Introduction
The current management of metastatic colorectal
cancer (CRC) involves chemotherapy and monoclonal antibodies
targeting vascular endothelial growth factor (VEGF; bevacizumab) or
epidermal growth factor receptor (EGFR; cetuximab and panitumumab).
However, when available standard therapies are unsuccessful,
patients may require additional treatment. The overall survival
rate of patients with metastatic CRC has increased in recent years
(1), and more patients for whom the
standard therapies have failed, with a promising performance status
may be candidates for further therapy (2).
Previous efforts to develop small-molecule kinase
inhibitors have been unsuccessful in CRC. Until 2012, regorafenib
was demonstrated to be the first small-molecule inhibitor with a
survival benefit in patients with metastatic CRC in which standard
therapy had failed in a randomized phase III study (3). Furthermore, regorafenib is an oral
multi-kinase inhibitor that targets several protein kinases
involved in tumor angiogenesis, such as VEGF receptor 1–3
(VEGFR1-3) and tyrosine kinase with immunoglobulin and epidermal
growth factor homology domain 2 (TIE2), oncogenesis, such as KIT,
RET, RAF1 and BRAF, and the tumor microenvironment, including
platelet-derived growth factor receptor and fibroblast growth
factor receptor (4). The successful
application of regorafenib suggested that small-molecule kinase
inhibitors may be available, effective and safe for treatment of
metastatic CRC. In addition, the most effective outcome of
regorafenib that may clinically benefit patients may lie in
combined targeting (3–5). Therefore, it is necessary to develop
additional combined targets for creating treatment options for
CRC.
Multiple signaling pathways have been implicated in
the development and progression of CRC, involving transmembrane
receptor tyrosine kinases (RKTs), such as EGFR, VEGFR, insulin-like
growth factor-1 receptor (IGF-1R) and MET, and downstream signaling
cascades (2,6). Furthermore, hepatocyte growth factor
(HGF) and its receptor, MET, are crucial in uncontrolled cell
survival, growth, angiogenesis and metastasis (7). Additionally, functional crosstalk
between MET and other TKRs has emerged as a major mechanism for
tumor progression and therapy resistance (8–10). The
activated MET-driven phosphoinositide 3-kinase (PI3K) signaling
pathway predicts poor survival in patients with CRC, which is
independent of their KRAS mutational status (11). Furthermore, research has indicated
that patients with MET amplification or protein expression were
significantly associated with poorer survival (12,13). In
CRC, amplification of MET (on chromosome 7q31) may occur; however,
the prevalence of MET amplification overall in CRC was as low as
~1% (14). Notably, MET
amplification may develop and drive resistance to anti-EGFR
therapies in CRC, highlighting the requirement for MET inhibitors
in patients who have exhibited resistance as a result of MET
amplification (14). For these
reasons, MET acts as one of the principal targets within
multitargeted small molecule tyrosine kinase inhibitors.
Aberrant MET signaling sequentially activates
various downstream signaling cascades, which is predominantly
mediated by the extracellular signal-regulated kinase
(ERK)-mitogen-activated protein kinase and PI3K-protein kinase B
(AKT) pathways (7). In multiple
downstream pathways, SRC family kinases (SFKs) are important
central mediators that interact with multiple TKRs, representing a
promising target in cancer therapy (15). For instance, activation of SRC has
been demonstrated to confer resistance to targeted therapies,
including anti-EGFR and anti-human epidermal growth receptor 2
(16,17). Although SRC activity serves as a key
downstream node, single-agent treatment of SRC inhibitor has been
demonstrated to have limited clinical benefit, and combination
regimens with targeted therapy have indicated more clinically
relevant effects (18). Generally,
bypass activation of MET is accompanied with downstream activation
of SRC (19). Furthermore, the
interaction of MET with SRC mediates resistance to targeted drugs
(20–22). Although the association between MET
and SRC has been demonstrated to cooperate intensely (20), few studies have focused on the effect
of dual inhibition of MET and SRC in targeted therapy.
Previous research has demonstrated that
cetuximab-induced MET and SRC activation was involved in the
resistance to cetuximab in colon cancer cells (22). Furthermore, single addition of MET
inhibitor, PHA-665752, or the multitargeted SFK inhibitor,
dasatinib, exerted certain antitumor effects in colon cancer cells
(22). Therefore, the present study
further investigated the role of MET activation in the
ligand-dependent and -independent HGF/MET pathway, the interaction
between MET and SRC and the mechanisms underlying the antitumor
effect of MET and SRC inhibitors in colon cancer cells, providing a
rationale for combinatorial inhibition of MET and SRC in therapy
targeting colon cancer.
Materials and methods
Cell culture and reagents
Colon cancer cells, HT-29 and HCT-116, were obtained
from the Type Culture Collection of the Chinese Academy of Sciences
(Shanghai, China). The two cell lines were cultured at 37°C with 5%
CO2 in RPMI-1640 medium (Gibco; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) supplemented with 10% fetal
bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc). HGF,
epidermal growth factor (EGF) and IGF-1 were purchased from R&D
Systems, Inc. (Minneapolis, MN, USA). PHA-665752 was purchased from
Sigma-Aldrich (Merck KGaA, Darmstadt, Germany). Dasatinib was
purchased from Selleck Chemicals (Houston, TX, USA). Antibodies
against MET (cat. no. 3127; 1:1.000), phosphorylated (p)-MET
(Tyr1234/1235; cat. no. 3077; 1:500), SRC (cat. no. 2110; 1:1,000),
p-SRC (Y416; cat. no. 6943; 1:500), AKT (cat. no. 9272; 1:1,000),
p-AKT (Ser473; cat. no. 9271; 1:1,000), EGFR (cat. no. 2646;
1:1,000), p-EGFR (Tyr1068; cat. no. 2234; 1:500), IGF-1R (cat. no.
3027; 1:1,000), p-IGF-1R (Tyr1131; cat. no. 3021; 1:500) and poly
(ADP-ribose) polymerase (PARP; cat. no. 9542; 1:1,000) were
obtained from Cell Signaling Technology, Inc., (Danvers, MA, USA).
Anti-β-actin (cat. no. sc-1616; 1:1,000), anti-ERK (cat. no.
sc-292838; 1:1,000), p-ERK1/ERK2 (Thr202/Tyr204; cat. no. sc-16982;
1:1,000), horseradish peroxidase-conjugated secondary goat
anti-rabbit (cat. no. sc-2385; 1:2,000) and goat anti-mouse (cat.
no. sc-2375; 1:2,000) antibodies were purchased from Santa Cruz
Biotechnology, Inc., (Dallas, TX, USA).
Cell viability assay
Cell viability was measured using an MTT assay.
HT-29 and HCT-116 cells were seeded in triplicate at 6,000
cells/well in 96-well plates and incubated at 37°C for 24 h in
RPMI-1640 medium supplemented with 10% FBS. Subsequently, cells
were treated with the indicated doses of HGF (5, 25 or 125 ng/ml),
PHA-665752 (0.2, 1.0 or 5.0 µM), dasatinib (0.1, 0.5 or 2.5 µM) or
a combination (0.1 µM dasatinib and 0.2 µM PHA) for 48 h at 37°C.
The same volume of dimethylsulfoxide (DMSO) was used as the
negative control. Following this, 25 µl MTT solution (5 mg/ml) was
added to each well for 4 h at 37°C, cell culture supernatants were
carefully removed and 200 µl DMSO was added. Finally, the optical
density was measured at a wavelength of 570 nm using a microplate
reader (Model 550; Bio-Rad Laboratories, Inc., Hercules, CA,
USA).
