Introduction
Alzheimer's disease (AD) is the most prevalent form
of dementia and is defined as a neurodegenerative disorder that
occurs in the elderly (especially those aged >60 years). AD is
the fourth most common cause of human mortality after
cardiovascular disease, cancer and stroke, and its incidence rate
is increasing due to an aging population worldwide. Thus, the
disease is a considerable social economic burden for families and
society (1,2). Clinically, AD is characterized by a
range of neurological symptoms, including memory deficit, cognitive
impairment and movement disorder (3). Pathologically, the hallmark of AD is an
accumulation of insoluble amyloid-β (Aβ) deposits within amyloid
plaques, mainly consisting of Aβ40 and Aβ42 peptides (4,5).
Deposition of Aβ is possibly related to an imbalance between
production and clearance of Aβ, with defective clearance considered
to be the leading cause (6). In
particular, Aβ-rich oligomers, protofibrils and fibrils, formed by
the self-assembly of Aβ, are potentially responsible for the
neurotoxic effects of Aβ in the pathogenesis of AD (7).
Previous studies have suggested different mechanisms
for the removal of Aβ from the brain (8,9).
Receptor-mediated transport of Aβ across the blood brain barrier
(BBB) to the periphery is considered to be a key mechanism for Aβ
removal, involving low-density lipoprotein receptor-related protein
(LRP), the receptor for advanced glycation end products (RAGE) and
the adenosine triphosphate-binding cassette transporter
p-glycoprotein (p-gp) (10).
Therefore, identifying cellular factors that affect Aβ production
and clearance aid in the development of novel therapeutics for the
treatment of AD. In addition, studies over the past decade have
suggested a key role of inflammation in the pathogenesis of AD
(11).
Results of past studies have suggested that AD is a
‘protein misfolding disorder’, characterized by misfolded and
aggregated proteins in the brain of AD patients (12,13). In
healthy individuals, this is typically prevented by heat shock
proteins (HSPs) acting as molecular chaperones. These integrate
with non-native proteins to facilitate their folding into a native
state, thus preventing misfolding and aggregation, particularly
under cell stress (14,15). However, a potential link between HSPs
and AD is not well understood. A number of past studies have
suggested a potential role of HSPs in AD pathogenesis, particularly
for HSP70 (16–18). In addition, neuroprotective
activities of HSP70 regarding the anti-aggregation and initiation
of Aβ clearance have been reported (19), suggesting that inducers of HSP70 may
be potential therapeutic targets in the treatment of AD.
The present study aimed to determine how raised
levels of HSP70 alleviate AD-related phenotypes in the pathogenesis
of AD. In particular, the effects of the non-toxic HSP-inducer
geranylgeranylacetone (GGA) (20) on
AD phenotypes were investigated via Y-maze, object recognition, and
Morris water maze tests in APP/PS1 mice. The pathological
characteristics related to these phenotypes were also evaluated
through western blot analysis and ELISA.
Materials and methods
Animals
Three-month-old male APP/PS1 double transgenic mice
[B6.Cg-Tg(APPswe, PSEN1dE9)85Dbo/Mmjax] (n=112), each weighing
25–35 g, were obtained from Jackson Laboratory (Bar Harbor, ME,
USA) and 3-month-old littermate male wild-type (WT) mice (n=24),
each weighing 25–35 g, also purchased from Jackson Laboratory, were
used as controls. The Tg mice express a chimeric mouse/human
amyloid precursor protein (APP695swe) and a mutant human presenilin
1 protein (PSEN1dE9) with an exon 9 deletion, both under the
control of murine prion promoter elements (21). Mice were kept under a 12-h light-dark
cycle at 23±1°C and 50–60% humidity with water and food available
ad libitum. For oral administration of GGA, APP/PS1 and WT
mice were administered GGA-supplemented chow from the age of 3 to
12 months (9 months of administration period), according to a
previous method (22). Mice were
divided into the following six groups (n=8 per group): WT, Tg
(APP/PS1 mice, model of AD), Tg+GGA200 (oral administration of GGA
200 mg/kg/day), Tg+GGA400 (oral administration of GGA 400
mg/kg/day), Tg+GGA800 (oral administration of GGA 800 mg/kg/day)
and WT+GGA800 (oral administration of GGA 800 mg/kg/day). To
investigate whether GGA affects MAPK signaling in APP/PS1 mice,
mice were divided into five groups (n=8): WT, Tg, Tg+GGA400,
Tg+GGA+quercetin (intraperitoneal injection of 100 mg/kg quercetin
3 times/week for 6 months, HSP70 inhibitor) and WT+GGA800.
