Introduction
Cardiac fibrosis refers to excessive accumulation of
collagen in the normal myocardium and significantly increased
collagen concentration or change of collagen composition in heart
tissue. Cardiac fibrosis is the inevitable process of a variety of
clinical cardiovascular diseases that have developed to the final
stage, which is the main performance of cardiac remodeling
(1,2). At present, it is known that cardiac
fibrosis is closely associated with arrhythmia, heart failure and
mortality due to sudden cardiac arrest (3–5). It is
generally known that activation and proliferation of cardiac
fibroblasts (CFs) and deposition of extracellular matrix (ECM)
secreted by CFs are the primary features of cardiac fibrosis
(1,6). Accumulation of ECM increases myocardial
hardness, reduces myocardial compliance and affects the normal
diastolic and systolic function of the heart (6). Therefore, cardiac fibrosis is a key
factor in cardiovascular disease outcomes, and CFs are important in
the process of cardiac fibrosis (6).
In the stimuli of myocardial infarction, an excessive pressure load
or neurohumoral factors and CFs begin with pathologic hyperplasia
and convert to myofibroblasts (6,7). Under
the mediation of a variety of cytokines, myofibroblasts can migrate
to the damaged area and proliferate rapidly to synthesize and
release a large amount of collagen I and III, which accumulates in
the myocardial interstitium and perivascular spaces (3). It may also promote crosslinking between
the ECM, and cause excessive deposition of collagen that leads to
cardiac fibrosis and provides a pathological basis to the
occurrence and development of cardiovascular diseases (6,8).
Therefore, in improving the prognosis of cardiovascular diseases
and inhibiting cardiac remodeling at the cellular level, CFs have
become an important target to control the progression of cardiac
fibrosis. Abrogation of CF transformation into myofibroblasts, and
the inhibition of CF proliferation and collagen synthesis may be a
strategy for suppressing cardiac fibrotic remodeling.
Naringenin (NRG) is a type of double hydrogen
flavonoid that predominantly exists in rutaceae citrus plants
(9). A number of studies have
confirmed that NRG demonstrates numerous biological effects on
oxidative stress, the inflammatory response, disorders of lipid
metabolism and apoptosis (9–11). Recently, the cardioprotective effects
of NRG have been the focus of a number of studies (12,13).
However, the mechanism underlying its effects remains unclear.
Therefore, the aim of the present study was to investigate the
effects and mechanism of NRG on the proliferation and collagen
synthesis of CFs induced by transforming growth factor β1 (TGF-β1)
from the cellular level to elucidate the mechanism of NRG against
cardiac fibrosis.
Materials and methods
Materials
Primary antibodies including anti-cyclin D1 (cat no.
2978), anti-cyclin dependent kinase 4 (anti-CDK4; cat no. 12790),
anti-CDK6 (cat no. 3136) and anti-CDK2 (cat no. 2546) were
purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA).
Primary anti-cyclin E2 antibody (cat. no. sc-22777) was purchased
from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA). Primary
anti-GAPDH antibody (cat. no. MB001) was purchased from Bioworld
Technology, Inc. (St. Louis Park, MN, USA). Cell counting kit-8
(CCK-8; cat. no. CK04-11) was purchased from Dojindo Molecular
Technologies, Inc. (Rockville, MD, USA). TRIzol reagent (cat. no.
15596018) was purchased from Invitrogen (Thermo Fisher Scientific,
Inc., Waltham, MA, USA). Transcriptor First Strand cDNA Synthesis
kit (cat no. 04896866001) was bought from Roche Diagnostics (Basel,
Switzerland). Bicinchoninic acid (BCA) protein assay kit (cat. no.
23227) was from Pierce (Thermo Fisher Scientific, Inc.). NRG (cat.
no. W530098) was purchased from Sigma-Aldrich (Merck KGaA,
Darmstadt, Germany).
Cell culture and treatment
A total of 70 neonatal Sprague-Dawley rats were
obtained from the Animal Experimental Center of Wuhan University
(Wuhan, China) and the Center for Disease Control and Prevention of
Hubei Province (Wuhan, China). Primary culture of neonatal rat CFs
was prepared from ventricles of 1–3 day-old rats via the
differential attachment method (1,2).
Briefly, following sacrifice, rat hearts were removed from the
thorax and immediately placed in 4°C PBS, and ventricles were
digested with 0.125% trypsin and 0.08% collagenase type II at 37°C
(5 times for 5 min each). However, in order to reduce the presence
of cell fragments and blood in cell samples, the first digestion
solution was discarded. Cells from the last four digestions were
collected, and incubated in Dulbecco's modified Eagle's
medium/Nutrient Mixture F-12 (DMEM/F12) medium (Hyclone; GE
Healthcare, Logan, UT, USA) supplemented with 10% fetal bovine
serum (FBS; Hyclone; GE Healthcare) at 37°C for 1 h. Subsequently,
unattached or weakly attached cells were discarded, but attached
cells were incubated in fresh DMEM/F12 supplemented with 10% FBS.
