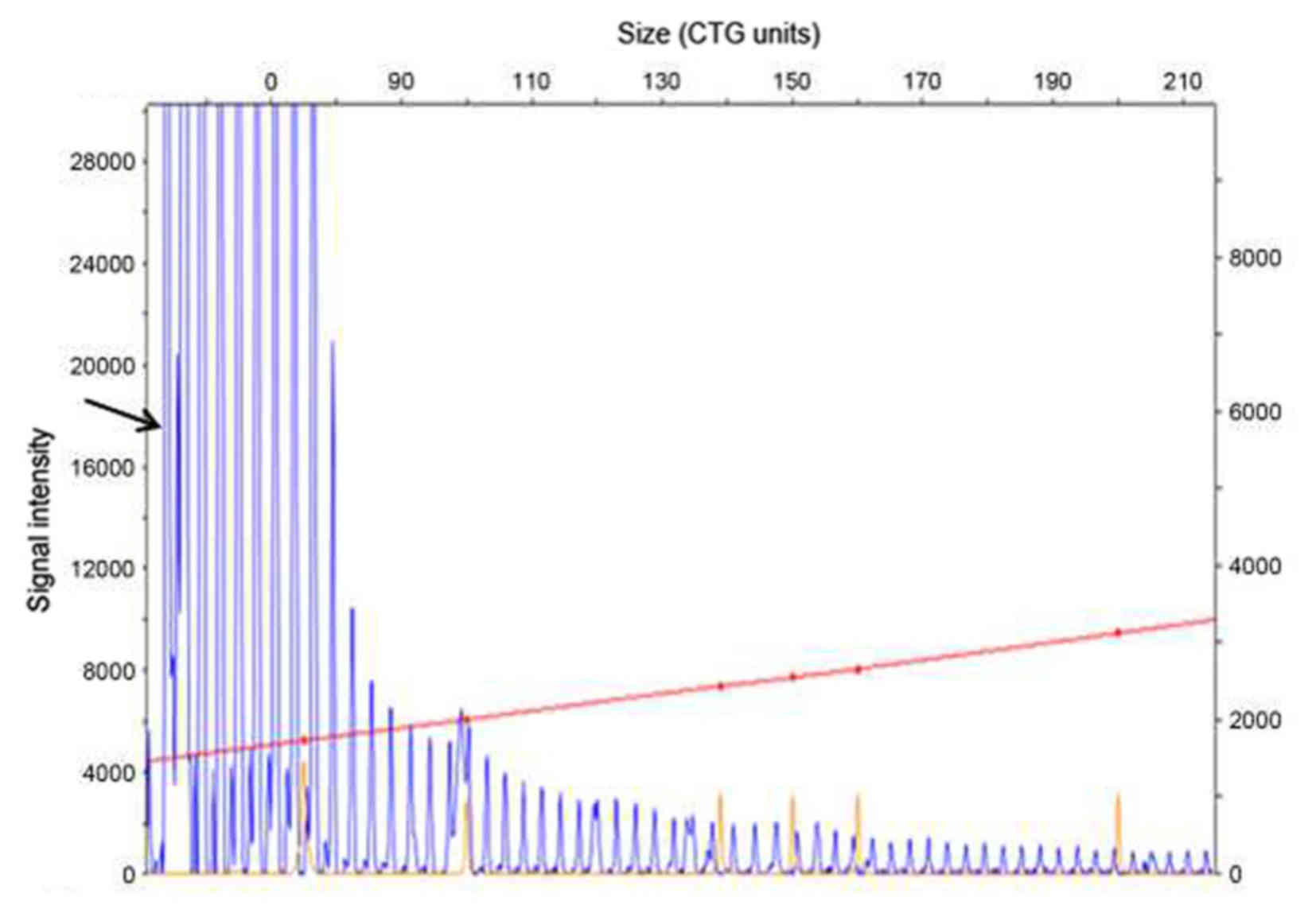
sequence within the mutant CTG repeat region (Fig. 4). Genetic analysis detected a single
allele in the P12 segment of the DMPK gene that included a
CTG expansion of 13 repeats (Fig.
5), as well as a three-base gradient fragment in the P134
segment including a CTG expansion of >600 repeats (0–50 repeats;
Fig. 6). The results of the genetic
analysis were consistent with the pathological characteristics of
DM type 1. Thus, the patient was diagnosed with DM type 1 and
orally treated with vitamin B1 10 mg, vitamin B12 500 µg and folic
acid 5 mg three times a day. The patient was discharged after 4
days of hospitalization. Over a 5-month follow-up period, the
patient experienced a marked improvement in muscular function and
was able to perform daily activities.
