Introduction
Colon cancer is one of the most common malignancies
in the gastrointestinal tract (1).
Every year, >1 million people are diagnosed with colorectal
cancer, accounting for ~10% of all cancer types (2). Treatments used for colon cancer
includes various combinations of surgery, radiation therapy,
chemotherapy and targeted therapy (3). Colon cancer that is confined within the
colon, and may be curable with surgery; however, cancer that has
spread widely typically has a poor prognosis and is usually not
curable (4). Therefore, it is
necessary to identify the molecular mechanism involved in
metastasis, and the identification of novel approaches for the
diagnosis and treatment of colon cancer is urgent.
Aplysia ras homolog I (ARHI) is a member of the ras
superfamily, with 55–62% homology to Ras and Rap (5). Notably, in contrast to Ras, ARHI acts
as a tumor suppressor in various cancer types by inhibiting cell
growth, motility and invasion (6–10). A
study by Wang et al (11)
reported that low expression of ARHI was observed in 61.7% of human
colon cancer specimens, and the ARHI expression level in colon
cancer tissues was markedly lower than that in the paired
non-cancerous tissues. However, the role of ARHI in colon cancer
development has not previously been reported.
The majority of mortalities as a result of colon
cancer are associated with metastasis (4). Epithelial-mesenchymal transition (EMT)
is a well-coordinated process that is necessary for metastasis of
epithelial cancer types (12,13). The
wnt/β-catenin signaling pathway has crucial roles in tumor
metastasis, and it is involved in regulating EMT (14–16).
In the present study, an in vitro
investigation was conducted to explore the functional role of ARHI
in colon cancer, focusing on the aspect of metastasis. Furthermore,
the molecular mechanism underlying the function of ARHI was
investigated.
Materials and methods
Cell culture and transfection
A human colon epithelial cell line (FHC) and four
colon cancer cell lines (LoVo, HCT116, HT-29 and SW620) were
purchased from American Type Culture Collection (Manassas, VA,
USA). Cells were cultured in Dulbecco's modified Eagle medium
(DMEM; Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA,
USA) supplemented with 10% fetal bovine serum (FBS; Invitrogen;
Thermo Fisher Scientific, Inc.) at 37°C in a humidified atmosphere
of 5% CO2. To activate the wnt/β-catenin signaling
pathway in HCT116 cells, the cells were treated with 20 mM lithium
chloride (LiCl; Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) for
24 h. Also, an ARHI-pcDNA3.1 plasmid (100 ng; Shenzhen Zhonghong
Boyuan Biological Technology Co., Ltd., Shenzhen, China) or empty
vector pcDNA3.1 plasmid (100 ng; Shenzhen Zhonghong Boyuan
Biological Technology Co., Ltd.) was transfected into the HCT116
cells using Lipofectamine 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.), according to the manufacturer's instructions.
Transfection with an empty vector was considered the control.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was isolated from FHC, LoVo, HCT116, HT-29
and SW620 cells using TRIzol Reagent (Invitrogen; Thermo Fisher
Scientific, Inc.). Total RNA (2 µg) was used as a template to
generate cDNA using a PrimeScript First Strand cDNA Synthesis kit
(Takara Bio, Inc., Otsu, Japan), according to the manufacturer's
instructions. The primers (Sangon Biotech Co., Ltd., Shanghai,
China) used are demonstrated in Table
I. qPCR was performed using a KiCqStart SYBR Green qPCR
ReadyMix (Sigma-Aldrich; Merck KGaA), according to the
manufacturer's instructions, on a Bio-Rad iQ5 Real-Time PCR system
(Bio-Rad Laboratories, Inc., Hercules, CA, USA). All samples were
analyzed in triplicate. Reactions were performed for 10 min at 94°C
followed by 40 cycles of 20 sec at 94°C and 1 min at 59°C. The
relative mRNA expression levels were calculated using the
2−ΔΔCq method (17) and
normalized to the control, β-actin.
 | Table I.Primers used in reverse
transcription-quantitative polymerase chain reaction. |
Table I.
Primers used in reverse
transcription-quantitative polymerase chain reaction.
|
| Primer sequence
(5′-3′) |
|---|
|
|
|
|---|
| Gene | Sense | Antisense |
|---|
| Aplysia ras homolog
I |
atgcctgttacccacactcc |
acgaaccaagcagcctagaa |
| E-cadherin |
tgcccagaaaatgaaaaagg |
gtgtatgtggcaatgcgttc |
| N-cadherin |
aggggaccttttcctcaaga |
tcaaatgaaaccgggctatc |
| Vimentin |
gagaactttgccgttgaagc |
tccagcagcttcctgtaggt |
| Wnt3a |
ctcccacaccgtcaggtact |
acgggacgagaggcttctat |
| β-catenin |
gaaaatccagcgtggacaat |
cctcaggattgcctttacca |
| Axin2 |
cccaggttgatcctgtgact |
aggtgtgtggaggaaaggtg |
| c-Myc |
ttcgggtagtggaaaaccag |
agcagctcgaatttcttcca |
| Cyclin D1 |
gaggaagaggaggaggagga |
agagatggaagggggaaaga |
| β-actin |
agagctacgagctgcctgac |
agcactgtgttggcgtacag |
Western blot analysis
FHC, LoVo, HCT116, HT-29 and SW620 cells were washed
with phosphate-buffered saline and lysed using cell lysis buffer
(Beyotime Institute of Biotechnology, Haimen, China). Protein
concentrations were determined by the Bradford method, using a
Bradford Protein Assay kit (Beyotime Institute of Biotechnology).
Equal amounts of proteins (20 ug) were separated by 10% SDS-PAGE.