Western blot analysis
The cells were washed twice with phosphate-buffered
saline (PBS), lysed in lysis buffer (1% Triton X-100, 50 mM
Tris-HCl pH 7.4, 150 mM NaCl, 10 mM EDTA, 100 mM NaF, 1 mM
Na3VO4, 1 mM PMSF, 2 µg/ml aprotinin) and
quantified using the BCA protein quantification kit (cat. no.
ab102536; Abcam). The cell lysates (30 µg protein/lane) were
separated by 8% SDS-PAGE, the samples (30 µg protein/lane) were
transferred to a nitrocellulose membrane (Immoblin-P, Millipore;
Merck KGaA). After blocking with 5% skim milk in tris-buffered
saline Tween-20 (TBST) buffer (10 mM Tris-HCl pH 7.4, 150 mM NaCl,
0.1% Tween-20) at room temperature for 1 h, antibodies against MET,
p-MET, SRC, p-SRC, AKT, p-AKT, EGFR, p-EGFR, IGF-1R, p-IGF-1R,
PARP, β-actin, anti-ERK and p-ERK1/ERK2 were added and incubated
overnight at 4°C. Following three washes with TBST buffer, the
membrane was incubated with secondary goat anti-rabbit and goat
anti-mouse antibodies for 30 min at room temperature followed by
three washes with TBST buffer. Finally, the protein bands were
detected with enhanced chemiluminescence reagent
(SuperSignal™ Western Pico Chemiluminescent Substrate;
Pierce; Thermo Fisher Scientific, Inc.) and scanned using the
Electrophoresis Gel Imaging Analysis System (DNR Bio-Imaging
Systems, Neve Yamin, Israel).
Small interfering RNA (siRNA)
transfections
The MET and SRC siRNA (5 nM) sequences from Shanghai
GenePharma Co., Ltd. (Shanghai, China) were as follows:
5′-GCCUGAAUGAUGACAUUCU-3′ and 5′-GGCUCCAGAUUGUCAACAAtt-3′,
respectively. Furthermore, the siRNA were transfected with
Lipofectamine® 2000 (Invitrogen; Thermo Fisher Scientific, Inc.),
according to the manufacturer's instructions (Invitrogen; Thermo
Fisher Scientific, Inc.). EGF or IGF-1 were added to the cells for
2 h at 37°C, 48 h after transfection and collected for further
study.
Plasmid construction
Flag-tagged wild type SRC plasmid was kindly
provided by Dr. Li Feng (Department of Cell Biology, China Medical
University, Shenyang, China). pcDNA3.1 was purchased from
Invitrogen (Thermo Fisher Scientific, Inc.) and used as a negative
control. Furthermore, cells were transfected with 2 µg plasmid or
empty control using Lipofectamine® 2000, according to the
manufacturer's instructions.
Co-immunoprecipitation
HCT-116 cells (8×106) were seeded into
100 mm plates and allowed to attach overnight at 37°C. The cells
were then washed twice with PBS and lysed in lysis buffer on ice.
Co-immunoprecipitation was performed using 200 µg lysates with 4 µl
of mouse anti-MET or control immunoglobulin G mixed with protein G
agarose beads (GE Healthcare Bio-Sciences, Pittsburgh, PA, USA).
The final mixture was gently rocked overnight at 4°C. Following
this, the beads were spun down for 1 min at 13,000 × g at 4°C and
washed four times for 1 min with lysis buffer (1% Triton X-100, 50
mM Tris-HCl pH 7.4, 150 mM NaCl, 10 mM EDTA, 100 mM NaF, 1 mM
Na3VO4, 1 mM PMSF and 2 µg/ml aprotinin).
Finally, 40 µl sampling buffer (Beyotime Institute of
Biotechnology, Haimen, China) was added, boiled at 95°C for 5 min
and subjected to western blot analysis.
Colony formation assay
Cells were seeded at 300 cells/well in 12-well
plates. Following culture for 24 h at 37°C in RPMI-1640 medium
supplemented with 10% FBS, cells were treated with either 0.2 µM
PHA-665752, 50 nM dasatinib or a combination (0.2 µM PHA-665752 and
50 nM dasatinib). The cells were then cultured for an additional 10
days at 37°C, then stained with Wright Giemsa, after which the
number of colonies was counted using a fluorescence microscope
(BX53; Olympus Corporation, Tokyo, Japan).
Flow cytometry assay
The cells were collected and washed twice for 5 min
with PBS. Following blocking in 70% ethanol at 4°C for 12 h, the
samples were washed twice for 5 min with PBS and incubated with 20
µg/ml RNase A (R4642; Sigma-Aldrich; Merck KGaA) for 30 min at 37°C
and 10 µg/ml propidium iodide for 30 min at room temperature in the
dark. Finally, the samples were evaluated by flow cytometry (BD
Accuri C6; BD Biosciences, San Jose, CA, USA), and analyzed with
WinMDI version 2.9 software (The Scripps Research Institute, La
Jolla, CA, USA).
Statistical analysis
Data were analyzed using SPSS version 21.0 software
(SPSS Corp., Chicago, IL, USA). All the values were expressed as
the mean ± standard deviation. Furthermore, the differences of the
results between the two groups were determined by Student's
t-tests. P<0.05 was considered to indicate a statistically
significant difference.
Results
Ligand-dependent activation of MET in
colon cancer cells
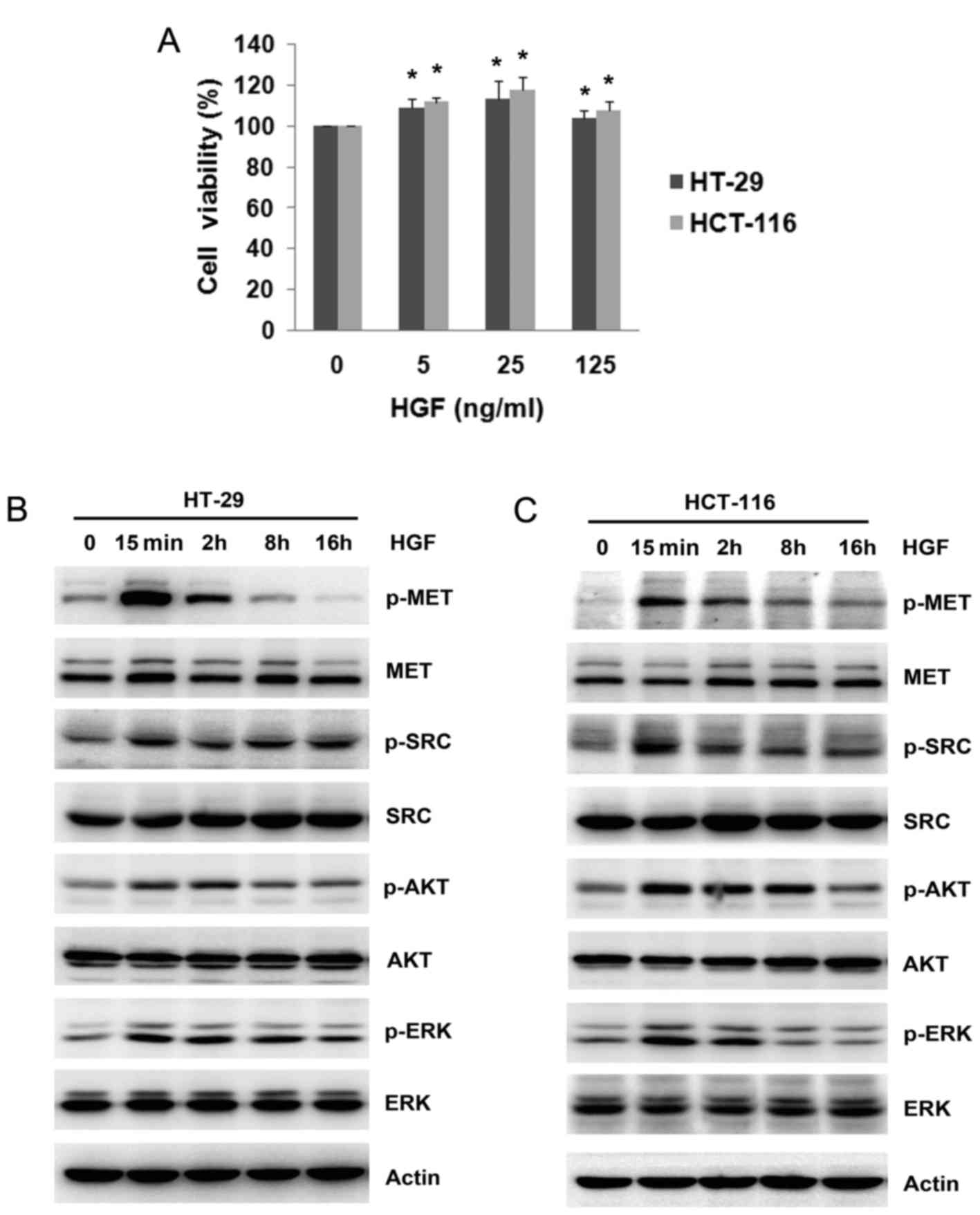
First, to ensure the HGF/MET axis was functional in
wild type and mutant RAS colon cancer cells, HT-29 (wild type RAS)
and HCT-116 (mutant RAS) cells were added to different doses of HGF
for 48 h. MTT assays revealed a significant increase in HGF cell
viability in colon cancer cells of 7.6–17.4% (P<0.05 vs. 0 ng/ml
HGF; Fig. 1A). Notably, it was
observed that the increase of cell viability induced by HGF was not
in a dose-dependent manner. The highest dose of HGF (125 ng/ml)
demonstrated a smaller increase in cell viability than the 25 ng/ml
HGF dose. Therefore, 25 ng/ml HGF was selected to study the effect
on cell viability. Subsequently, the expression of MET
phosphorylation and successive downstream cascades was evaluated by
western blotting. As expected, robust activation of MET on Tyr
1234/1235 was observed to peak at 15 min after HGF addition in both
cell lines, and decreased gradually after this. Phosphorylation of
SRC, AKT and ERK was elevated following the addition of HGF and
this was accompanied by MET activation (Fig. 1B and C). These results indicated that
a ligand-dependent activation of MET by HGF promoted cell viability
in colon cancer cells.