After the confluence of CFs reached 80%, CFs were digested using
0.25% trypsin and then passaged. Passages 2–4 were used for
subsequent experiments in the present study. The cells were
cultured in a 6-well plate when they were used for flow cytometry
analysis. The cells were stimulated with TGF-β1 or PBS to induce CF
transformation into myofibroblasts (1). In the present study, to detect the
appropriate concentration of NRG, the effects of several
concentrations (0, 10, 20, 30, 40 and 50 µM) of NRG on α-SMA mRNA
levels were evaluated following exposure for 48 h. Next, with the
appropriate concentration, the cells were treated with different
times (0, 12, 24, 48 and 72 h) to choose the appropriate treatment
time. Upon the appropriate concentration and time of NRG, the cells
were divided into the following groups: PBS + dimethyl sulfoxide
(DMSO), PBS + naringenin, TGF-β1 + DMSO and TGF-β1 + naringenin.
The use of animals in the present study was approved by the Ethics
Committee of The First People's Hospital of Yueyang (Yueyang,
China).
Immunofluorescence staining
In order to detect the number of CFs that expressed
α-smooth muscle actin (α-SMA) protein, CFs were stained for the
marker α-SMA (1). Cells were washed
three times with PBS, fixed with 4% paraformaldehyde at room
temperature for 15 min and permeabilized in 0.2% Triton X-100 in
PBS at room temperature for 20 min. Furthermore, they were blocked
with 5% bovine serum albumin (cat. no. A3733; Sigma-Aldrich; Merck
KGaA) in PBS at room temperature for 30 min and then stained with
anti-α-SMA antibody (cat. no. ab3280; Abcam, Cambridge, MA, USA;
1:100 dilution) overnight. The cells were incubated with a green
fluorescence-marked secondary antibody (cat. no. AS1112; Aspen
Biological Co., Ltd., Wuhan, China; 1:100 dilution) for 1 h at room
temperature and then counterstained with DAPI for 8 min at room
temperature. Finally, the cells were covered with mounting medium
and kept in the dark at 4°C.
Cell proliferation assay
Cell proliferation assay was performed according to
the manufacturer's instructions of the CCK-8. Initially, the cell
suspension was inoculated in a 96-well plate (100 µl/well) and
incubated until the confluence of CFs was 60–70%. Following
incubation with 30 µM NRG at 37°C for 48 h, CFs were then incubated
with CCK-8 solution (10 µl/well) at 37°C for 2 h and the optical
density of each well was measured at 450 nm using a microplate
reader (Tecan Infinite M200; Tecan Group Ltd., Mannedorf,
Switzerland). CFs with culture medium and CCK8, without PBS/TGF-h1
or NRG/DMSO treatment, were used as the control.
Flow cytometry analysis
Each group of ~1×106 CFs, including PBS +
DMSO, PBS + NRG, TGF-β1 + DMSO and TGF-β1 + NRG, was cultivated in
a 6-well plate, collected, fixed in 70% ice-cold ethanol and,
maintained at 4°C overnight. CFs were washed with PBS and
centrifuged for 2 min at 716 × g at room temperature, and the
supernatant was discarded. The cell pellet was vortexed in 500 µl
PBS left behind to avoid clumping of cells. 500 µl RNase (cat. no.
10109142001; Sigma-Aldrich; Merck KGaA) was added and the cells
were subsequently stained with propidium iodide (PI) (cat. no.
P3566; Invitrogen; Thermo Fisher Scientific, Inc.) in the dark for
45 min. To avoid clumps, the cells were transferred through meshed
blue-capped falcon tubes prior to analysis using a FACSCalibur
system (BD Biosciences, Franklin Lakes, NJ, USA).
Reverse transcription-quantitative
polymerase chain reaction assay
CFs were collected and the total mRNA was extracted
using TRIzol reagent according to the manufacturer's instructions.
cDNA synthesis was performed using the Transcriptor First Strand
cDNA synthesis kit (cat. no. 04896866001; Roche Diagnostics),
according to the manufacturer's protocol. Gene differences were
detected using SYBR green (cat. no. 04913850001; Roche Diagnostics)
and GAPDH gene expression was used as an internal control. The
reaction conditions were as follows: 95°C for 10 min, followed by
40 cycles of 95°C for 15 sec and 60°C for 30 sec, then 95°C for 15
min and 60°C for 1 min. The expression levels of each gene were
quantified through the analysis of the Cq value and standard curve
(14). The sequences of the primer
pairs were exhibited as follows: ki67: 5′-TAGAGGATCTGCCTGGCTTC-3′
(forward) and 5′-TGTCCTTGGTTGGTTCCTCC-3′ (reverse); proliferating
cell nuclear antigen (PCN A): 5′-CAACTTGGAATCCCAGAACAGGAG-3′
(forward) and 5′-TAAGGTCCCGGCATATACGTGC-3′ (reverse); α-SMA:
5′-GCTATTCAGGCTGTGCTGTC-3′ (forward) and 5′-GGTAGTCGGTGAGATCTCGG-3′
(reverse); connective tissue growth factor (CTG F):
5′-GGAAGACACATTTGGCCCTG-3′ (forward) and 5′-GCAATTTTAGGCGTCCGGAT-3′
(reverse); collagen I: 5′-GAGCGGAGAGTACTGGATCGA-3′ (forward) and
5′-CTGACCTGTCTCCATGTTGCA-3′ (reverse); collagen III:
5′-TGCCATTGCTGGAGTTGGA-3′ (forward) and
5′-GAAGACATGATCTCCTCAGTGTTGA-3′ (reverse) and GAP DH:
5′-GGGTGATGCTGGTGCTGAGTATGT-3′ (forward) and
5′-CAGTGGATGCAGGGATGATGTTCT-3′ (reverse).