Furthermore, mice (WT and APP/PS1) were also divided into six
groups (n=8): WT, Tg, Tg+GGA400, Tg+GGA+quercetin, Tg+GGA+SB-203580
(p38 inhibitor, intracerebroventricular injection of SB-203580 10
nmol/day for 9 months); Tg+GGA+PD98059 (ERK inhibitor,
intracerebroventricular injection of PD98059 10 nmol for 9 months).
The animal protocol was approved by the Ethics of Animal
Experiments Committee at Jiaotong University School of Medicine
(Shanghai, China).
Morris water maze (MWM) test
An MWM test was performed in order to evaluate
memory function, as previously described (23). Briefly, a circular water tank (100 cm
diameter, 40 cm height) was divided into quadrants and filled with
water (23±1°C) to a depth of 15.5 cm. The water was made opaque by
the addition of white tempera powder to the pool. A transparent
escape platform (10 cm diameter, 20 cm height) was hidden 1 cm
below the water surface and positioned at the midpoint of one
quadrant. Each mouse received daily training for six days using a
single hidden platform in successive quadrants. Latency to escape
from the water maze (finding the submerged platform) was recorded
for each trial. On day seven, mice were subjected to a probe test
in which the platform was removed and each mouse was permitted to
swim freely for 60 sec. Data were recorded using SMART 2.5 video
tracking software (Harvard Apparatus, Holliston, MA, USA).
Y-maze spontaneous alternation
test
A Y-maze spontaneous alternation test was performed
in order to evaluate recognition memory, as described previously
(24). The Y-maze was made of
black-painted wood and each arm of the maze was 50×10×20 cm. Each
mouse was released in the middle of the apparatus and permitted to
move through the maze for 8 min. The series of arm entries were
visually observed. Spontaneous alteration was defined as successive
entries into each of the three arms in overlapping triplet sets.
Alteration behavior (%) was calculated as the ratio of the number
of alternations to the total number of arm entries.
Object recognition test
An object recognition test was carried out as a
second measure of recognition memory, as described previously
(25). Briefly, mice were kept in a
test chamber (25×25×40 cm) overnight under a 12-h light-dark cycle
at controlled temperature (23±1°C) and humidity (50–60%) with water
and food ad libitum, prior to training. During training, two
identical objects (round filter units, 33 mm diameter, 27 mm
height) were placed in the chamber and mice were permitted to
explore for 10 min. The following day, one of the objects was
replaced with a new object (plastic cone, 25 mm diameter, 30 mm
height). The object recognition index was defined as the percentage
of time spent sniffing or touching the novel object with the nose
in a 5 min period. All training and test trials were video recorded
and analyzed using EthoVision XT8.5 (Noldus Information Technology,
Wageningen, Netherlands).
Sandwich ELISA (sELISA) of Aβ
Levels of Aβ40 and Aβ42 peptides in mouse brain
tissue from all groups were determined as described previously
(26). The mice were anaesthetized
by intraperitoneal injection with 1% pentobarbital sodium (50
mg/kg, Sumitomo Dainippon Pharmaceutical Co., Ltd, Osaka, Japan)
and decapitated to obtain brain tissues. Briefly, brain hemispheres
were homogenized in 50 mM Tris-hydrochloride (HCl) buffer (pH 7.6)
supplemented with 150 mM sodium chloride, then centrifuged. A total
of 0.5 M guanidine-HCl was added to the supernatants (soluble
fractions), while precipitates were solubilized by sonication in 6
M guanidine-HCl, then centrifuged at 20,000 × g for 1 h at 4°C to
remove insoluble material. The quantity of Aβ40 and Aβ42 in each
fraction was determined by sELISA, according to the manufacturer's
protocol (cat. no. 27721; Immuno-Biological Laboratories, Inc.,
Minneapolis, MN, USA). The experiments were performed 3 times.