Ligand-independent activation of MET
by EGF and IGF-1
Apart from ligand activation by HGF, MET may be
activated in the absence of ligand binding by crosstalk with other
growth factor receptors (9,10). To further demonstrate the
ligand-independent activation of MET in wild type and mutant RAS
colon cancer cells, 25 ng/ml EGF or IGF-1 was added to HT-29 and
HCT-116 cells at 15 min and 2, 8 and 16 h. In contrast to acute
activation of EGFR and IGF-1R signaling and downstream cascades
(including SRC, AKT and ERK) after EGF and IGF-1 addition, MET
phosphorylation appeared to be activated later, at 2 h after the
addition of EGF or IGF-1 (Fig.
2A-D). Furthermore, no differences in MET expression were
evident by western blotting. To demonstrate the role of MET on
ligand-independent activation, the expression of MET was inhibited
by siRNA before the addition of EGF or IGF-1. As expected, the
ligand-independent activation of MET stimulated by EGF or IGF-1 was
evidently attenuated (Fig. 2E and
F). These results demonstrated that the MET pathway cross
talked with the EGFR and IGF-1R pathways in colon cancer cells.
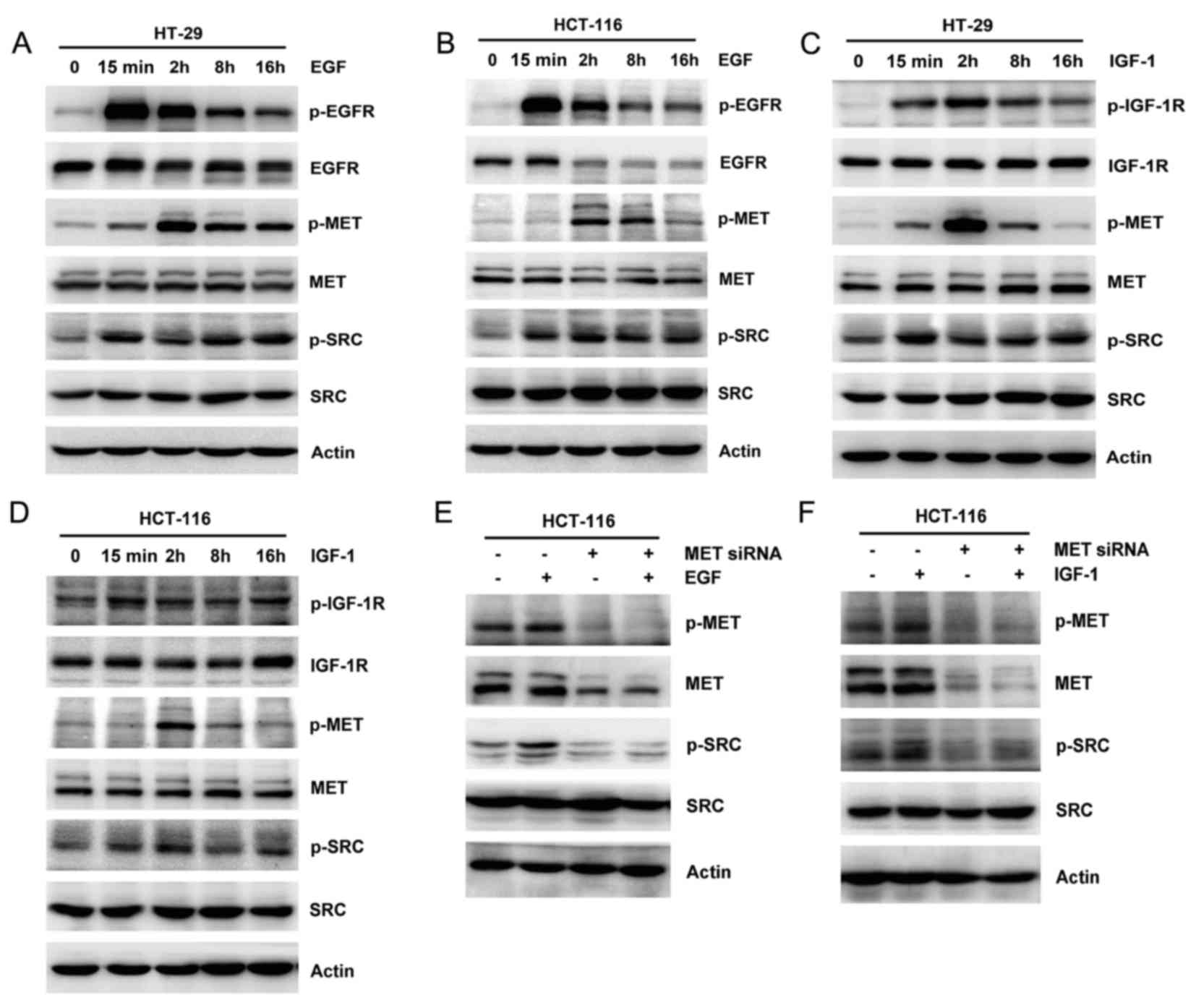 | Figure 2.Effect of EGF and IGF-1 on MET
activation. EGF (25 ng/ml) was added to (A) HT-29 and (B) HCT-116
cells and the expression and phosphorylation of EGFR, MET, SRC, AKT
and ERK was assessed by western blotting. IGF-1 (25 ng/ml) was
added to (C) HT-29 and (D) HCT-116 cells. The expression and
phosphorylation of IGF-1R, MET, SRC, AKT and ERK were observed by
western blotting. HCT-116 cells were transiently transfected with
scramble control or MET siRNA, and then 25 ng/ml (E) EGF or (F)
IGF-1 was added for 2 h. The expression and phosphorylation of MET
and SRC were studied by western blotting. Actin was used as a
loading control. EGF, epidermal growth factor; EGFR, epidermal
growth factor receptor; AKT, protein kinase B; ERK, extracellular
signal-regulated kinase; IGF-1, insulin-like growth factor-1;
IGF-1R, insulin-like growth factor-1 receptor; siRNA, small
interfering RNA; p, phosphorylated. |
MET inhibition exhibits a limited
anti-viability effect in colon cancer cells
Since the HGF/MET pathway contributed to increased
cellular viability, including cross talk with other growth factor
receptor pathways, the present study aimed to assess the
anti-viability activity of MET inhibition in colon cancer cells.