Western blot analysis
Proteins were extracted from CFs using
radioimmunoprecipitation assay lysis buffer (cat. no. P0013B;
Beyotime Institute of Biotechnology, Haimen, China), and the
protein concentration was determined using a BCA protein assay kit
(cat. no. 23227; Pierce; Thermo Fisher Scientific, Inc.). Proteins
were loaded (10 µg/well) and separated using 10% SDS-PAGE at 120 V
for 120 min. Subsequently, the proteins were transferred onto
polyvinyl difluoride transfer membranes at 4°C and the membranes
were blocked for 60 min with freshly prepared 5% skimmed milk in
Tris-buffered saline with Tween 20 (TBST) at room temperature.
Following this, the membranes were incubated with various primary
antibodies, including cyclin D1 (1:1,000), CDK4 (1:1,000), CDK6
(1:1,000), cyclin E2 (1:500), CDK2 (1:1,000) and GAPDH (1:10,000)
overnight at 4°C with gentle shaking. Membranes were washed three
times with TBST, and incubated with either HRP-conjugated goat
anti-mouse (cat. no. 074-1806; Kirkegaard and Perry Laboratories,
Inc., Gaithersburg, MD, USA; 1:10,000 dilution) or goat anti-rabbit
(cat. no. 074-1506; Kirkegaard and Perry Laboratories, Inc.;
1:10,000 dilution) secondary antibodies for 60 min at room
temperature with shaking. Following four washes of the membranes,
images were captured on films, which were placed in LumiGLO
solution (cat. no. 7003; Cell Signaling Technology, Inc.) for 1 min
at room temperature. Following development, the images were placed
into an automatic image analyzer to determine the expression levels
of the proteins as well as the reference gray scale values. GAPDH
was used as a loading control and three independent experiments
were performed.
Statistical analysis
Data are expressed as the mean ± standard error of
the mean. Statistical analyses were performed using SPSS version
13.0 software (SPSS, Inc., Chicago, IL, USA). Comparisons between
each group were performed using one-way analysis of variance
followed by Fisher's least significant difference test. P<0.05
was considered to indicate a statistically significant
difference.
Results
Appropriate concentration and
treatment time of NRG in CFs induced by TGF-β1
Initially, to detect the appropriate concentration
of NRG, the effects of several concentrations of it were measured
on the α-SMA mRNA level following exposure for 48 h. It was
revealed that induction of α-SMA expression was significantly
inhibited when NRG was added at a concentration of 30 µM, compared
with that when 0, 10 or 20 µM NRG was added (Fig. 1A). Additionally, it was demonstrated
that 30 µM NRG inhibited α-SMA expression in a time-dependent
manner. Compared with the 12 and 24 h groups, the minimum
expression of α-SMA was revealed at the 48 and 72 h time points
(Fig. 1B). Therefore, 30 µM NRG with
48 h treatment time was selected for subsequent experiments.