The separated proteins were then transferred to polyvinylidene
fluoride membranes (EMD Millipore, Billerica, MA, USA). The
membranes were blocked with 3% bovine serum albumin (BSA;
Sigma-Aldrich; Merck KGaA) at 4°C overnight, and immunoblotted with
the following primary antibodies at 37°C for 2 h: Rabbit polyclonal
antibody for ARHI (1:400; ab107051; Abcam, Cambridge, MA, USA);
rabbit polyclonal antibody for E-cadherin (1:800; ab15148; Abcam);
rabbit polyclonal antibody for N-cadherin (1:400; ab18203; Abcam);
rabbit polyclonal antibody for vimentin (1:500; ab137321; Abcam);
rabbit polyclonal antibody for wnt3a (1:500; ab28472; Abcam);
rabbit polyclonal antibody for Axin 2 (1:1,000; ab107613; Abcam);
rabbit polyclonal antibody for β-catenin (1:400; sc-7199; Santa
Cruz Biotechnology, Inc., Dallas, TX, USA); rabbit polyclonal
antibody for c-Myc (1:500; sc-788; Santa Cruz Biotechnology, Inc.);
rabbit polyclonal antibody for cyclin D1 (1:400; sc-718; Santa Cruz
Biotechnology, Inc.); and rabbit polyclonal antibody for β-actin
(1:1,000; ab8227; Abcam). Subsequently, the membranes were washed
with Tris-buffered saline-Tween 20 three times and then incubated
with goat anti-rabbit immunoglobulin G conjugated with horseradish
peroxidase (1:2,000; A00098; GenScript Co., Ltd., Nanjing, China)
at 37°C for 1 h. Signals were detected using an enhanced
chemiluminescent western blotting substrate (Pierce; Thermo Fisher
Scientific, Inc.). The band density was quantified with ImageJ 1.37
software (National Institutes of Health, Bethesda, MD, USA). The
protein level was normalized to β-actin.
Cell invasion assay
Matrigel (BD Biosciences, Franklin Lakes, NJ, USA)
was loaded into the upper well of Transwell chambers (8-µm pore
size; Corning, Inc., Corning, NY, USA) and incubated at 37°C for 30
min. HCT116 cells (5×104 cells/ml) were resuspended in
DMEM supplemented with 0.5% BSA, and were loaded into the upper
well of the Transwell chamber. The lower well was filled with 1 ml
DMEM supplemented with 10% FBS. Following incubation at 37°C for 12
h, the non-invaded cells on the top of the membrane were removed
using a cotton swab. The cells invading on the lower face of the
membrane were fixed with 95% ethanol and stained with hematoxylin
(Beyotime Institute of Biotechnology) at room temperature for 10
min. The stained cells were then counted under an inverted
microscope (TS100; Nikon Corp., Tokyo, Japan) at a magnification of