Knockdown of MET by siRNA led to significant inhibition of cell
viability in HT-29 (77.8±6.18 vs. 100%; P=0.012) and HCT-116
(82.9±4.45% vs. 100%; P=0.011; Fig.
3A) cells compared with scramble controls. Furthermore, a
selective MET inhibitor, PHA-665752, was used; however, this
induced no significant inhibition to cell viability (Fig. 3B). The anti-viability activity of
PHA-665752 was dose-dependent, with the highest dose of PHA-665752
(5 µM) having inhibitory rates of 34.5 and 25.5% in HT-29 and
HCT-116 cells, respectively (Fig.
3B). Subsequently, the change of the signaling pathway after
the addition of PHA-665752 was examined. The single use of
PHA-665752 markedly inhibited the basal level of MET
phosphorylation, as predicted (Fig. 3C
and D). However, the basal phosphorylation levels of SRC, AKT
or ERK were not inhibited by PHA-665752 (Fig. 3C and D). Pretreatment with PHA-665752
markedly suppressed the activation and/or phosphorylation of MET,
SRC, AKT or ERK stimulated by HGF (Fig.
3E). Altogether, MET inhibition led to the inhibition of cell
viability in colon cancer cells.
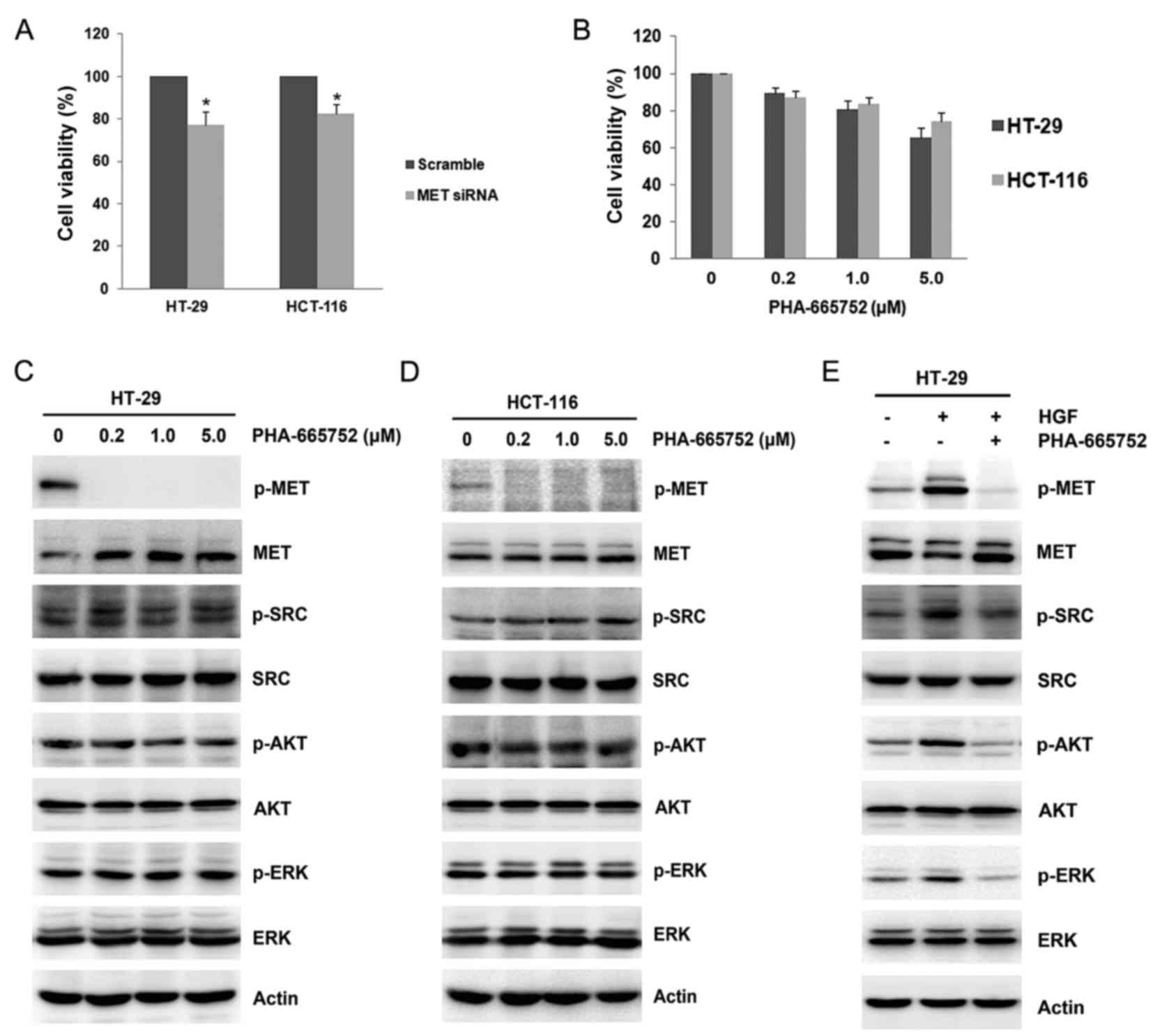 | Figure 3.MET inhibition by siRNA or inhibitor.
(A) HT-29 and HCT-116 cells were transiently transfected with
scramble control or MET siRNA for 48 h in 96-well plates. Cell
viability was assessed by MTT assay. (B) Increasing concentrations
of PHA-665752 (0.2, 1.0 and 5.0 µM) were added to HT-29 and HCT-116
cells, and cell viability was assessed after 48 h. Data are
presented as the mean ± standard deviation of three independent
experiments. Increasing concentrations of PHA-665752 (0.2, 1.0 and
5.0 µM) were added to (C) HT-29 and (D) HCT-116 cells for 24 h. The
expression and phosphorylation of MET, SRC, AKT and ERK were
studied by western blotting. (E) HCT-116 cells were pretreated with
0.2 µM PHA-665752 for 24 h after which they were stimulated with 25
ng/ml HGF for 2 h. The expression and phosphorylation of MET, SRC,
AKT and ERK were analyzed by western blotting. Actin was used as a
loading control. *P<0.05 vs. scramble control. siRNA, small
interfering RNA; AKT, protein kinase B; ERK, extracellular
signal-regulated kinase; p, phosphorylated. |
SRC activation is essential for
ligand-dependent and ligand-independent MET activation
As SRC activation was commonly accompanied by
ligand-dependent and -independent activation of MET, and SRC was
the key mediator downstream of the MET pathway, the interaction
between MET and SRC in colon cancer cells was examined. First, a
co-immunoprecipitation assay was performed, which revealed a
physical interaction between MET and SRC in HCT-116 cells (Fig. 4A). Knockdown of MET by siRNA
decreased SRC phosphorylation without altering its total expression
(Fig. 2E and F). To explore the role
of SRC on MET activity, siRNA was used to knockdown SRC expression
and wild type SRC plasmid was used to enhance SRC expression.
Similarly, SRC knockdown resulted in a decrease of MET
phosphorylation (Fig. 4B); while
overexpression of SRC with a plasmid harboring an activated wild
type SRC was sufficient to directly phosphorylate MET without
altering its total expression (Fig.