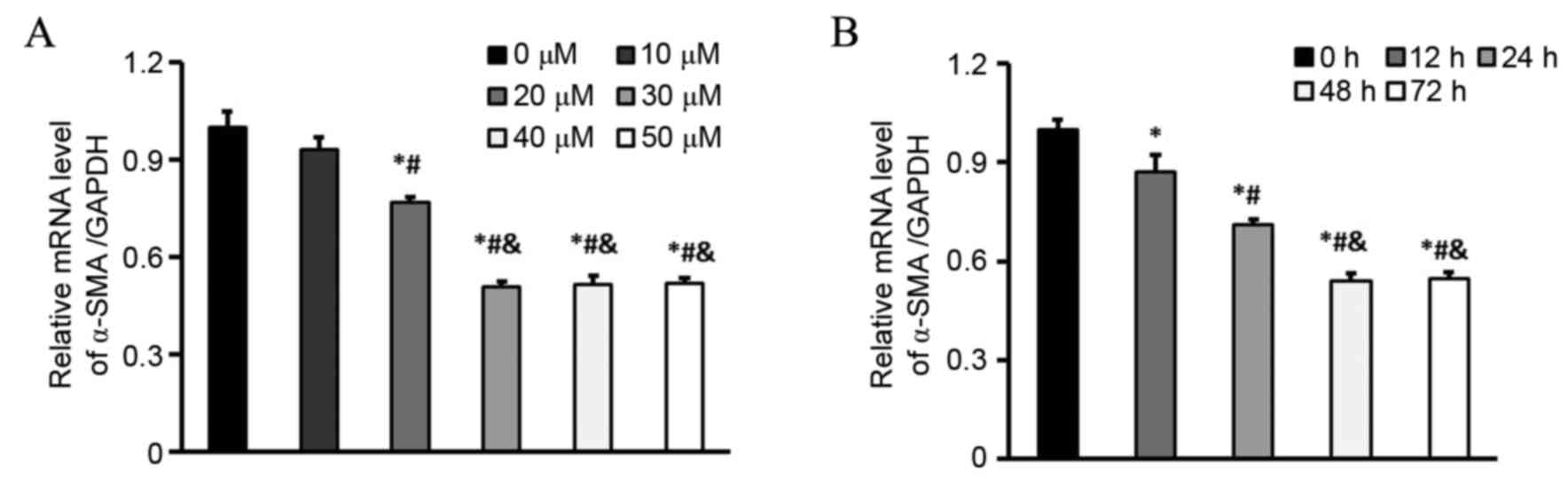 | Figure 1.Appropriate concentration and
treatment time of NRG in CFs induced by TGF-β1. (A) mRNA levels of
α-SMA in CFs treated with 0, 10, 20 30, 40 and 50 µM NRG and
stimulated with 5 ng/ml TGF-β1 for 48 h (n=6 samples per group).
*P<0.05 vs. 0 µM, #P<0.05 vs. 10 µM and
&P<0.05 vs. 20 µM. (B) mRNA levels of α-SMA in
CFs treated with 30 µM NRG and stimulated with 5 ng/ml TGF-β1 for
0, 12, 24, 48, and 72 h (n=6 samples per group). *P<0.05 vs. 0
h, #P<0.05 vs. 12 h and &P<0.05 vs.
24 h. NRG, naringenin; CF, cardiac fibroblast; TGF-β1, transforming
growth factor β1; α-SMA, α-smooth muscle actin. |
NRG treatment inhibits transformation
and proliferation of CFs
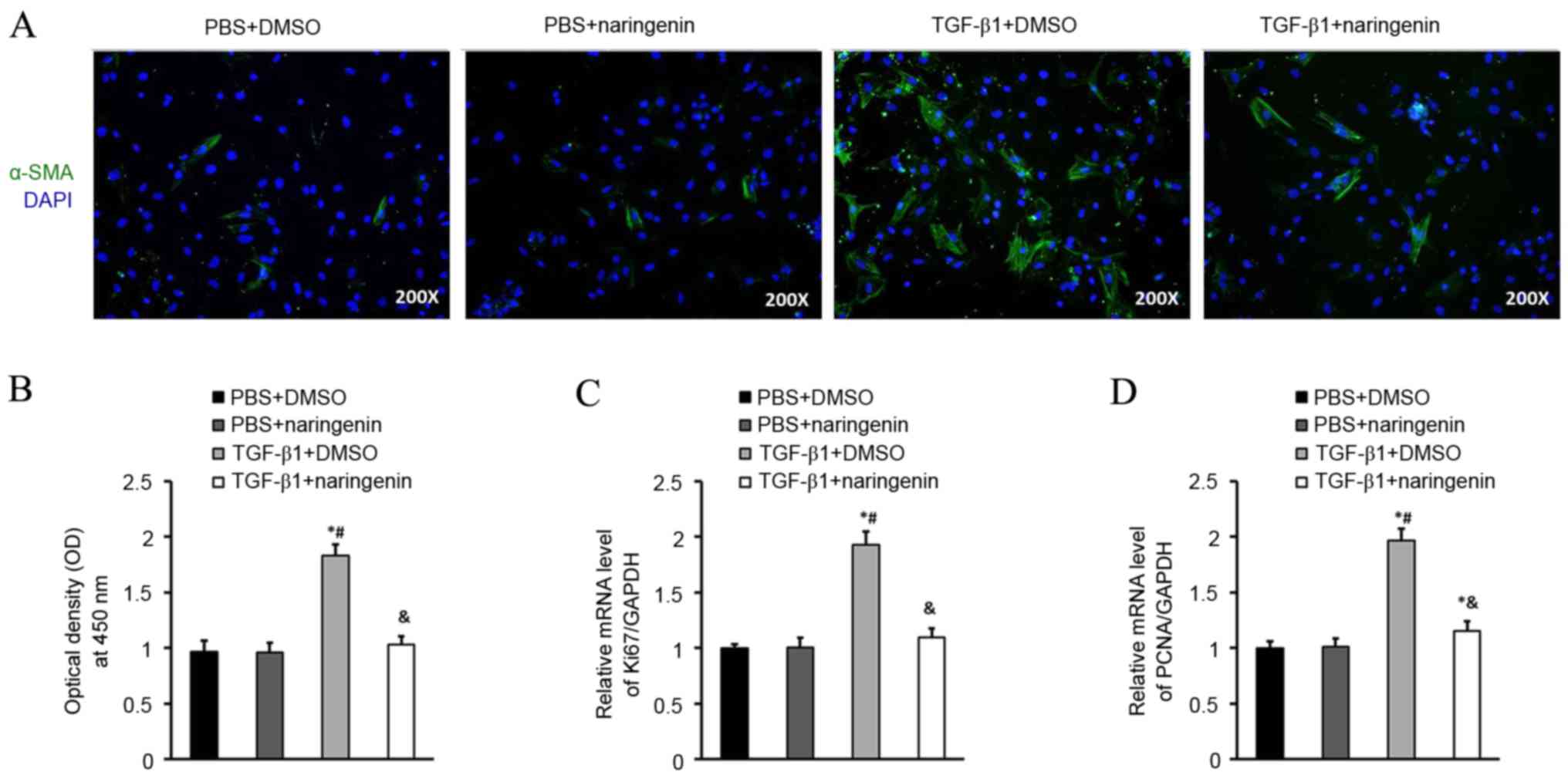
Transformation of CFs into cardiac myofibroblasts
was investigated with α-SMA. The expression of α-SMA protein
detected via immunofluorescence staining was markedly inhibited in
the NRG-treated group, which indicated that NRG treatment for 48 h