×400.
Cell adhesion assay
Fibronectin solutions (100 µl; Sigma-Aldrich; Merck
KGaA) were loaded into 96-well plates and incubated at 37°C for 2
h. The plates were blocked with 1% BSA (Sigma-Aldrich; Merck KGaA)
at 37°C for 2 h. HCT116 cells (3×105 cells/ml) were
resuspended in DMEM without serum, and were loaded into the 96-well
plates. Following incubation at 37°C for 2 h, the adhesive cells
were fixed with 4% paraformaldehyde for 30 min and stained with
0.5% crystal violet (Beyotime Institute of Biotechnology) for 2 h
at room temperature. A total of 100 µl SDS solution (2%;
Sigma-Aldrich; Merck KGaA) was added into each well to dissolve the
crystals. The absorbance at 570 nm was measured using a microplate
reader (Bio-Rad Laboratories, Inc.).
Statistical analysis
All data were expressed as the mean ± standard
deviation. Statistical analysis was performed using SPSS v. 19.0
statistical software (IBM Corp., Armonk, NY, USA). The difference
between two groups was determined by Student's t-tests, and the
difference among multiple groups was determined by one-way analysis
of variance followed by the least significant difference test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
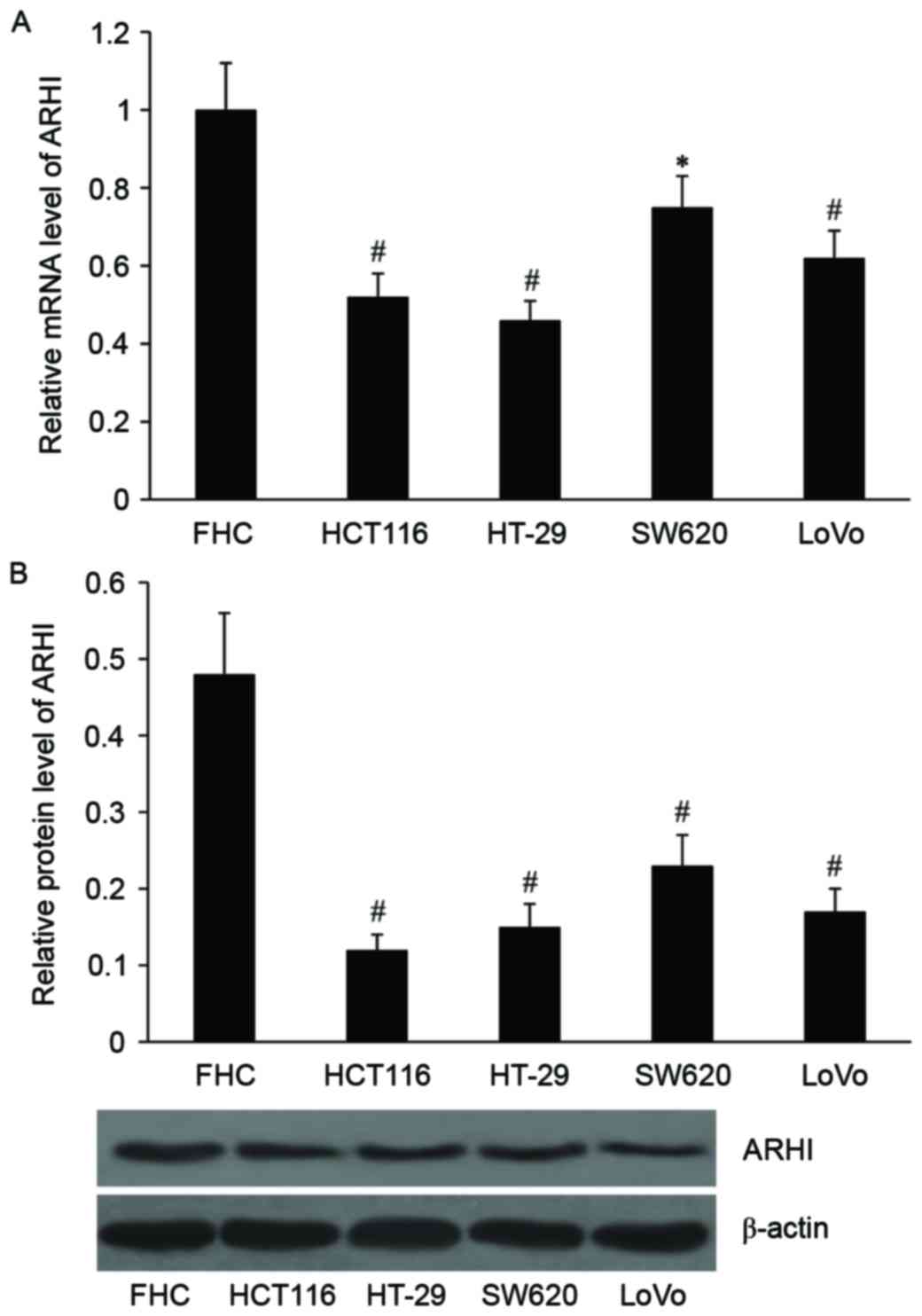
Expression of ARHI in colon cancer
cell lines
The expression levels of ARHI mRNA and protein in a
human colon epithelial cell line (FHC) and four colon cancer cell
lines (LoVo, HCT116, HT-29 and SW620) were detected. As
demonstrated in Fig. 1, the relative
mRNA expression level was significantly decreased in SW620
(P<0.05), HCT116, HT-29 and LoVo cells (all P<0.01) compared
with the normal colon epithelial cell line, FHC. The protein
expression levels of ARHI were also significantly decreased in the
four colon cancer cell lines compared with the normal colon
epithelial cell line (P<0.01).
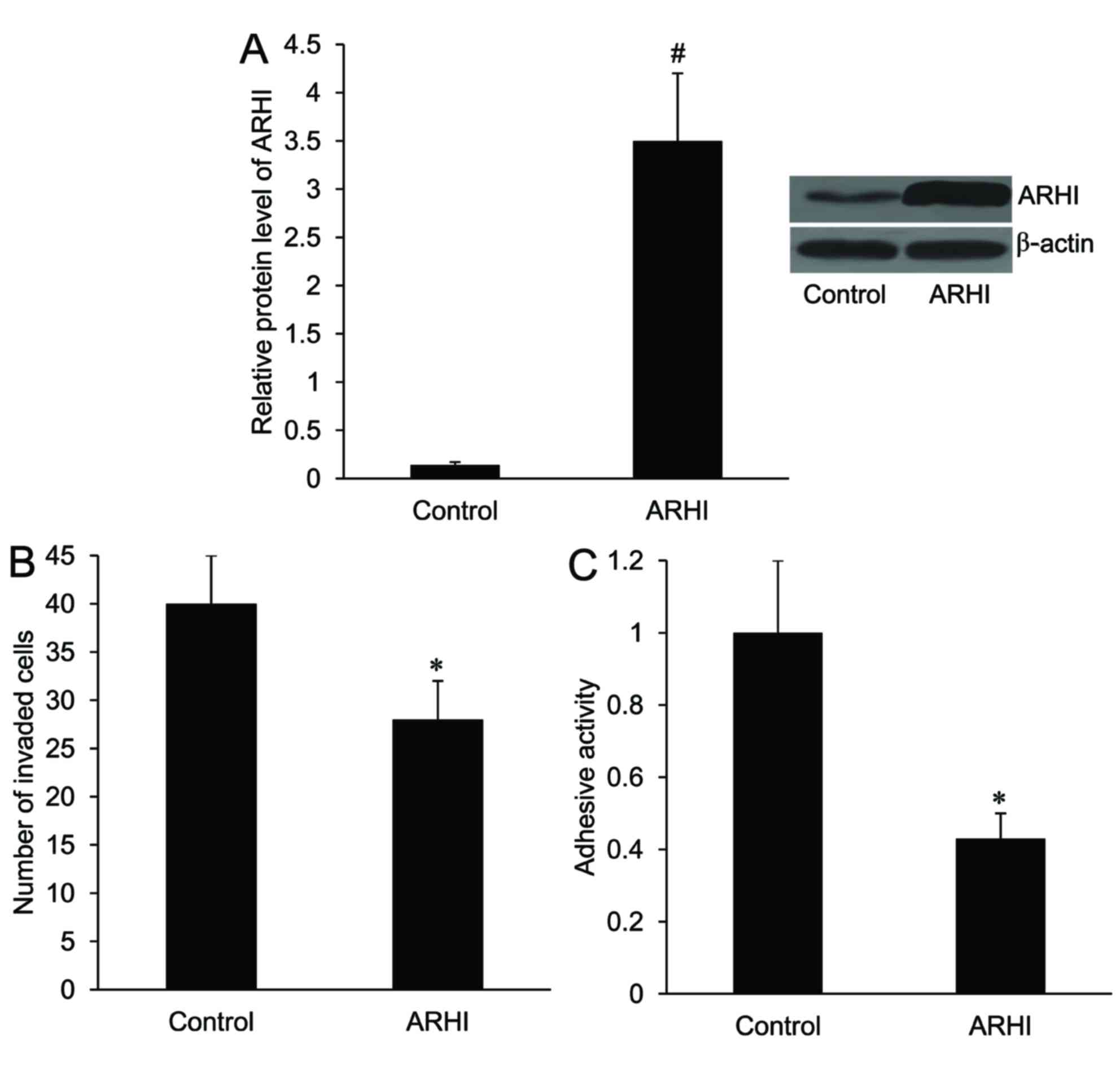
Effect of ARHI on HCT116 cell invasion
and adhesion
An ARHI overexpression plasmid was created and
transfected into HCT116 cells. As demonstrated in Fig. 2A, the relative protein expression
level of ARHI was significantly increased in HCT116 cells following
transfection with the ARHI-pcDNA3.1 plasmid compared with the
control (P<0.01). Furthermore, the number of invaded cells and
the adhesion activity of HCT116 cells were significantly decreased
in the ARHI overexpression group compared with the control group
(P<0.05; Fig. 2B and C).