4C). Furthermore, the addition of the multitargeted SFK
inhibitor, dasatinib, at different concentrations inhibited the
basal level of MET phosphorylation, accompanied by decreased
phosphorylation of AKT and ERK in HT-29 and HCT-116 cells (Fig. 4D and E). These results suggested that
MET may be a direct target of SRC in colon cancer cells.
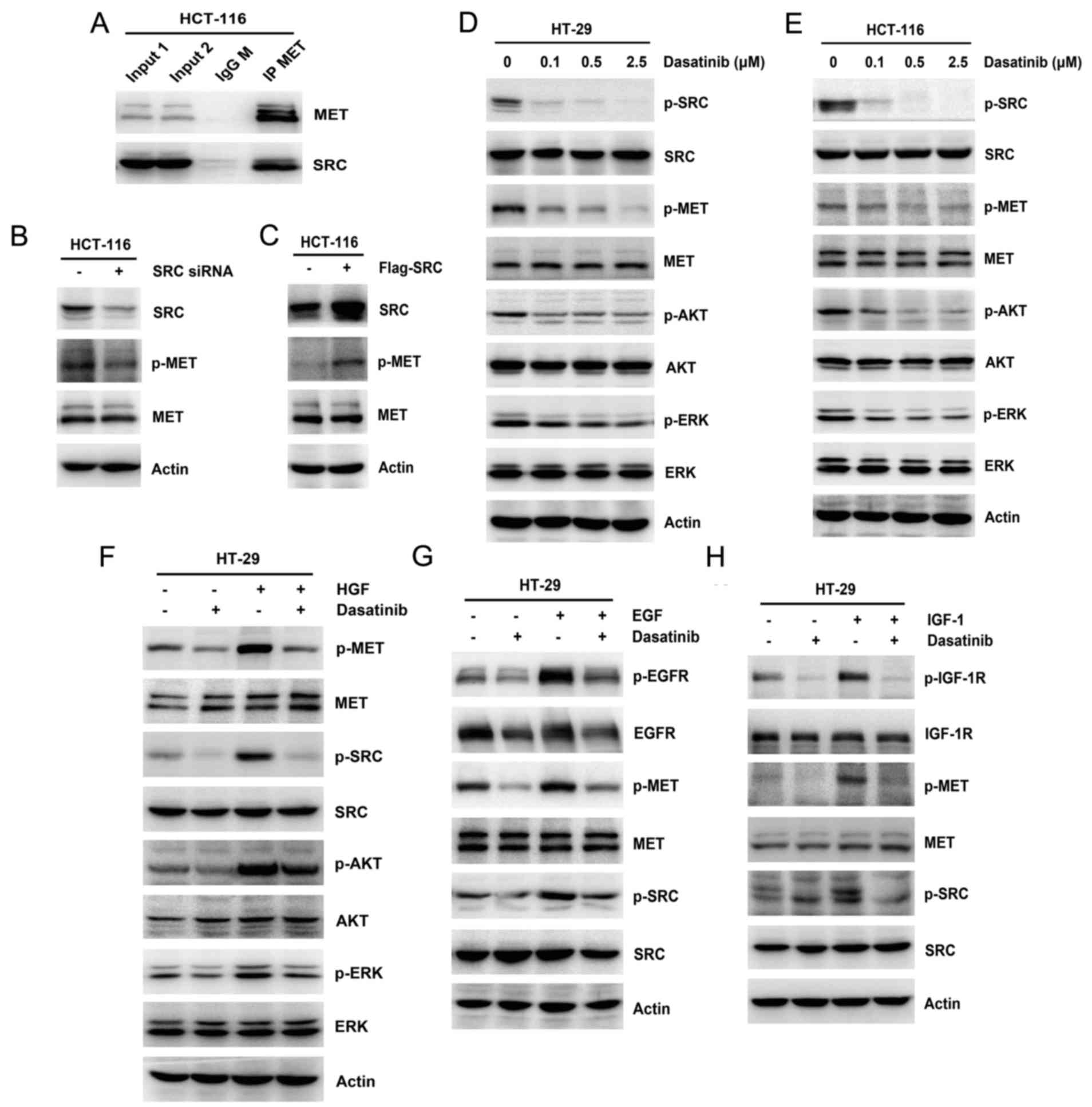 | Figure 4.Regulation of SRC on MET and
non-ligand mediated MET activation. (A) Whole-cell extracts from
HCT-116 cells were immunoprecipitated with anti-MET antibody. The
immunoprecipitates were probed with MET and SRC antibodies. The
input represents cell lysates that were not subjected to
immunoprecipitation. Control immunoprecipitation was performed
using IgG M. HCT-116 cells were transiently transfected with
scramble control and (B) SRC siRNA for 48 h or (C) flag-tagged wild
type SRC plasmid for 24 h. The expression of SRC, MET and p-MET was
studied by western blotting. Increasing concentrations of dasatinib
(0.1, 0.5 and 2.5 µM) were added to (D) HT-29 and (E) HCT-116
cells, and the expression and phosphorylation of SRC, MET, AKT and
ERK was analyzed by western blotting. (F) HT-29 cells were
pretreated with 0.1 µM dasatinib for 24 h, then stimulated with 25
ng/ml HGF for 2 h. The expression and phosphorylation of MET, SRC,
AKT and ERK was examined by western blotting. HT-29 cells were
pretreated with 0.1 µM dasatinib for 24 h, then stimulated with 25
ng/ml (G) EGF or (H) IGF-1 for 2 h. The expression and
phosphorylation of EGFR, IGF-1R, MET and SRC was studied by western
blotting. Actin was used as a loading control. IgG M,
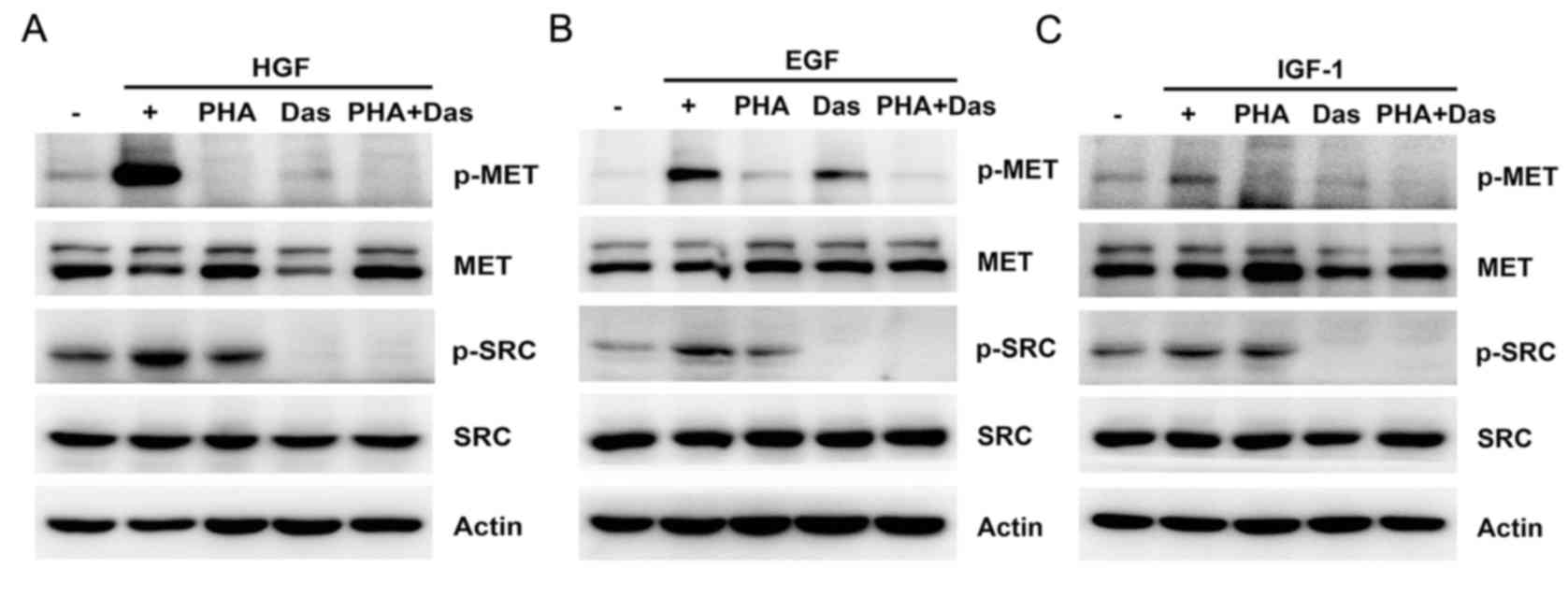
immunoglobulin G mouse; siRNA, small interfering RNA; p,
phosphorylated; AKT, protein kinase B; ERK, extracellular
signal-regulated kinase; HGF, hepatocyte growth factor; EGF,
epidermal growth factor; EGFR, epidermal growth factor receptor;
IGF-1, insulin-like growth factor-1; IGF-1R, insulin-like growth
factor-1 receptor. |
To examine the potential role of SRC activity in
delayed MET activation, pretreatment with dasatinib was used before
stimulating cells with HGF, EGF or IGF-1, respectively. Under these
conditions, pretreatment with dasatinib reduced growth
factor-induced MET activation and led to a partial decrease in the
phosphorylation of AKT and ERK in HT-29 cells compared with growth
factor stimulation (Fig. 4F-H).