Immunoblotting
Whole proteins were prepared from the brain tissues
of all 12-month-old WT and APP/PS1 mice in 200 µl lysis buffer (30
mM Tris-HCl pH 8.0, 150 mM NaCl, 1% NP-40, 1 mM
phenylmethylsulfonyl fluoride and protease inhibitor cocktail; all
from Sigma-Aldrich; Merck KGaA, Darmstadt, Germany), and total
protein concentration was determined with a Bio-Rad Protein Assay
kit (Bio-Rad Laboratories, Inc., Hercules, CA, USA). Then samples
(50 µg protein) were added to 10% SDS-PAGE gels at a ratio of 1:4,
boiled for 5 min, and transferred for 2 h to PVDF membranes. After
blocking for 1 h with 5% non-fat dry milk at room temperature in
PBS, membranes were incubated overnight at room temperature with
the following primary antibodies: ERK (cat. no. 9102; Cell
Signaling Technology, Inc., Danvers, MA, USA), p-ERK (cat. no.
9101; Cell Signaling Technology, Inc.), p38 (cat. no. sc-7972;
Santa Cruz Biotechnology, Inc., Dallas, TX, USA), p-p38 (cat. no.
sc-7973; Santa Cruz Biotechnology, Inc.), JNK (cat. no. sc-137019;
Santa Cruz Biotechnology, Inc.), p-JNK (cat. no. sc-293136; Santa
Cruz Biotechnology, Inc.), LRP1 (cat. no. sc-16168; Santa Cruz
Biotechnology, Inc.), RAGE (cat. no. sc-8230; Santa Cruz
Biotechnology, Inc.), p-gp (cat. no. sc-13131; Santa Cruz
Biotechnology, Inc.) at a dilution of 1:2,000. Membranes were then
incubated for 45 min with goat anti-rabbit horseradish
peroxidase-coupled secondary antibodies (cat. no. sc-3836; Santa
Cruz Biotechnology, Inc.; 1:3,000) at room temperature. The bound
antibodies were detected using an enhanced chemiluminescence
detection kit (Thermo Fisher Scientific, Inc., Waltham, MA, USA).
For gel loading control, membranes were re-probed with a monoclonal
α-tubulin antibody (cat. no. T5168; 1:1,000; Sigma-Aldrich; Merck