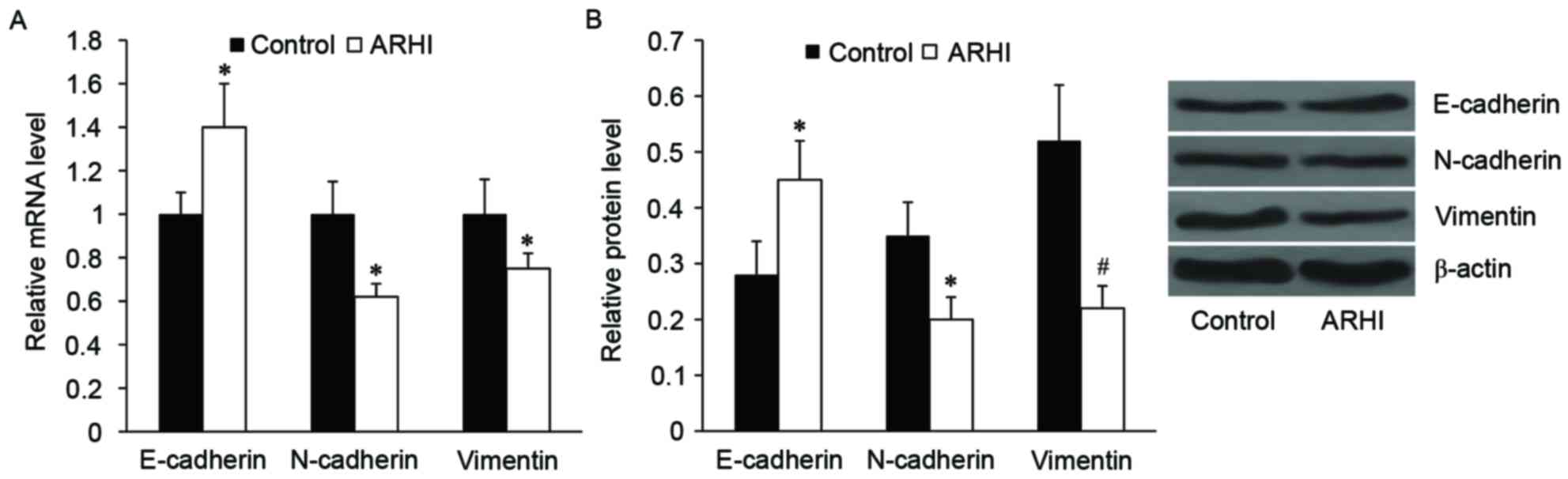
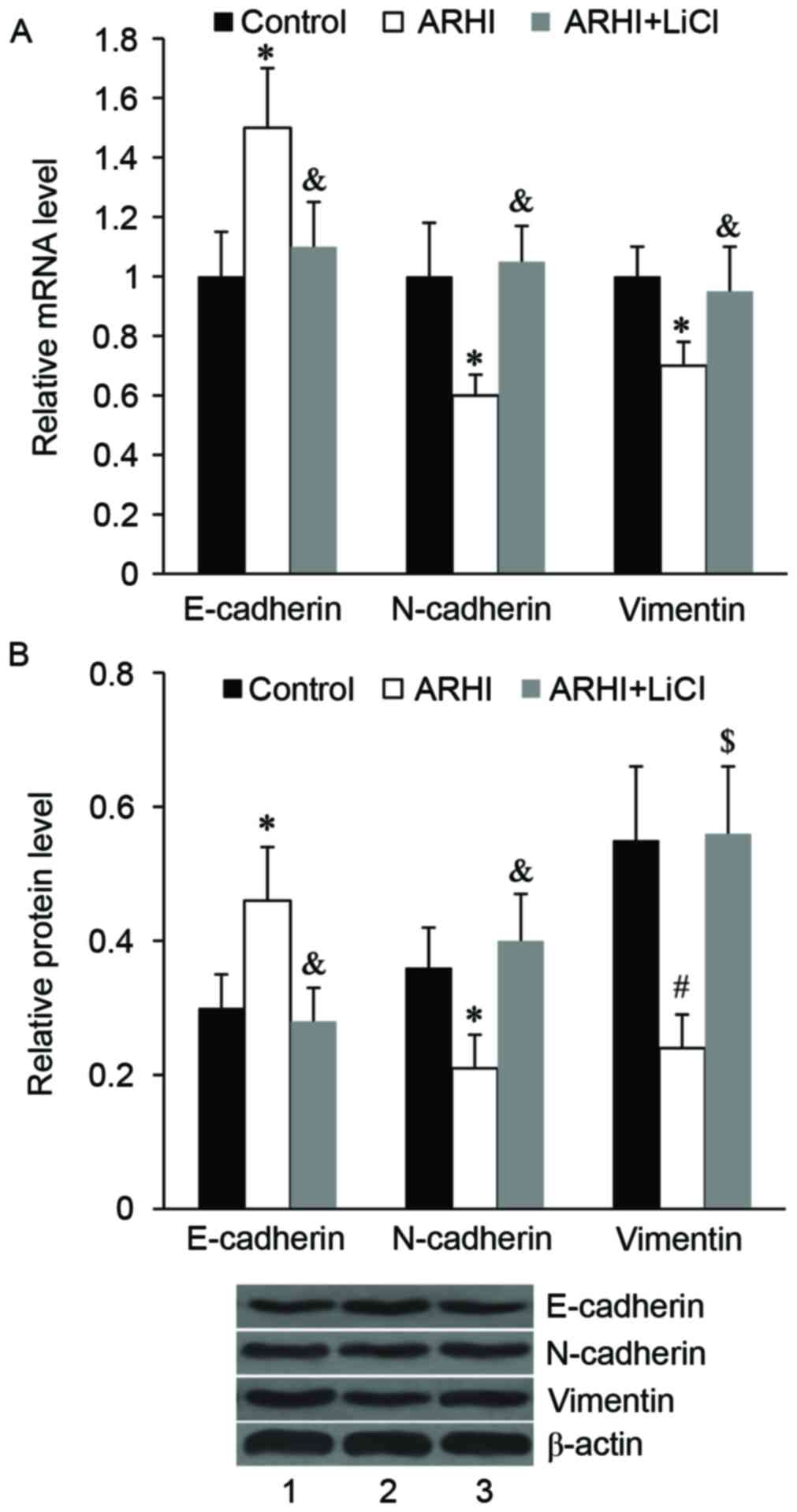
Effect of ARHI on EMT markers in
HCT116 cells
Subsequently, the effect of ARHI on EMT-like
phenotypic changes in HCT116 cells was examined. As determined by
RT-qPCR and western blot analysis, the relative mRNA and protein
expression levels of epithelial marker, E-cadherin, was
significantly increased in the ARHI overexpression group compared
with the control group (P<0.05; Fig.
3). By contrast, the relative mRNA and protein expression
levels of mesenchymal markers, including N-cadherin and vimentin,
were significantly decreased in the ARHI overexpression group
compared with the control group (P<0.05; Fig. 3).
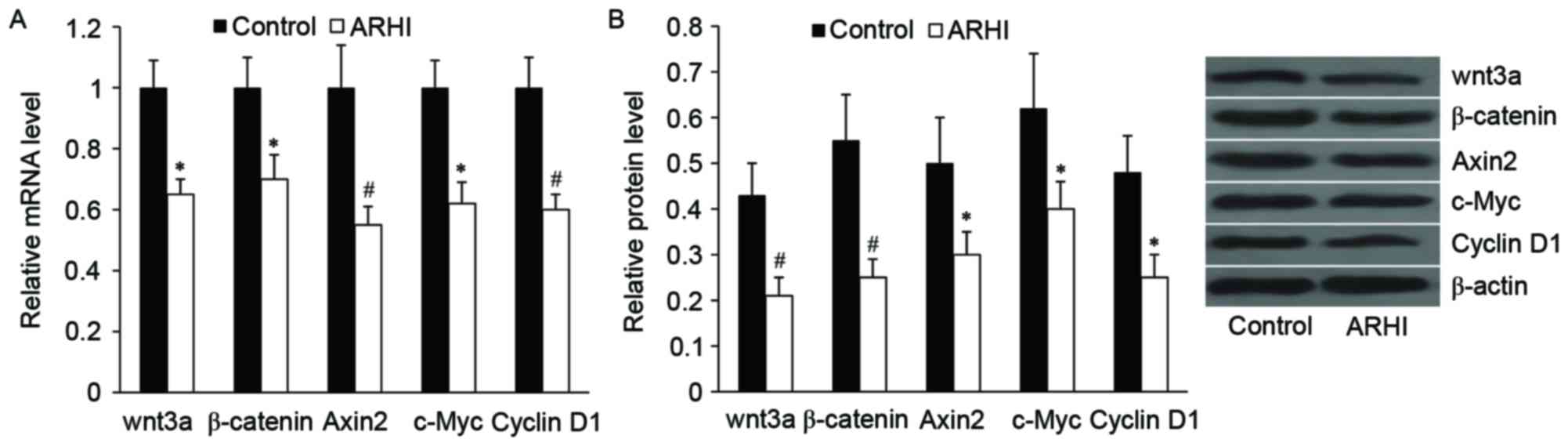
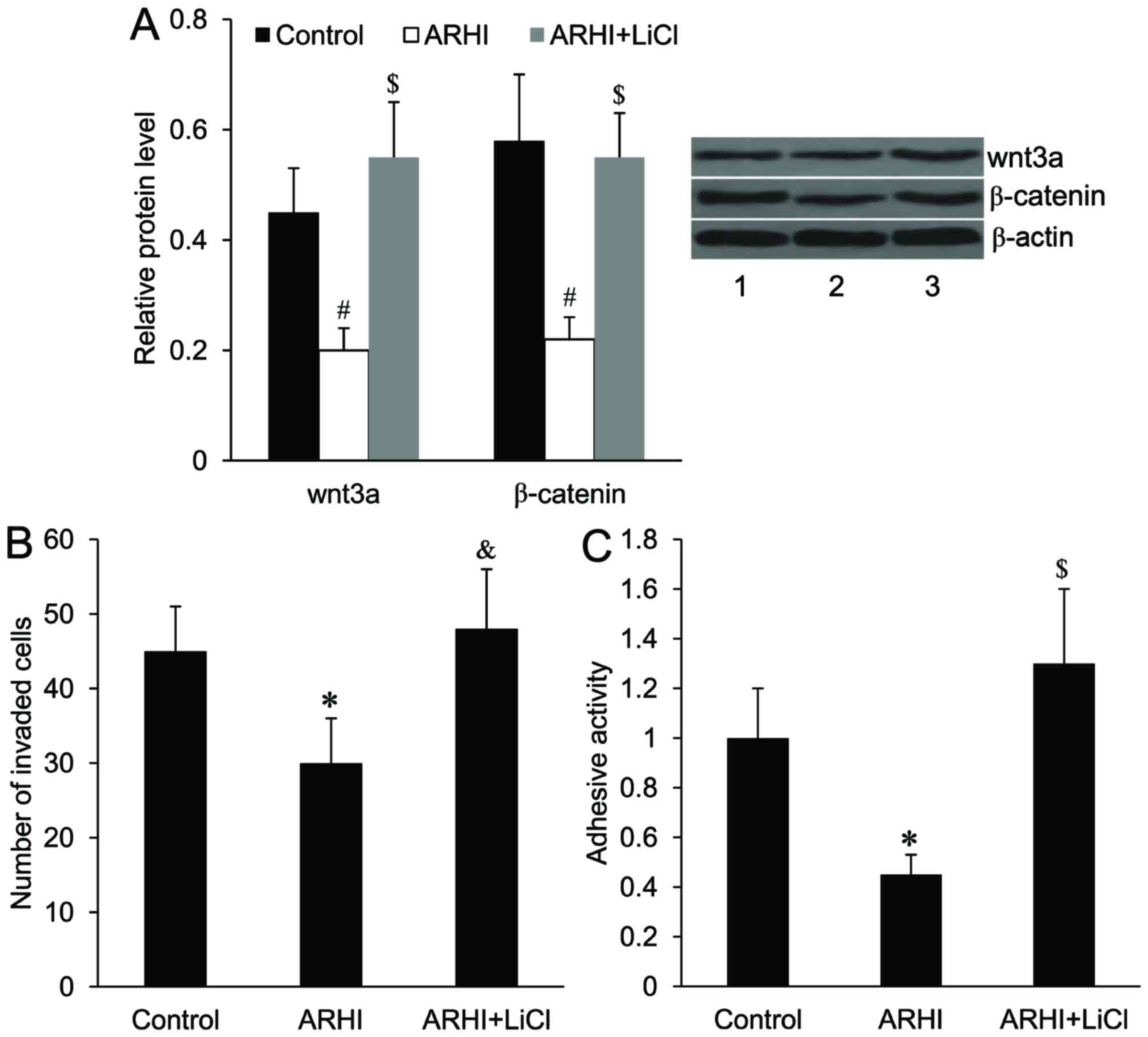
Effect of ARHI on wnt/β-catenin
signaling in HCT116 cells
To investigate the effect of ARHI on wnt/β-catenin
signaling in HCT116 cells, the alterations of wnt3a and β-catenin,
as well as the important downstream targets of wnt/β-catenin
signaling, including Axin 2, c-Myc and cyclin D1, at the mRNA and
protein levels were examined. As demonstrated in Fig. 4, ARHI overexpression significantly
downregulated the relative mRNA and protein expression levels of
wnt3a, β-catenin, Axin 2, c-Myc and cyclin D1 (P<0.05).