Discussion
DM type 1 is one of the most prevalent hereditary
neuromuscular disorders and follows a pattern of autosomal dominant
inheritance. It typically affects skeletal and smooth muscle, as
well as the endocrine and central nervous systems (5,6,13). The disorder is characterized by
progressive myopathy, myotonia and the involvement of multiple
organs. Although the pathogenesis of DM type 1 is not fully
understood, it is associated with a trinucleotide CTG repeat
expansion in the 3′untranslated region of the DMPK gene
(chromosome 19q13) (13,14).
Previous studies have focused on determining the
pathogenetic mechanisms underlying the effects of the trinucleotide
CTG repeat expansion on multi-systemic dysfunction (15,16). The
CTG triplet repeat expansion in the DMPK gene may cause
nuclear localization of mutant mRNA. The mutant mRNA may then form
RNA foci and sequestration of interacting RNA-binding proteins
(15) Toxic repeat RNA sequences may
potentially alter the regular expression of genes and the splicing
process and may induce the abnormal expression of neuromuscular
proteins, resulting in systemic manifestations including myotonia,
muscle wasting, weakness and histopathology, cardiac conduction
defects, cataracts and insulin resistance (17,18).
DM type 1 is categorized into four distinct clinical
forms: Adult-onset, congenital, childhood-onset and late-onset
oligosymptomatic (19). The
prognosis of DM type 1 is associated with the age of onset
(20). Patients with childhood-onset
DM typically experience poorer outcomes and have higher mortality
rates, whereas patients with adult-onset DM generally exhibit a
more favorable prognosis (20).
Adult-onset DM type 1, as documented in the present report, is the
most prevalent form of the disorder (21). The primary clinical manifestations of
DM type 1 are myotonia, muscle weakness and amyotrophy of the
skeletal muscles. Myotonia is the most frequent symptom and
typically manifests as difficulty in actively relaxing the thumb
and/or fingers following contraction and may cause hypoventilation
in some cases, due to stiffness of the respiratory or throat
muscles. Amyotrophy initially affects the hand and forearm muscles
but typically progresses to involve the head and facial muscles,
and myotonia may present concomitantly with amyotrophy or precede
it by a few years (22). The
involvement of multiple organ systems may also complicate the
pathophysiology of DM type 1 (23).
For example, peristalsis in the gastrointestinal tract may become
abnormal and potentially lead to abnormal rectum peristalsis,
spasmodic colic and delayed emptying of the gallbladder, which
ultimately results in cholelithiasis (17). Furthermore, endocrinal involvement in
DM type 1 may lead to alopecia, impaired glucose tolerance, genital
hypoplasia, sexual dysfunction and menstrual disorders (24). Central nervous system abnormalities
observed in patients with DM type 1 are structural and functional;
patients typically present with hypophrenia and somnolence, and
brain MRI scans indicate a thickened skull, narrowed sella,
encephalatrophy and regional or diffuse white-matter changes
(25). In addition, patients with DM
type 1 are predisposed to develop cataracts and/or undergo retinal
degenerative changes (21).
Diagnosis of DM type 1 is based on a combination of
clinical manifestations, family history, EMG results, muscle
histopathology and genetic testing. In the present case, the
patient presented with chronic, progressive dysarthria, which
initially obscured diagnosis. However a few years later, symptoms
of myotonia, muscle weakness and amyotrophy became evident. No
relevant family history of DM-associated pathologies was noted. A
brain MRI scan identified multifocal hyperintense lesions in the
white matter of the bilateral parietal and frontal lobes,
suggestive of demyelination. As cases of DM type 1 with lesions in
the white matter are rare (26),
this was an unexpected result. It is not yet universally accepted
that there is an association between white matter lesions and the
severity of cognitive impairment (27). However, the present results suggest
that conducting routine brain MRI scans in all patients with DM
type 1 may be useful in diagnosing this disease, even in the
absence of clear neurological manifestations.
It is important to differentiate DM type 1 from
congenital myotonia (also known as myotonia congenital). There are
two forms of congenital myotonia caused by mutations in the
chloride voltage gated channel 1 gene located on chromosome 7q35,
which encodes the skeletal muscle chloride channel CIC-1 (28). The clinical presentation of
congenital myotonia differs from that of DM type 1 in that it is
typically accompanied by hypermyotrophy and systemic involvement
other than that of the skeletal muscles is uncommon (29).