was able to reduce the number of CFs that expressed α-SMA protein
induced by TGF-β1 (Fig. 2A). CF
proliferation was investigated via CCK-8 assay (Fig. 2B). Subsequently, the effect of NRG on
proliferation of CFs was further assessed by the mRNA levels of
ki67 and PCNA (Fig. 2C and D). It
was revealed that the mRNA levels of ki67 and PCNA were
significantly inhibited in CFs following treatment with NRG
compared with the TGF-β1 group (P<0.05), which indicated that
NRG treatment was able to inhibit the proliferation of CFs compared
with the TGF-β1 group.
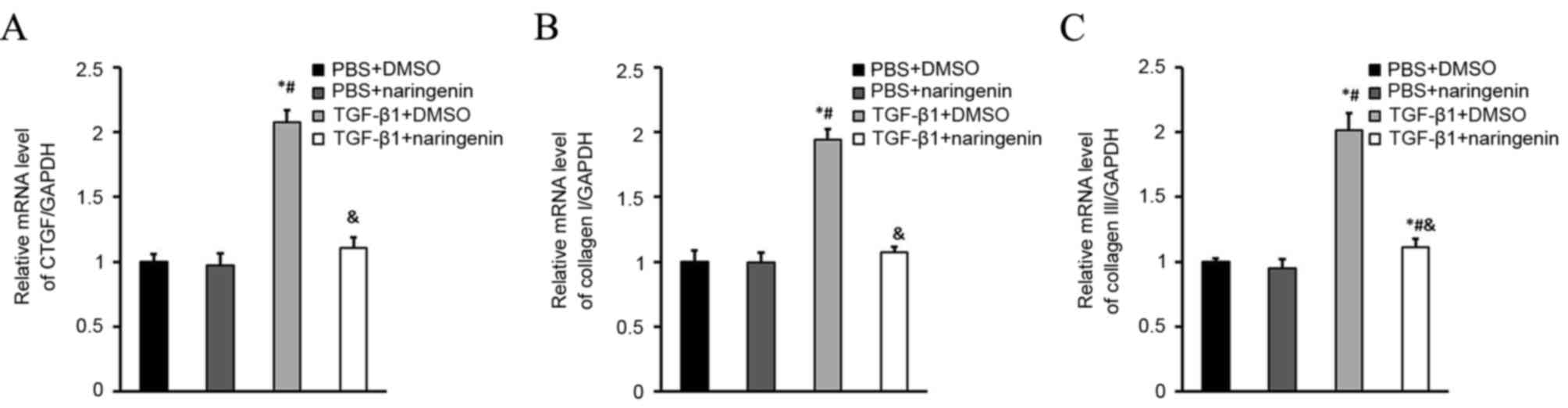
NRG treatment inhibits collagen
synthesis of CFs
Collagen synthesis of cardiac myofibroblasts is
perceived to be important in the development of cardiac fibrosis
(15,16). Therefore, the role of NRG on collagen
synthesis was detected via the mRNA levels of CTGF, collagen I and
collagen III. It was revealed that NRG significantly decreased the
mRNA levels of CTGF, collagen I and collagen III in CFs induced by
TGF-β1 (Fig. 3).