These results suggested that SRC activation was required for
ligand-dependent and ligand-independent MET activation.
Combinational inhibition of MET and
SRC exerts increased antitumor effects
Although dasatinib partly repressed the growth
factor-induced activation of MET, AKT and ERK, the inhibitory
effect was not complete. Therefore, the present study focused on
the mechanisms of dual inhibition of MET and SRC. Initially, the
concurrent inhibition of MET and SRC was observed in
ligand-dependent and -independent MET activation. As expected,
pretreatment with MET and SRC inhibitors markedly inhibited the
activation of MET and SRC stimulated by growth factors compared to
single inhibition in HCT-116 cells (Fig.
5). Following this, the effect of combined treatment with MET
and SRC inhibitors was evaluated. Combination treatment resulted in
a significant decrease in cell viability compared with single agent
treatment in HT-29 cells (56.5±5.2% in combination treatment vs.
85.9±4.4% in PHA-665752 treatment; P=0.001; and 56.5±5.2% in
combination treatment vs. 66.9±5.4% in dasatinib treatment;
P=0.008) and in HCT-116 cells (63.7±7.6% in combination treatment
vs. 86.8±3.8% in PHA-665752 treatment; P=0.006; and 63.7±7.6% in
combination treatment vs. 74.3±3.9% in dasatinib treatment;
P=0.033; Fig. 6A). Similarly,
colonies that formed in the presence of the combined treatment were
markedly smaller and fewer in number than those that grew with
either of the single agent treatments (Fig. 6B). These results demonstrated that
the combination of MET with SRC inhibitor enhanced the
anti-viability effects of these treatments alone.
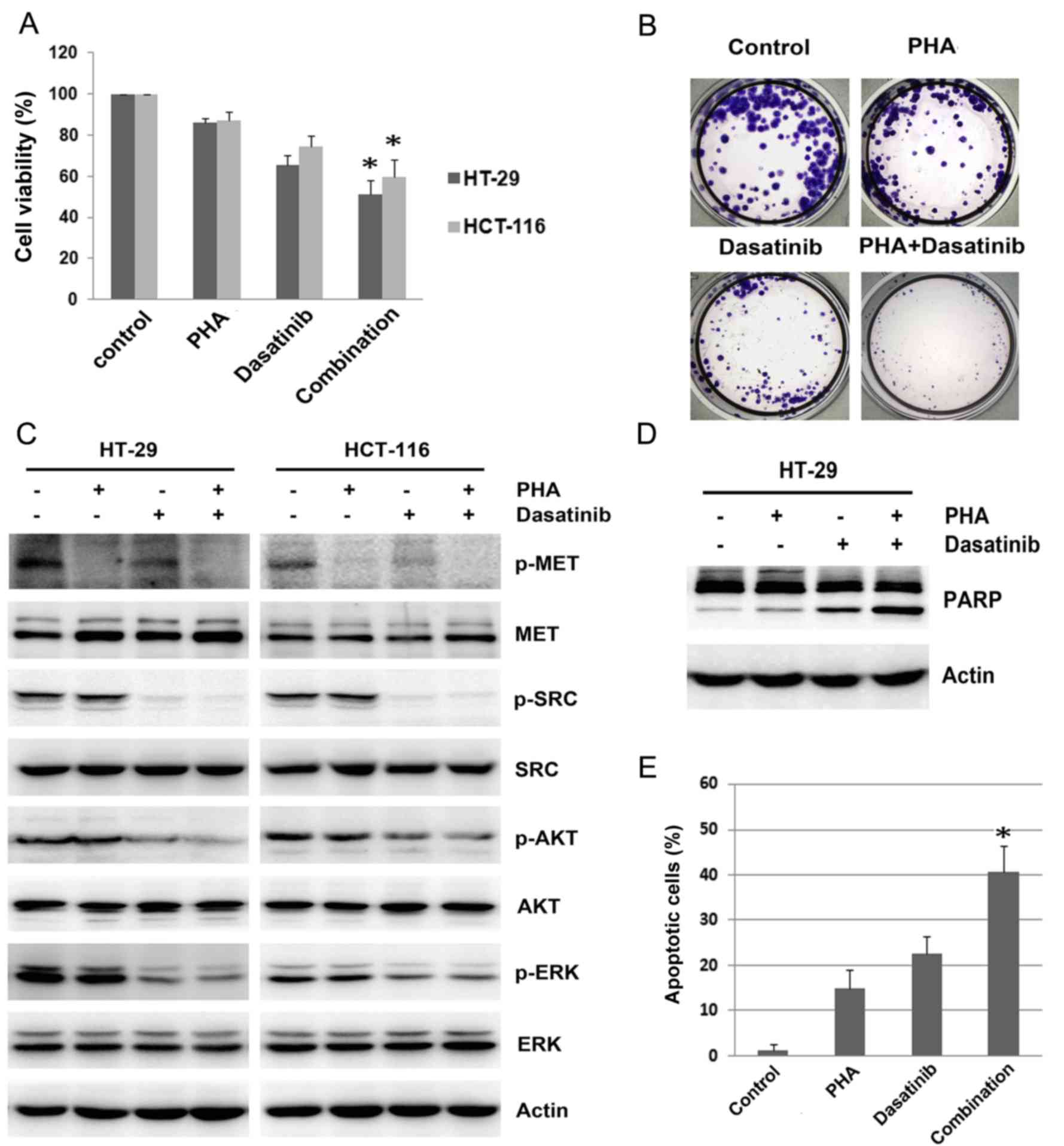 | Figure 6.Effect of combining MET and SRC
inhibition on cell viability. (A) HT-29 and HCT-116 cells were
treated with 0.1 µM dasatinib, 0.2 µM PHA or combination for 48 h,
and cell viability was assessed by MTT assay. (B) HCT-116 cells
were treated with 50 nM dasatinib, 0.2 µM PHA or combination for 10
days, and the clones were counted. (C) HT-29 and HCT-116 cells were
treated with 0.1 µM dasatinib, 0.2 µM PHA or combination for 24 h.
The expression and phosphorylation of MET, SRC, AKT and ERK was
analyzed by western blotting. (D) HT-29 cells were treated with 0.1
µM dasatinib, 0.2 µM PHA or combination for 48 h, and the
expression of PARP was detected by western blotting. (E) HT-29
cells were treated with 0.1 µM dasatinib, 0.2 µM PHA or combination
for 48 h. The proportion of apoptotic cells was examined by flow
cytometric assay. Actin was used as a loading control for western
blotting. Data are presented as the mean ± standard deviation from
three separate experiments where appropriate. *P<0.05 vs.