KGaA). The band densities were quantified with Image Pro Plus 4.5
software (Media Cybernetics, Inc., Rockville, MD, USA). The
experiments were performed 3 times.
Statistical analysis
Data are presented as the mean ± standard error of
the mean, and were analyzed using SPSS 16.0 software (SPSS, Inc.,
Chicago, IL, USA). One or two way analysis of variance followed by
a Tukey test was performed to assess differences between >three
groups. A Student's t-test for unpaired results was performed to
evaluate differences between two groups. P<0.05 was considered
to indicate a statistically significant difference.
Results
Orally administered GGA improves
cognitive function in APP/PS1 mice
AD-related phenotypes become apparent in
12-month-old APP/PS1 mice, therefore APP/PS1 and wild-type mice
were orally administered GGA from the age of 3 to 12 months (9
months of administration period), according to a previous method
(22). Y-maze and object recognition
tests were then performed to examine potential dose-dependent
effects of GGA on spatial learning and memory in APP/PS1 mice at
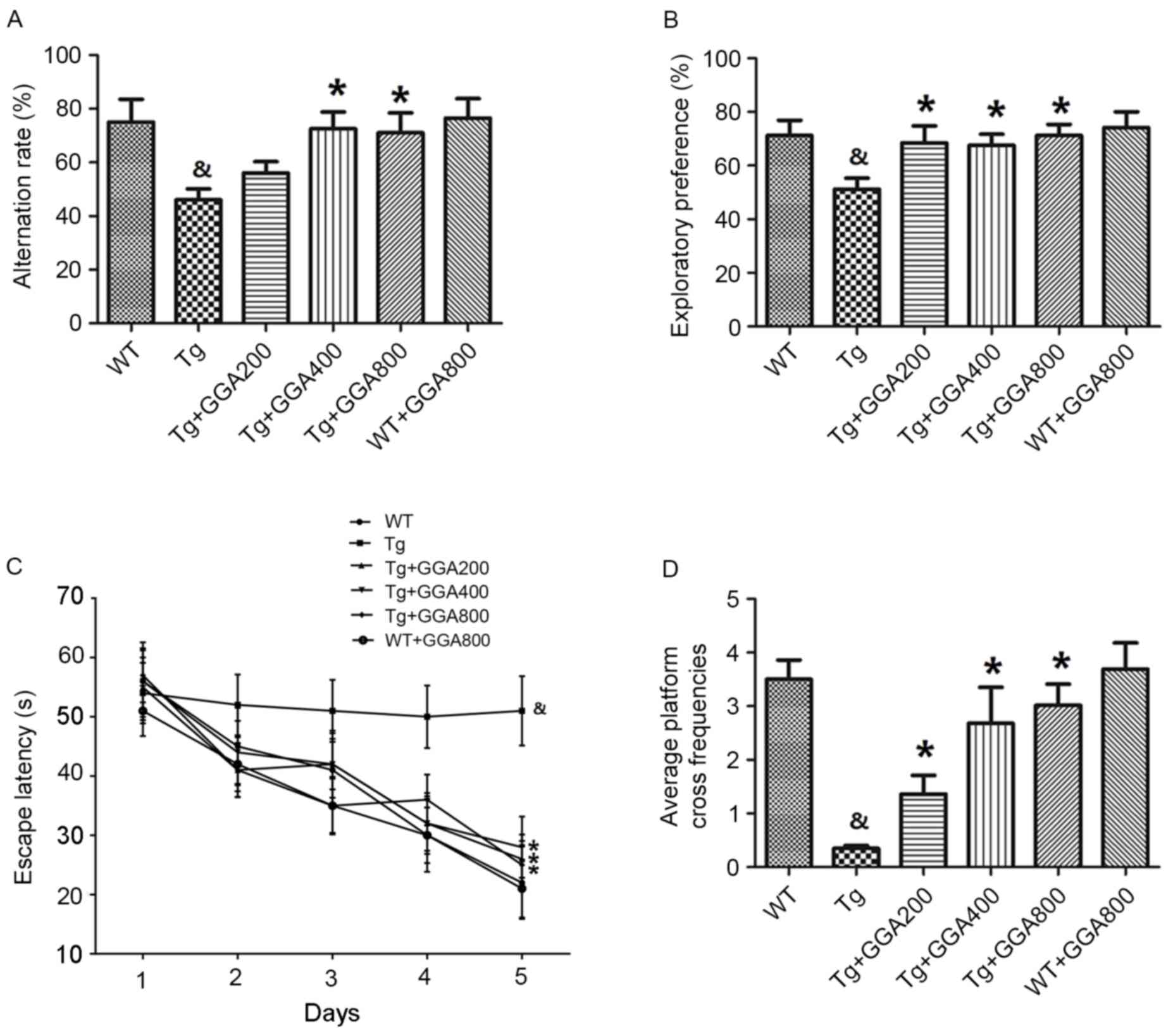
the end of the administration period. Results of the Y-maze
demonstrated that the alternation rates of APP/PS1 mice were
significantly lower than those of WT mice (P<0.05), and that GGA
administration (400 and 800 mg/kg/day) significantly increased the
alternation rates of APP/PS1 mice, relative to untreated APP/PS1
mice (P<0.05; Fig. 1A).
Similarly, in the object recognition test, APP/PS1 mice exhibited a
significantly lower exploratory preference for a novel object,
compared with WT mice, and all doses of orally administered GGA
(200–800 mg/kg/day) significantly increased the exploratory
preference of APP/PS1 mice, relative to untreated APP/PS1 mice
(P<0.05; Fig. 1B).