Activation of wnt/β-catenin signaling
attenuates the effect of ARHI on HCT116 cell invasion and
adhesion
To activate wnt/β-catenin signaling in HCT116 cells,
the cells were treated with LiCl. As demonstrated in Fig. 5A, the relative protein expression
levels of wnt3a and β-catenin were significantly increased in
HCT116 cells overexpressing ARHI following LiCl treatment compared
with HCT116 cells overexpressing ARHI without LiCl treatment
(P<0.01). It was demonstrated that the suppressed cell invasive
ability and cell adhesion activity induced by ARHI overexpression
were significantly reversed following LiCl treatment (P<0.05 and
P<0.01, respectively; Fig. 5B and
C).
Activation of wnt/β-catenin signaling
attenuates the effect of ARHI on EMT in HCT116 cells
The results of RT-qPCR and western blot analyses
demonstrated that ARHI overexpression significantly increased the
mRNA and protein expression levels of E-cadherin compared with the
control (P<0.05); however, this effect was significantly
attenuated by LiCl treatment (P<0.05; Fig. 6). The suppressive effect of ARHI on
N-cadherin and vimentin mRNA and protein expression was also
significantly reversed by LiCl treatment (P<0.05; Fig. 6).
Discussion
ARHI has been suggested to be a tumor suppressor in
various cancer cells (7,8,10,18). A
study by Li et al (19)
reported that ARHI expression was significantly downregulated in
breast cancer cells in comparison to normal breast tissues. Similar
results were demonstrated in ovarian, renal, gastric and colon
cancer cells (10,11,20,21).
Loss of ARHI expression is associated with the decreased ability to
inhibit cell growth, thus contributing to the development of breast
and ovarian cancer (6).
Re-expression of ARHI may suppress the clonogenic growth of breast
and ovarian cancer cells via downregulation of cyclin D1 promoter
activity and inducing p21(WAF1/CIP1) expression
(18). Furthermore, overexpression
of ARHI is associated with the motility and invasiveness of glioma
and lung cancer cells (7,8). However, the function of ARHI in colon
cancer is unclear. The present study, for the first time, detected
the expression of ARHI in human colon cancer cell lines. Consistent
with the results in colon cancer tissues and other cancer cells, it
was demonstrated that ARHI expression was significantly
downregulated in a series of human colon cancer cell lines compared
with the normal colon epithelial cell line. These results indicated
that ARHI may be involved in the development of colon cancer.