In the present case report, the patient presented
with multiple organ system involvement. Endocrinal involvement
manifested as exiguous hair and bilateral mammary gland
hyperplasia, whereas smooth muscle involvement was indicated by a
4-year history of intermittent diarrhea, potentially related to
decreased gastrointestinal peristalsis and emptying. The ECG also
identified a first-degree atrioventricular block and the brain MRI
scan confirmed the presence of multifocal white matter lesions. The
clinical symptoms in the present case were atypical. Although EMG
identified myotonic discharges in all the extremities, a muscle
biopsy failed to identify the characteristic pathologies of DM type
1. The absence of a family history of DM type 1 further inhibited
the diagnosis. However, genetic analysis ultimately confirmed the
diagnosis of DM type 1, indicating the value of genetic analysis as
a diagnostic tool.
There are limited therapeutic options available for
patients with DM type 1. Previous studies have indicated that
membrane-depressant drugs, including phenytoin sodium and
carbamazepine, may alleviate the symptoms of myotonia by promoting
the activity of sodium pumps, leading to a reduction in
intramembranous sodium concentration and an increase in resting
membrane potential (30,31). In addition, physical training may aid
the maintenance of normal muscle functions (22). Cardiac arrhythmia is a primary cause
of mortality in patients with DM type 1 and thus requires stringent
monitoring during treatment. Cataracts associated with DM type 1
may be treated using conventional surgical strategies. In patients
that exhibit endocrinal involvement, lifestyle changes, such as
diet and exercise (8), are generally
adequate to relieve symptoms. Recent progress in the understanding
of the underlying molecular mechanisms involved in myotonic
dystrophy have generated new approaches for DM type 1. Thus, future
therapeutic strategies may employ gene therapy to treat genetic
disorders such as DM.
References
|
1
|
Glaser AM, Johnston JH, Gleason WA and
Rhoads JM: Myotonic dystrophy as a cause of
colonicpseudoobstruction: Not just another constipated child. Clin
Case Rep. 3:424–426. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Finsterer J, Karpatova A, Rauschka H,
Loewe-Grgurin M, Frank M and Gencik M: Myotonic dystrophy 2
manifesting with non-alcoholic and non-hepatitic liver cirrhosis.
Acta Clin Belg. 70:432–435. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Omond KJ and Byard RW: Forensic
considerations in cases of myotonic dystrophy at autopsy. J
Forensic Sci. Feb 7–2017.(Epub ahead of print). View Article : Google Scholar
|
|
4
|
Tschuppert S and Gerding H: Myotonic
dystrophy with reticular maculopathy as first ocular symptom. Klin
Monbl Augenheilkd. 232:568–569. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Finsterer J and Rudnik-Schöneborn S:
Myotonic dystrophies: Clinical presentation, pathogenesis,
diagnostics and therapy. Fortschr Neurol Psychiatr. 83:9–17.
2015.(In German). View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Thornton CA: Myotonic dystrophy. Neurol
Clin. 32705–719. (viii)2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mathieu J, Allard P, Potvin L, Prévost C
and Bégin P: A 10-year study of mortality in a cohort of patients
with myotonic dystrophy. Neurology. 52:1658–1662. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Smith CA and Gutmann L: Myotonic dystrophy
type 1 management and therapeutics. Curr Treat Options Neurol.
18:522016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Arandel L, Espinoza M Polay, Matloka M,
Bazinet A, De Dea Diniz D, Naouar N, Rau F, Jollet A, Edom-Vovard
F, Mamchaoui K, et al: Immortalized human myotonic dystrophy muscle
cell lines to assess therapeutic compounds. Dis Model Mech.
10:487–497. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Joyce NC, Oskarsson B and Jin LW: Muscle
biopsy evaluation in neuromuscular disorders. Phys Med Rehabil Clin
N Am. 23:609–631. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang SH, Zhu L, Wu ZH, Zhang Y, Tang GQ,
Jiang YZ, Li MZ, Bai L and Li XW: Effect of muscle-fiber type on
glycogenin-1 gene expression and its relationship with the
glycolytic potential and pH of pork. Genet Mol Res. 12:3383–3390.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kamsteeg EJ, Kress W, Catalli C, Hertz JM,
Witsch-Baumgartner M, Buckley MF, van Engelen BG, Schwartz M and
Scheffer H: Best practice guidelines and recommendations on the
molecular diagnosis of myotonic dystrophy types 1 and 2. Eur J Hum
Gene. 20:1203–1208. 2012. View Article : Google Scholar
|
|
13
|
Wissocque L, Brigadeau F, Richardson M,
Boulé S, Kouakam C, Polge AS, Marquié C and Klug D: Impairment of
global and regional longitudinal strains in patients with myotonic
dystrophy type 1. Int J Cardiol. 191:46–47. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gallais B, Montreuil M, Gargiulo M, Eymard
B, Gagnon C and Laberge L: Prevalence and correlates of apathy in
myotonic dystrophy type 1. BMC Neurol. 15:1482015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gladman JT, Mandal M, Srinivasan V and
Mahadevan MS: Age of onset of RNA toxicity influences phenotypic
severity: Evidence from an inducible mouse model of myotonic
dystrophy (DM1). PLoS One. 8:e729072013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Santoro M, Masciullo M, Silvestri G,
Novelli G and Botta A: Myotonic dystrophy type 1: Role of CCG CTC
and CGG interruptions within DMPK alleles in the pathogenesis and
molecular diagnosis. Clin Genet. Dec 19–2016.(Epub ahead of
print).