PHA-665752 alone or dasatinib alone. PHA, PHA-665752; AKT, protein
kinase B; ERK, extracellular signal-regulated kinase; PARP, poly
(ADP-ribose) polymerase; p, phosphorylated. |
Western blotting revealed that combined treatment
with PHA-665752 and dasatinib further decreased phosphorylation of
MET, SRC, AKT and ERK in HT-29 and HCT-116 cells (Fig. 6C). Additionally, it was revealed that
a combination of PHA-665752 and dasatinib significantly enhanced
apoptosis in HT-29 cells, as detected by PARP cleavage and the
increased the proportion of apoptotic cells (P<0.05; Fig. 6D and E). The proportion of apoptotic
cells was 40.6±6.9% in the combination treatment group, which was
significantly more than the proportion of apoptotic cells after
single PHA-665752 (14.8±4.2%; P=0.003) or dasatinib (22.6±4.1%;
P=0.004) treatment in HT-29 cells. These results demonstrated that
dual inhibition of MET and SRC exerted superior proliferation
inhibition and apoptosis enhancement than either agent alone.
Discussion
In targeted therapy, numerous studies have suggested
that combining inhibition of tyrosine kinases may be a beneficial
therapeutic strategy (3,23). To date, few therapeutic targeting
options are available, particularly in patients with CRC with
mutated RAS. In the present study, the important role of MET
activation in ligand-dependent activation by HGF and
ligand-independent activation by EGF and IGF-1 was investigated in
colon cancer cells. It was demonstrated that SRC was the key factor
that mediated the interaction of MET with EGFR or IGF-1R.
Therefore, combined treatment with MET and SRC inhibitors exhibited
increased inhibition of cell viability and apoptosis, which
provided novel insights into dual targeting of MET and SRC in colon
cancer cells.
HGF/MET signaling is essential in aberrant processes
of cell survival, growth, angiogenesis and metastasis (24). Aberrant MET activation may mediate
resistance to targeted therapies, and ultimately result in
treatment failure (22,25). The results of the present study
demonstrated that HGF promoted cell viability to a greater extent
at a lower concentration of 25 ng/ml compared to a higher
concentration of 125 ng/ml, while colon cancer cells may
predominantly exhibit manifestation of epithelial-to-mesenchymal
transition in a higher concentration of HGF (26). In addition, MET signaling was
involved in an intricate network of cross-signaling through
multiple ligand-independent mechanisms. The results of the present
study demonstrated that the main growth factors, including EGF and
IGF-1, induced delayed MET activation in colon cancer cells.
HGF/MET signaling is essential for the maintenance of colon cancer
stem cells (27). Therefore,
targeting MET signaling may be an important therapeutic strategy in
CRC.
Recent studies have clearly indicated that targeting
HGF/MET may have potential activity in several groups of cancer
patients either alone or with inhibitors of other signaling
pathways (28,29). In colon cancer cells, inhibition of
MET by siRNA or inhibitors demonstrated certain anti-viability
effects, with ~20% cell viability inhibition. However, the MET
inhibitor, PHA-665752, failed to inhibit autophosphorylation of
SRC, AKT and ERK in HT-29 and HCT-116 cells, while PHA-665752
effectively abrogated activation of SRC, AKT and ERK stimulated by
HGF. Thus, the sustained activation of downstream MET signaling may
cause limited anti-viability effects for the MET inhibitor in colon
cancer.
SRC has been demonstrated to be a key downstream
transducer of MET-driven tumor growth (30) and the activity of SRC is important
for the growth of CRC cells (31).
The results of the present study demonstrated that there was a
mutually strong interaction between MET and SRC in colon cancer
cells. SRC effectively influenced the activation of MET through the
physical complex of MET and SRC, according to the
co-immunoprecipitation assay and transfection experiments with
siRNA sequences or overexpression plasmids. Although the SRC
inhibitor, dasatinib, reduced MET autophosphorylation and decreased
MET phosphorylation by stimulating factors, the inhibition was not
complete. In addition, SRC inhibitors have been demonstrated to be
effective in certain solid tumors; however, sustained MET
activation may mediate resistance to SRC inhibition in head and
neck, and gastric cancer (32,33).
Additionally, in gefitinib-resistant lung cancer cells with MET
amplification, SRC activation is more dependent on MET signaling
(34). In these cases, combinatorial
treatment using targeted SRC and MET inhibitors may exhibit
synergistic cytotoxic effects.
Therefore, combinational treatment using MET and SRC
inhibitors was performed in colon cancer cells. The results
indicated that the mutual interaction between MET and SRC was
strongly linked in colon cancer cells. Furthermore, the combination
of PHA-665752 and dasatinib inhibited cell growth by decreasing
cell viability and increasing apoptosis. The combined treatment
prevented ligand-dependent and ligand-independent activation of MET
and SRC, and further decreased basal phosphorylation of SRC, AKT
and ERK in the two cell types. These results indicated that
combinatorial inhibition of MET and SRC may be essential to
effectively suppress activation of the HGF/MET pathway.
Furthermore, combined therapy demonstrated the same antitumor
effect, particularly in RAS mutant colon cancer cells.
In conclusion, the present study demonstrated that
MET and SRC are essential in ligand-dependent and
ligand-independent MET activation. Thus, combined inhibition of MET
and SRC may possess enhanced cytotoxicity, providing a strong basis
for assessing the therapeutic value of concurrent inhibition of MET
and SRC in CRC.
Acknowledgements
The present study was supported by grants from the
Chinese National Foundation of National Sciences Grants (grant nos.
81401938 and 81372546), the National Science and Technology Major
Project of the Ministry of Science and Technology of China (grant
no. 2013JX09303002) and the Science and Technology Plan Project of
Liaoning Province (grant nos. 2014225013 and 2014021089).
References
|
1
|
Cremolini C, Loupakis F, Antoniotti C,
Lupi C, Sensi E, Lonardi S, Mezi S, Tomasello G, Ronzoni M,
Zaniboni A, et al: FOLFOXIRI plus bevacizumab versus FOLFIRI plus
bevacizumab as first-line treatment of patients with metastatic
colorectal cancer: Updated overall survival and molecular subgroup
analyses of the open-label, phase 3 TRIBE study. Lancet Oncol.
16:1306–1315. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Maurel J and Postigo A: Prognostic and
predictive biomarkers in colorectal cancer. From the preclinical
setting to clinical practice. Curr Cancer Drug Targets. 15:703–715.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Grothey A, Van Cutsem E, Sobrero A, Siena
S, Falcone A, Ychou M, Humblet Y, Bouché O, Mineur L, Barone C, et
al: Regorafenib monotherapy for previously treated metastatic
colorectal cancer (CORRECT): An international, multicentre,
randomised, placebo-controlled, phase 3 trial. Lancet. 381:303–312.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wilhelm SM, Dumas J, Adnane L, Lynch M,
Carter CA, Schütz G, Thierauch KH and Zopf D: Regorafenib (BAY
73–4506): A new oral multikinase inhibitor of angiogenic, stromal
and oncogenic receptor tyrosine kinases with potent preclinical
antitumor activity. Int J Cancer. 129:245–255. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sartore-Bianchi A, Siena S, Tonini G,
Bardelli A and Santini D: Overcoming dynamic molecular
heterogeneity in metastatic colorectal cancer: Multikinase
inhibition with regorafenib and the case of rechallenge with
anti-EGFR. Cancer Treat Rev. 51:54–62. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Winder T and Lenz HJ: Vascular endothelial
growth factor and epidermal growth factor signaling pathways as
therapeutic targets for colorectal cancer. Gastroenterology.