NRG treatment induces CF cell cycle
arrest in the G0/G1-phase
In order to elucidate the mechanism of CF
proliferation regulated by NRG, the effect of NRG on cell cycle
progression was evaluated. CFs under subconfluent culture
conditions were synchronized by serum starvation, which caused cell
cycle arrest at the G0/G1-phase, and then treated with the
indicated stimuli, labeled with PI and analyzed by flow cytometry
(17,18). It was revealed that the S-phase
population was significantly increased and the G0/G1-phase was
significantly decreased in the TGF-β1 + DMSO group compared with
the PBS groups, whereas the G0/G1-phase of CFs in the TGF-β1 group
treated with NRG was significantly increased and the S-phase
population was significantly lower compared with all other groups.
These results indicated that NRG treatment induces CF cell cycle
arrest in the G0/G1-phase (Table
I).
 | Table I.NRG treatment induces cardiac
fibroblast cell cycle arrest in the G0/G1-phase (%). |
Table I.
NRG treatment induces cardiac
fibroblast cell cycle arrest in the G0/G1-phase (%).
| Stage | PBS + DMSO | PBS + NRG | TGF-β1 + DMSO | TGF-β1 + NRG |
|---|
| G0/G1 | 77.32±3.11 | 76.08±3.32 |
65.641±2.90a,b |
86.53±2.64a–c |
| S | 15.61±1.18 | 16.31±0.93 |
25.87±1.47a,b |
10.19±0.78a–c |
| G2/M |
7.07±0.34 |
7.61±0.73 |
8.74±0.69a |
3.27±0.48a–c |
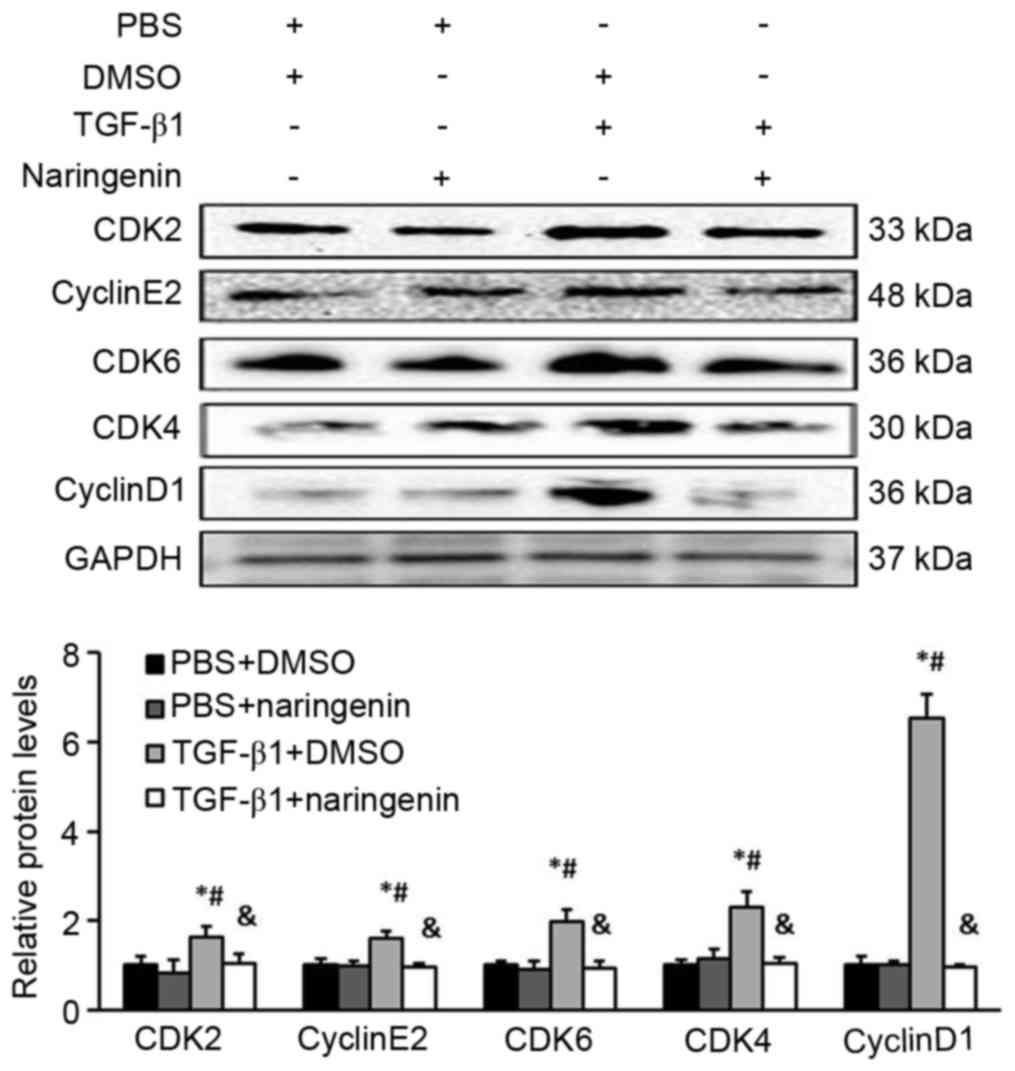
NRG inhibits CFs entering the S-phase
by downregulating the cyclin D1-CDK4/6 and cyclin E2-CDK2
complexes
The G0/G1 to S-phase transition is primarily
regulated by the cyclin D1-CDK4/6 and cyclin E2-CDK2 complexes
(19,20). To investigate the status of these
complexes in NRG-treated CFs induced by TGF-β1, the protein levels
of individual cyclins and CDKs were measured by western blot
analysis. It was revealed that the expression levels of cyclin D1,
CDK4, CDK6, cyclin E2 and CDK2 decreased significantly in CFs
treated with NRG and TGF-β1 compared with the TGF-β1 group, which
indicated that NRG repressed the G1/S-phase transition, in part, by
downregulating the cyclin D1-CDK4/6 and cyclin E2-CDK2 complexes
(Fig. 4).