|
|
17
|
Lee JE, Bennett CF and Cooper TA: RNase
H-mediated degradation of toxic RNA in myotonic dystrophy type 1.
Proc Natl Acad Sci USA. 109:pp. 4221–4226. 2012; View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Meola G and Cardani R: Myotonic
dystrophies: An update on clinical aspects, genetic, pathology, and
molecular pathomechanisms. Biochim Biophys Acta. 1852:594–606.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
De Antonio M, Dogan C, Hamroun D, Mati M,
Zerrouki S, Eymard B, Katsahian S and Bassez G: French Myotonic
Dystrophy Clinical Network: Unravelling the myotonic dystrophy type
1 clinical spectrum: A systematic registry-based study with
implications for disease classification. Rev Neurol (Paris).
172:572–580. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gagnon C, Kierkegaard M, Blackburn C,
Chrestian N, Lavoie M, Bouchard MF and Mathieu J: Participation
restriction in childhood phenotype of myotonic dystrophy type 1: A
systematic retrospective chart review. Dev Med Child Neurol.
59:291–296. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Udd B and Krahe R: The myotonic
dystrophies: Molecular, clinical, and therapeutic challenges.
Lancet Neurol. 11:891–905. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Turner C and Hilton-Jones D: Myotonic
dystrophy: Diagnosis, management and new therapies. Curr Opin
Neurol. 27:599–606. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Groh WJ, Groh MR, Saha C, Kincaid JC,
Simmons Z, Ciafaloni E, Pourmand R, Otten RF, Bhakta D, Nair GV, et
al: Electrocardiographic abnormalities and sudden death in myotonic
dystrophy type 1. N Engl J Med. 358:2688–2697. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Meola G and Sansone V: Cerebral
involvement in myotonic dystrophies. Muscle Nerve. 36:294–306.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kornblum C, Reul J, Kress W, Grothe C,
Amanatidis N, Klockgether T and Schröder R: Cranial magnetic
resonance imaging in genetically proven myotonic dystrophy type 1
and 2. J Neurol. 251:710–714. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Liu L, Liu HM, Liu ZJ, Zhang LW, Gu WH and
Wang RB: Myotonic dystrophy type 1 associated with white matter
hyperintense lesions: Clinic, imaging, and genetic analysis. Chin
Med J (Engl). 128:1412–1414. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Minnerop M, Weber B, Schoene-Bake JC,
Roeske S, Mirbach S, Anspach C, Schneider-Gold C, Betz RC,
Helmstaedter C, Tittgemeyer M, et al: The brain in myotonic
dystrophy 1 and 2: Evidence for a predominant white matter disease.
Brain. 134:3530–3546. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Portaro S, Altamura C, Licata N, Camerino
GM, Imbrici P, Musumeci O, Rodolico C, Camerino D Conte, Toscano A
and Desaphy JF: Clinical, Molecular, and functional
characterization of CLCN1 mutations in three families with
recessive myotonia congenita. Neuromolecular Med. 17:285–296. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lakraj AA, Miller G, Vortmeyer AO, Khokhar
B, Nowak RJ and DiCapua DB: Novel mutations in the CLCN1 gene of
myotonia congenita: 2 case reports. Yale J Biol Med. 86:101–106.
2013.PubMed/NCBI
|
|
30
|
Kurihara T: New classification and
treatment for myotonic disorders. Intern Med. 44:1027–1032. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sechi GP, Traccis S, Durelli L, Monaco F
and Mutani R: Carbamazepine versus diphenylhydantoin in the
treatment of myotonia. Eur Neurol. 22:113–118. 1983. View Article : Google Scholar : PubMed/NCBI
|