138:2163–2176. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gherardi E, Birchmeier W, Birchmeier C and
Woude G Vande: Targeting MET in cancer: Rationale and progress. Nat
Rev Cancer. 12:89–103. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mitra AK, Sawada K, Tiwari P, Mui K, Gwin
K and Lengyel E: Ligand-independent activation of c-Met by
fibronectin and α(5)β(1)-integrin regulates ovarian cancer invasion
and metastasis. Oncogene. 30:1566–1576. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Dulak AM, Gubish CT, Stabile LP, Henry C
and Siegfried JM: HGF-independent potentiation of EGFR action by
c-Met. Oncogene. 30:3625–3635. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Varkaris A, Gaur S, Parikh NU, Song JH,
Dayyani F, Jin JK, Logothetis CJ and Gallick GE: Ligand-independent
activation of MET through IGF-1/IGF-1R signaling. Int J Cancer.
133:1536–1546. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lee J, Jain A, Kim P, Lee T, Kuller A,
Princen F, In-Gu Do, Kim SH, Park JO, Park YS, et al: Activated
cMET and IGF1R-driven PI3K signaling predicts poor survival in
colorectal cancers independent of KRAS mutational status. PLoS One.
9:e1035512014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Peng Z, Zhu Y, Wang Q, Gao J, Li Y, Li Y,
Ge S and Shen L: Prognostic significance of MET amplification and
expression in gastric cancer: A systematic review with
meta-analysis. PLoS One. 9:e845022014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Guo B, Cen H, Tan X, Liu W and Ke Q:
Prognostic value of MET gene copy number and protein expression in
patients with surgically resected non-small cell lung cancer: A
meta-analysis of published literatures. PLoS One. 9:e993992014.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bardelli A, Corso S, Bertotti A, Hobor S,
Valtorta E, Siravegna G, Sartore-Bianchi A, Scala E, Cassingena A,
Zecchin D, et al: Amplification of the MET receptor drives
resistance to anti-EGFR therapies in colorectal cancer. Cancer
Discov. 3:658–673. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kim LC, Song L and Haura EB: Src kinases
as therapeutic targets for cancer. Nav Rev Clin Oncol. 6:587–595.
2009. View Article : Google Scholar
|
|
16
|
Stabile LP, He G, Lui VW, Thomas S, Henry
C, Gubish CT, Joyce S, Quesnelle KM, Siegfried JM and Grandis JR:
c-Src activation mediates erlotinib resistance in head and neck
cancer by stimulating c-Met. Clin Cancer Res. 19:380–392. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang S, Huang WC, Li P, Guo H, Poh SB,
Brady SW, Xiong Y, Tseng LM, Li SH, Ding Z, et al: Combating
trastuzumab resistance by targeting SRC, a common node downstream
of multiple resistance pathways. Nat Med. 17:461–469. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lieu C and Kopetz S: The SRC family of
protein tyrosine kinases: A new and promising target for colorectal
cancer therapy. Clin Colorectal Cancer. 9:89–94. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Vergani E, Vallacchi V, Frigerio S, Deho
P, Mondellini P, Perego P, Cassinelli G, Lanzi C, Testi MA,
Rivoltini L, et al: Identification of MET and SRC activation in
melanoma cell lines showing primary resistance to PLX4032.
Neoplasia. 13:1132–1142. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mueller KL, Hunter LA, Ethier SP and
Boemer JL: Met and c-Src cooperate to compensate for loss of
epidermal growth factor receptor kinase activity in breast cancer
cells. Cancer Res. 68:3314–3322. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Booth L, Roberts JL, Tavallai M, Webb T,
Leon D, Chen J, McGuire WP, Poklepovic A and Dent P: The afatinib
resistance of in vivo generated H1975 lung cancer cell clones is
mediated by SRC/ERBB3/c-KIT/c-MET compensatory survival signaling.
Oncotarget. 7:19620–19630. 2016.PubMed/NCBI
|
|
22
|
Song N, Liu S, Zhang J, Liu J, Xu L, Liu Y
and Qu X: Cetuximab-induced MET activation acts as a novel
resistance mechanism in colon cancer cells. Int J Mol Sci.
15:5838–5851. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Llovet JM, Villanueva A, Lachenmayer A and
Finn RS: Advances in targeted therapies for hepatocellular
carcinoma in the genomic era. Nat Rev Clin Oncol. 12:408–424. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Corso S and Giordano S: Cell-autonomous
and non-cell-autonomous mechanisms of HGF/MET-driven resistance to
targeted therapies: From basic research to a clinical perspective.
Cancer Discov. 3:978–992. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Liska D, Chen CT, Bachleitner-Hofmann T,
Christensen JG and Weiser MR: HGF rescues colorectal cancer cells
from EGFR inhibition via MET activation. Clin Cancer Res.
17:472–482. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chen QY, Jiao DM, Wu YQ, Chen J, Wang J,
Tang XL, Mou H, Hu HZ, Song J, Yan J, et al: MiR-206 inhibits
HGF-induced epithelial-mesenchymal transition and angiogenesis in
non-small cell lung cancer via c-Met /PI3k/Akt/mTOR pathway.
Oncotarget. 7:18247–18261. 2016.PubMed/NCBI
|
|
27
|
Luraghi P, Reato G, Cipriano E, Sassi F,
Orzan F, Bigatto V, De Bacco F, Menietti E, Han M, Rideout WM III,
et al: MET signaling in colon cancer stem-like cells blunts the
therapeutic response to EGFR inhibitors. Cancer Res. 74:1857–1869.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Smith MR, Sweeney CJ, Corn PG, Rathkopf
DE, Smith DC, Hussain M, George DJ, Higano CS, Harzstark AL, Sartor
AO, et al: Cabozantinib in chemotherapy-pretreated metastatic
castration-resistant prostate cancer: Results of a phase II
nonrandomized expansion study. J Clin Oncol. 32:3391–3399. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Scagliotti G, von Pawel J, Novello S,
Ramlau R, Favaretto A, Barlesi F, Akerley W, Orlov S, Santoro A,
Spigel D, et al: Phase III multinational, randomized, double-blind,
placebo-controlled study of tivantinib (ARQ 197) plus erlotinib
versus erlotinib alone in previously treated patients with locally
advanced or metastatic nonsquamous non-small-cell lung cancer. J
Clin Oncol. 33:2667–2674. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bertotti A, Bracco C, Girolami F, Torti D,
Gastaldi S, Galimi F, Medico E, Elvin P, Comoglio PM and Trusolino
L: Inhibition of Src impairs the growth of met-addicted gastric
tumors. Clin Cancer Res. 16:3933–3943. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Emaduddin M, Bicknell DC, Bodmer WF and
Feller SM: Cell growth, global phosphotyrosine elevation, and c-Met
phosphorylation through Src family kinases in colorectal cancer
cells. Proc Natl Acad Sci USA. 105:pp. 2358–2362. 2008; View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sen B, Peng S, Saigal B, Williams MD and
Johnson FM: Distinct interactions between c-Src and c-Met in
mediating resistance to c-Src inhibition in head and neck cancer.
Clin Cancer Res. 17:514–524. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Okamoto W, Okamoto I, Yoshida T, Okamoto
K, Takezawa K, Hatashita E, Yamada Y, Kuwata K, Arao T, Yanagihara
K, et al: Identification of c-Src as a potential therapeutic target
for gastric cancer and of MET activation as a cause of resistance
to c-Src inhibition. Mol Cancer Ther. 9:1188–1197. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yoshida T, Okamoto I, Okamoto W, Hatashita
E, Yamada Y, Kuwata K, Nishio K, Fukuoka M, Jänne PA and Nakagawa
K: Effects of Src inhibitors on cell growth and epidermal growth
factor receptor and MET signaling in gefitinib-resistant non-small
cell lung cancer cells with acquired MET amplification. Cancer Sci.
101:167–172. 2010. View Article : Google Scholar : PubMed/NCBI
|