Convincing evidence has been provided that cancer cell invasion and
adhesion are related to cancer progression and therapy efficacy
(22). Subsequently, the present
study utilized gain-of-function experiments on the HCT116 cell
line, which demonstrated the lowest ARHI expression among these
colon cell lines, to investigate the effect of ARHI on colon cancer
cell invasion and adhesion. To the best of our knowledge, the
present study provided the first evidence that ARHI could inhibit
colon cancer cell invasion and adhesion, thus contributing to the
suppression of colon cell metastasis.
EMT is a process in which epithelial cells lose
their cell polarity and cell-cell adhesion, and gain the invasive
and metastatic properties to become mesenchymal stem cells
(23). EMT has been demonstrated to
occur in the initiation of metastasis for cancer progression
(12,13,24–26).
Loss of E-cadherin is considered to be a fundamental event in EMT
(27). N-cadherin and vimentin are
mesenchymal cell markers, and they are closely related to cell
invasion (28,29). In the present study, western blot
analysis for E-cadherin, N-cadherin and vimentin protein expression
was performed to investigate the effect of ARHI on EMT in colon
cancer cells. It was demonstrated that ARHI overexpression led to
the increased expression of E-cadherin and the decreased expression
of N-cadherin and vimentin. These findings indicated that ARHI
could inhibit EMT in colon cancer cells.
The wnt/β-catenin signaling pathway has critical
roles in embryonic development and carcinogenesis (30–34). It
has been demonstrated that the wnt/β-catenin signaling pathway
regulates EMT in cancer (35–37). In
a study by Yu et al (21), it
was indicated that ARHI could induce apoptosis in renal cancer
cells and it exerted its effect via the β-catenin signaling
pathway. In the present study, it was demonstrated that ARHI
overexpression suppressed the wnt/β-catenin signaling pathway by
downregulating the expression of wnt3a, β-catenin, as well as Axin
2, c-Myc and cyclin D1, which are important downstream targets of
wnt/β-catenin signaling. Furthermore, it was observed that the
activation of wnt/β-catenin signaling in colon cancer cells by LiCl
treatment could attenuate the effect of ARHI on EMT, and colon
cancer cell invasion and adhesion. These results indicated that
ARHI inhibits colon cancer cell invasion, adhesion and EMT, at
least partially, via the suppression of the wnt/β-catenin signaling
pathway.
In conclusion, the present study was the first to
determine the effects of ARHI on colon cancer cell invasion and
adhesion. Results demonstrated that ARHI could inhibit colon cancer
cell invasion and adhesion through suppressing EMT, and these
effects were achieved, at least partially, via the suppression of
the wnt/β-catenin signaling pathway. The present study provided the
molecular basis for the role of ARHI in colon cancer, and may help
in developing novel therapeutic approaches for colon cancer.
References
|
1
|
Bartczak-Tomczyk M, Sałagacka A, Mirowski
M, Jeleń A and Balcerczak E: Quantitative analysis of FJ 194940.1
gene expression in colon cancer and its association with
clinicopathological parameters. Contemp Oncol (Pozn). 17:45–50.
2013.PubMed/NCBI
|
|
2
|
Cunningham D, Atkin W, Lenz HJ, Lynch HT,
Minsky B, Nordlinger B and Starling N: Colorectal cancer. Lancet.
375:1030–1047. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gulbake A, Jain A, Jain A, Jain A and Jain
SK: Insight to drug delivery aspects for colorectal cancer. World J
Gastroenterol. 22:582–599. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cunningham D, Atkin W, Lenz HJ, Lynch HT,
Minsky B, Nordlinger B and Starling N: Colorectal cancer. Lancet.
375:1030–1047. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yu Y, Xu F, Peng H, Fang X, Zhao S, Li Y,
Cuevas B, Kuo WL, Gray JW, Siciliano M, et al: NOEY2 (ARHI), an
imprinted putative tumor suppressor gene in ovarian and breast
carcinomas. Proc Natl Acad Sci USA. 96:214–219. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Luo RZ, Fang X, Marquez R, Liu SY, Mills
GB, Liao WS, Yu Y and Bast RC: ARHI is a Ras-related small
G-protein with a novel N-terminal extension that inhibits growth of
ovarian and breast cancers. Oncogene. 22:2897–2909. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chen J, Shi S, Yang W and Chen C:
Over-expression of ARHI decreases tumor growth, migration, and
invasion in human glioma. Med Oncol. 31:8462014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wu X, Liang L, Dong L, Yu Z and Fu X:
Effect of ARHI on lung cancer cell proliferation, apoptosis and
invasion in vitro. Mol Biol Rep. 40:2671–2678. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bao JJ, Le XF, Wang RY, Yuan J, Wang L,
Atkinson EN, LaPushin R, Andreeff M, Fang B, Yu Y and Bast RC Jr:
Reexpression of the tumor suppressor gene ARHI induces apoptosis in
ovarian and breast cancer cells through a caspase-independent
calpain-dependent pathway. Cancer Res. 62:7264–7272.
2002.PubMed/NCBI
|
|
10
|
Tang HL, Hu YQ, Qin XP, Jazag A, Yang H,
Yang YX, Yang XN, Liu JJ, Chen JM, Guleng B and Ren JL: Aplasia ras
homolog member I is downregulated in gastric cancer and silencing
its expression promotes cell growth in vitro. J Gastroenterol
Hepatol. 27:1395–1404. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang W, Chen L, Tang Q, Fan Y, Zhang X and
Zhai J: Loss of ARHI expression in colon cancer and its clinical
significance. Contemp Oncol (Pozn). 18:329–333. 2014.PubMed/NCBI
|
|
12
|
Thiery JP: Epithelial-mesenchymal
transitions in tumour progression. Nat Rev Cancer. 2:442–454. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Brabletz T, Hlubek F, Spaderna S,
Schmalhofer O, Hiendlmeyer E, Jung A and Kirchner T: Invasion and
metastasis in colorectal cancer: Epithelial-mesenchymal transition,
mesenchymal-epithelial transition, stem cells and beta-catenin.