An MWM test was also performed to evaluate spatial
memory in APP/PS1 mice. Mice were trained daily for six days and
the time taken to reach a hidden platform (escape latency) was
recorded. As presented in Fig. 1C,
APP/PS1 mice exhibited a significantly longer escape latency,
compared with WT mice (P<0.05), and all doses of GGA
significantly reduced the escape latency in APP/PS1 mice, relative
to untreated APP/PS1 mice (P<0.05). In addition, the
significantly lower platform cross frequencies in APP/PS1 mice,
relative to WT mice (P<0.05), were significantly increased by
all doses of GGA, relative to untreated APP/PS1 mice (P<0.05;
Fig. 1D). However, oral
administration of GGA did not affect spatial learning and memory in
WT mice (Fig. 1A-D). These data
suggest that oral administration of GGA (200–800 mg/kg/day) causes
significant alleviation of cognitive deficit in APP/PS1 mice.
Orally administered GGA reduces Aβ
levels in APP/PS1 mice
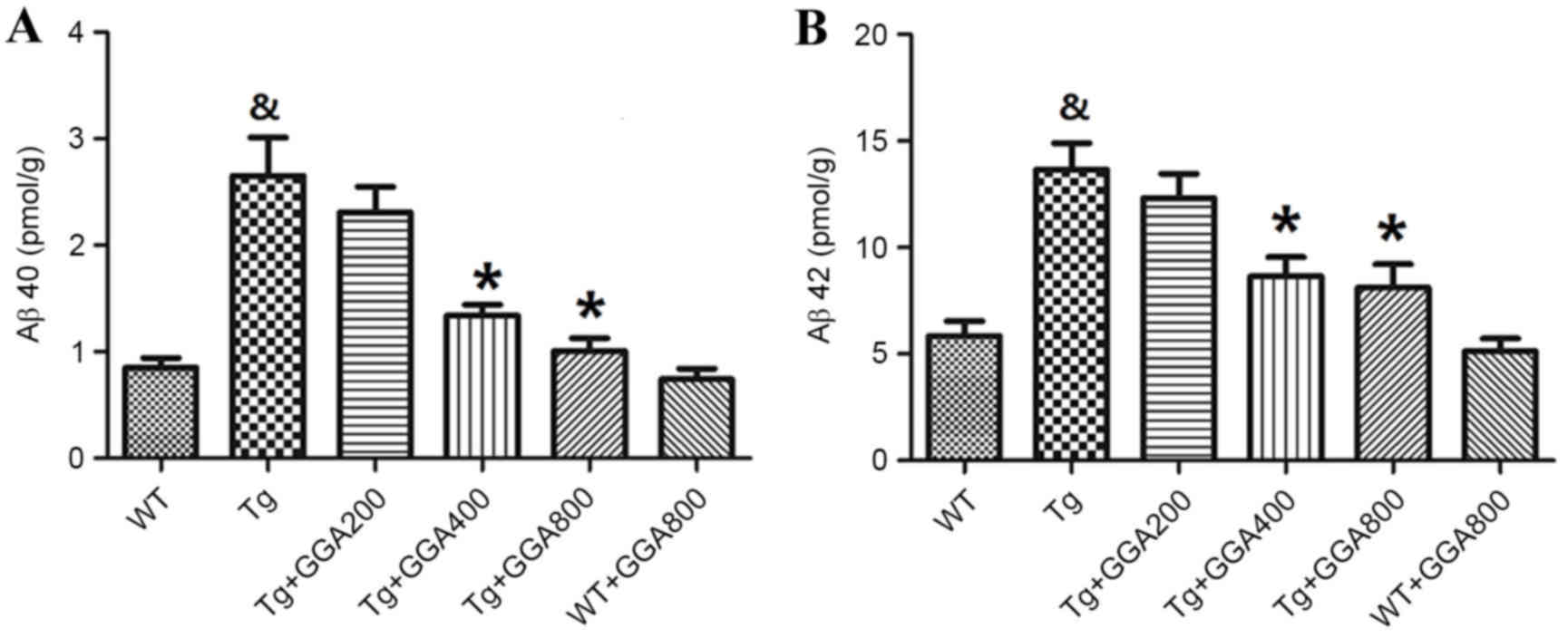
Based on the cytoprotective activity of HSP70
overexpression against Aβ neurotoxicity in mice (19), the present study evaluated the
effects of HSP70 inducer GGA on the levels of Aβ in APP/PS1 mice by
ELISA. As presented in Fig. 2A and
B, the levels of soluble Aβ40 and Aβ42 did not differ
significantly between WT mice and GGA-administered WT mice, while
Aβ40 and Aβ42 levels were significantly higher in APP/PS1 mice,
relative to untreated WT mice (P<0.05). In turn, oral
administration of GGA (400 and 800 mg/kg/day) significantly
reversed the elevated levels of soluble Aβ40 and Aβ42 in the brain
of APP/PS1 mice (P<0.05), suggesting that oral administration of
GGA may lower Aβ levels in APP/PS1 mice.