Discussion
In the present study, the effects and the probable
mechanism of NRG on CFs stimulated with TGF-β1 in vitro were
investigated. It was revealed that NRG had the following effects:
i) Inhibited CF transformation into cardiac myofibroblasts; ii)
inhibited the proliferation of CFs; iii) inhibited the synthesis of
CTGF, collagen I and III in CFs stimulated with TGF-β1; iv) induced
CF cell cycle arrest in the G0/G1-phase; and v) repressed the
G1/S-phase transition of CFs partly via downregulating the cyclin
D1-CDK4/6 and cyclin E2-CDK2 complexes following TGF-β1 induction.
Although further studies are required, to the best of our knowledge
the present study is the first to link the anti-proliferative
effect of NRG on CFs with the inhibition of DNA synthesis via G0/G1
arrest, indicating that NRG may be used as a therapeutic agent to
regulate cardiac fibrosis in the future.
Cardiac fibrosis is associated with various
physiological causes, including CF proliferation, inflammation and
hypertension (4). Among these
causes, the transformation of CFs into cardiac myofibroblasts and
activated proliferation are known to be important events in the
development of cardiac fibrosis, which may promote pathological
hypertrophy and fibrosis and subsequently result in cardiac
remodeling and cardiac dysfunction (6). Therefore, inhibiting CF proliferation
is considered important for treating cardiovascular diseases. In
the present study, the anti-proliferative effect of NRG was
examined on CF proliferation. NRG significantly inhibited
TGF-β1-induced CF proliferation, transformation into cardiac
myofibroblasts and the synthesis of CTGF, collagen I and III of
CFs. Notably, the anti-proliferative effect of NRG was associated
with the inhibition of DNA synthesis via G0/G1 cell cycle arrest.
The cell cycle consists of four sequential phases including G0/G1,
S, G2 and M. It regulates cellular proliferation by a highly
controlled process involving a complex cascade of cellular events,
including regulatory factor cyclins and CDKs (21,22). The
cyclin D1-CDK4/6 and cyclin E2-CDK2 complexes are important
mediators of the cell cycle transition from the G0/G1 to S-phase
(19,20). In the present study, NRG not only
inhibited cyclin D1 and cyclin E2 expression, it also inhibited
CDK2/4 and CDK6 expression. Similar to the present study, Lee et
al (21) demonstrated previously
that (2S)-NRG inhibited the platelet-derived growth factor
(PDGF)-BB-induced proliferation of vascular smooth muscle cells via
a G0/G1 arrest, did not affect the signaling pathways associated
with PDGF-Rβ, protein kinase B, extracellular signal-regulated
kinase 1/2 or phospholipase C-γ1 but downregulated the expression
of cyclin D and E, CDK2, CDK4 and reduced retinoblastomaprotein
phosphorylation. This indicated that (2S)-NRG may be valuable as a
therapeutic agent for managing atherosclerosis and/or vascular
restenosis.
In conclusion, the results of the present study
demonstrate that NRG was able to inhibit TGF-β1-induced
proliferation of CFs via G0/G1 arrest, resulting in the
downregulated expression of cyclin D1-CDK4/6 and cyclin E2-CDK2.
Therefore, the results indicated that NRG may be used as a novel
treatment strategy due to its anti-fibrotic effect in CFs by
regulating proliferation, transformation and collagen synthesis of
CFs in response to pathological stress. However, the detailed
mechanism of how NRG regulates cardiac remodeling remains to be
demonstrated in animal models. Additionally, detailed regulatory
mechanisms of cyclins and CDKs or other signaling pathways, which
are associated with the process of NRG affecting cardiac fibrosis,
require further study.
Acknowledgements
The authors would like to thank all members of the
Department of Cardiology at The First People Hospital of Yueyang
(Yueyang, China) for their expert technical assistance and
advice.