Cells Tissues Organs. 179:56–65. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Nelson WJ and Nusse R: Convergence of Wnt,
beta-catenin, and cadherin pathways. Science. 303:1483–1487. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tang B, Tang F, Wang Z, Qi G, Liang X, Li
B, Yuan S, Liu J, Yu S and He S: Overexpression of CTNND1 in
hepatocellular carcinoma promotes carcinous characters through
activation of Wnt/β-catenin signaling. J Exp Clin Cancer Res.
35:822016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Qi L, Sun B, Liu Z, Cheng R, Li Y and Zhao
X: Wnt3a expression is associated with epithelial-mesenchymal
transition and promotes colon cancer progression. J Exp Clin Cancer
Res. 33:1072014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yu Y, Xu F, Peng H, Fang X, Zhao S, Li Y,
Cuevas B, Kuo WL, Gray JW, Siciliano M, et al: NOEY2 (ARHI), an
imprinted putative tumor suppressor gene in ovarian and breast
carcinomas. Proc Natl Acad Sci USA. 96:214–219. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Li Y, Liu M, Zhang Y, Han C, You J, Yang
J, Cao C and Jiao S: Effects of ARHI on breast cancer cell
biological behavior regulated by microRNA-221. Tumour Biol.
34:3545–3554. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lu Z, Luo RZ, Peng H, Rosen DG, Atkinson
EN, Warneke C, Huang M, Nishmoto A, Liu J, Liao WS, et al:
Transcriptional and posttranscriptional down-regulation of the
imprinted tumor suppressor gene ARHI (DRAS3) in ovarian cancer.
Clin Cancer Res. 12:2404–2413. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yu J, Kong CZ, Zhang Z, Zhan B and Jiang
ZM: Aplasia Ras homolog member I expression induces apoptosis in
renal cancer cells via the β-catenin signaling pathway. Mol Med
Rep. 11:475–481. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Krakhmal NV, Zavyalova MV, Denisov EV,
Vtorushin SV and Perelmuter VM: Cancer invasion: Patterns and
mechanisms. Acta Naturae. 7:17–28. 2015.PubMed/NCBI
|
|
23
|
Kalluri R and Weinberg RA: The basics of
epithelial-mesenchymal transition. J Clin Invest. 119:1420–1428.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Thiery JP, Acloque H, Huang RY and Nieto
MA: Epithelial-mesenchymal transitions in development and disease.
Cell. 139:871–890. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Drasin DJ, Robin TP and Ford HL: Breast
cancer epithelial-to-mesenchymal transition: Examining the
functional consequences of plasticity. Breast Cancer Res.
13:2262011. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bates RC and Mercurio AM: The
epithelial-mesenchymal transition (EMT) and colorectal cancer
progression. Cancer Biol Ther. 4:365–370. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Prieto-García E, Díaz-García CV,
García-Ruiz I and Agulló-Ortuño MT: Epithelial-to-mesenchymal
transition in tumor progression. Med Oncol. 34:1222017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Mendez MG, Kojima S and Goldman RD:
Vimentin induces changes incell shape, motility, and adhesion
during the epithelial to mesenchymal transition. FASEB J.
24:1838–1851. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhang X, Liu G, Kang Y, Dong Z, Qian Q and
Ma X: N-cadherin expression is associated with acquisition of EMT
phenotype and with enhanced invasion in erlotinib-resistant lung
cancer cell lines. PLoS One. 8:e576922013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Li J, Ji L, Chen J, Zhang W and Ye Z:
Wnt/β-Catenin signaling pathway in skin carcinogenesis and therapy.
Biomed Res Int. 2015:9648422015.PubMed/NCBI
|
|
31
|
Sokol SY: Spatial and temporal aspects of
Wnt signaling and planar cell polarity during vertebrate embryonic
development. Semin Cell Dev Biol. 42:78–85. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sebio A, Kahn M and Lenz HJ: The potential
of targeting Wnt/β-catenin in colon cancer. Expert Opin Ther
Targets. 18:611–615. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Holland JD, Klaus A, Garratt AN and
Birchmeier W: Wnt signaling in stem and cancer stem cells. Curr
Opin Cell Biol. 25:254–264. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Tanaka SS, Kojima Y, Yamaguchi YL,
Nishinakamura R and Tam PP: Impact of WNT signaling on tissue
lineage differentiation in the early mouse embryo. Dev Growth
Differ. 53:843–856. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Shan S, Lv Q, Zhao Y, Liu C, Sun Y, Xi K,
Xiao J and Li C: Wnt/β-catenin pathway is required for epithelial
to mesenchymal transition in CXCL12 over expressed breast cancer
cells. Int J Clin Exp Pathol. 8:12357–12367. 2015.PubMed/NCBI
|
|
36
|
Ghahhari NM and Babashah S: Interplay
between microRNAs and WNT/β-catenin signalling pathway regulates
epithelial-mesenchymal transition in cancer. Eur J Cancer.
51:1638–1649. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Li H, Wang Z, Zhang W, Qian K, Liao G, Xu
W and Zhang S: VGLL4 inhibits EMT in part through suppressing
Wnt/β-catenin signaling pathway in gastric cancer. Med Oncol.
32:832015. View Article : Google Scholar : PubMed/NCBI
|