References
|
1
|
Yi X, Li X, Zhou Y, Ren S, Wan W, Feng G
and Jiang X: Hepatocyte growth factor regulates the TGF-β1-induced
proliferation, differentiation and secretory function of cardiac
fibroblasts. Int J Mol Med. 34:381–390. 2014.PubMed/NCBI
|
|
2
|
Zhou Y, Yi X, Wang T and Li M: Effects of
angiotensin II on transient receptor potential melastatin 7 channel
function in cardiac fibroblasts. Exp Ther Med. 9:2008–2012. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Li M, Yi X, Ma L and Zhou Y: Hepatocyte
growth factor and basic fibroblast growth factor regulate atrial
fibrosis in patients with atrial fibrillation and rheumatic heart
disease via the mitogen-activated protein kinase signaling pathway.
Exp Ther Med. 6:1121–1126. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhou YM, Li MJ, Zhou YL, Ma LL and Yi X:
Growth differentiation factor-15 (GDF-15), novel biomarker for
assessing atrial fibrosis in patients with atrial fibrillation and
rheumatic heart disease. Int J Clin Exp Med. 8:21201–21207.
2015.PubMed/NCBI
|
|
5
|
Stempien-Otero A, Kim DH and Davis J:
Molecular networks underlying myofibroblast fate and fibrosis. J
Mol Cell Cardiol. 97:153–161. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Travers JG, Kamal FA, Robbins J, Yutzey KE
and Blaxall BC: Cardiac fibrosis: The fibroblast awakens. Circ Res.
118:1021–1040. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Moore-Morris T, Cattaneo P, Puceat M and
Evans SM: Origins of cardiac fibroblasts. J Mol Cell Cardiol.
91:1–5. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
van Putten S, Shafieyan Y and Hinz B:
Mechanical control of cardiac myofibroblasts. J Mol Cell Cardiol.
93:133–142. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Pinho-Ribeiro FA, Zarpelon AC, Mizokami
SS, Borghi SM, Bordignon J, Silva RL, Cunha TM, Alves-Filho JC,
Cunha FQ, Casagrande R and Verri WA Jr: The citrus flavonone
naringenin reduces lipopolysaccharide-induced inflammatory pain and
leukocyte recruitment by inhibiting NF-κB activation. J Nutr
Biochem. 33:8–14. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Xing BH, Yang FZ and Wu XH: Naringenin
enhances the efficacy of human embryonic stem cell-derived
pancreatic endoderm in treating gestational diabetes mellitus mice.
J Pharmacol Sci. 131:93–100. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang F, Dong W, Zeng W, Zhang L, Zhang C,
Qiu Y, Wang L, Yin X, Zhang C and Liang W: Naringenin prevents
TGF-β1 secretion from breast cancer and suppresses pulmonary
metastasis by inhibiting PKC activation. Breast Cancer Res.
18:382016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhang N, Yang Z, Yuan Y, Li F, Liu Y, Ma
Z, Liao H, Bian Z, Zhang Y, Zhou H, et al: Naringenin attenuates
pressure overload-induced cardiac hypertrophy. Exp Ther Med.
10:2206–2212. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chtourou Y, Slima AB, Makni M, Gdoura R
and Fetoui H: Naringenin protects cardiac
hypercholesterolemia-induced oxidative stress and subsequent
necroptosis in rats. Pharmacol Rep. 67:1090–1097. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Roche P and Czubryt MP: Transcriptional
control of collagen I gene expression. Cardiovasc Hematol Disord
Drug Targets. 14:107–120. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li AH, Liu PP, Villarreal FJ and Garcia
RA: Dynamic changes in myocardial matrix and relevance to disease:
Translational perspectives. Circ Res. 114:916–927. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Langan TJ and Chou RC: Synchronization of
mammalian cell cultures by serum deprivation. Methods Mol Biol.
761:75–83. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lin X, Yang X, Li Q, Ma Y, Cui S, He D,
Lin X, Schwartz RJ and Chang J: Protein tyrosine phosphatase-like A
regulates myoblast proliferation and differentiation through MyoG
and the cell cycling signaling pathway. Mol Cell Biol. 32:297–308.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lundberg AS and Weinberg RA: Functional
inactivation of the retinoblastoma protein requires sequential
modification by at least two distinct cyclin-cdk complexes. Mol
Cell Biol. 18:753–761. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lauper N, Beck AR, Cariou S, Richman L,
Hofmann K, Reith W, Slingerland JM and Amati B: Cyclin E2: A novel
CDK2 partner in the late G1 and S phases of the mammalian cell
cycle. Oncogene. 17:2637–2643. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lee JJ, Yi H, Kim IS, Nhiem NX, Kim YH and
Myung CS: (2S)-naringenin from Typha angustata inhibits vascular
smooth muscle cell proliferation via a G0/G1 arrest. J
Ethnopharmacol. 139:873–878. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bendris N, Lemmers B and Blanchard JM:
Cell cycle, cytoskeleton dynamics and beyond: The many functions of
cyclins and CDK inhibitors. Cell Cycle. 14:1786–1798. 2015.
View Article : Google Scholar : PubMed/NCBI
|