Orally administered GGA may stimulate
Aβ clearance in APP/PS1 mice
As a removal mechanism of Aβ from the brain is
receptor-mediated transport of Aβ across the BBB into the
periphery, the current study evaluated the effect of orally
administered GGA on the level of proteins involved in
receptor-mediated Aβ elimination in APP/PS1 mice, namely LRP-1,
RAGE and p-gp, by western blot analysis. As depicted in Fig. 3A, the level of LRP-1 was
significantly lower in the brain of APP/PS1 mice relative to WT
mice (P<0.05), and all doses of orally administered GGA
significantly reversed the lower levels of LRP-1 in APP/PS1 mice
(P<0.05). However, the significantly increased levels of RAGE
and lower levels of p-gp in APP/PS1 mice, compared with WT mice
(P<0.05), were not altered by GGA administration (Fig. 3B and C). These results, suggest that
oral administration of GGA may in part stimulate Aβ clearance
through upregulation of LRP-1.
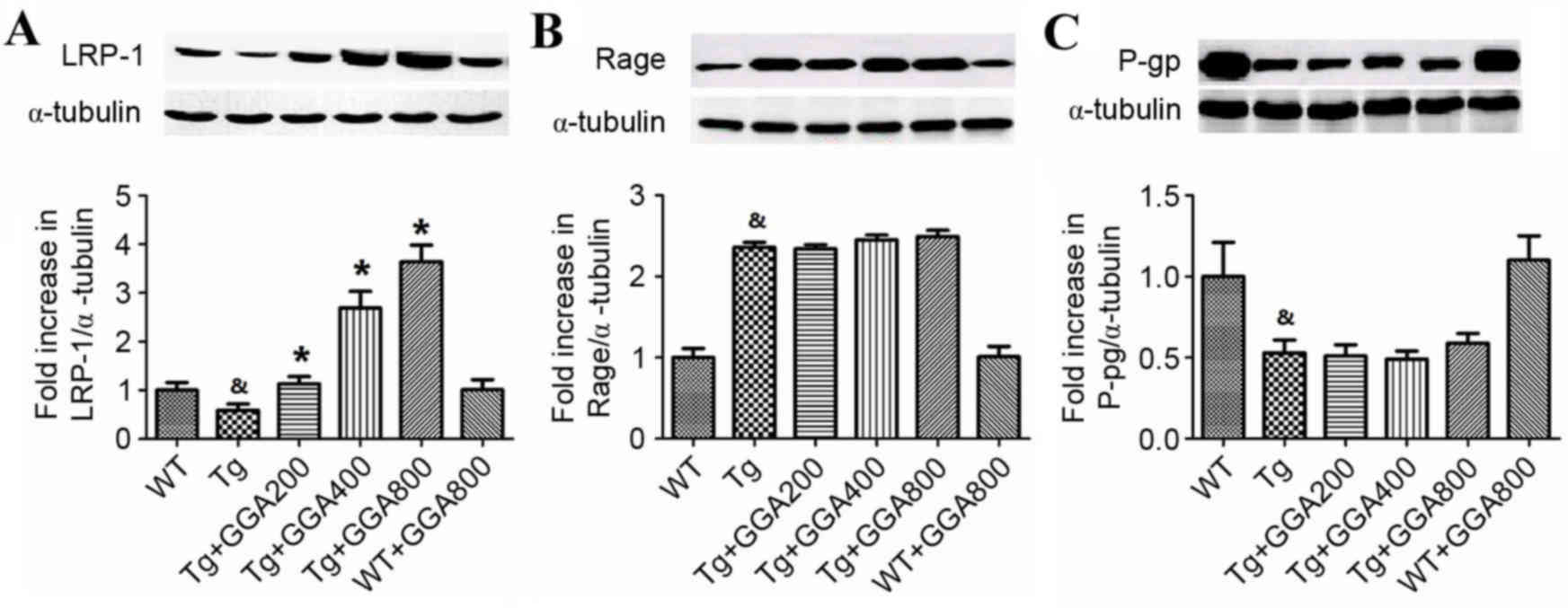 | Figure 3.Effect of orally administered GGA on
the levels of Aβ clearance proteins in APP/PS1 mice. Whole cell
extracts were prepared from the brains of GGA-treated WT and
APP/PS1 mice prior to western blot analysis to measure the levels
of (A) LRP-1, (B) RAGE and (C) p-gp expression. α-Tubulin was used
as an internal standard. &P<0.05 vs. untreated WT
mice, *P<0.05 vs. untreated Tg mice. GGA, geranylgeranylacetone;
Aβ, amyloid-β; APP/PS1, transgenic mouse strain; WT, wild type; Tg,
transgenic; LRP-1, lipoprotein receptor-related protein 1; RAGE,
receptor for advanced glycation end products; p-gp,
p-glycoprotein. |
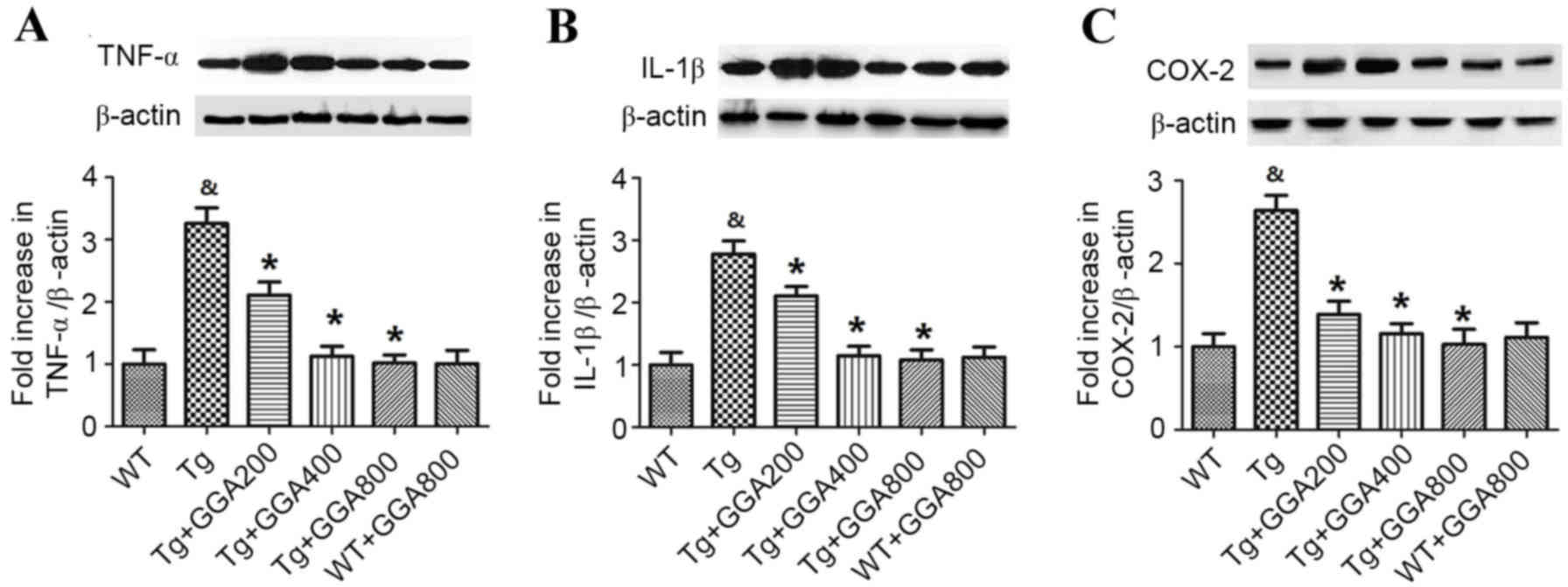
Orally administered GGA on
inflammation in the brain of APP/PS1 mice
Based on the role of inflammation in AD
pathogenesis, the current study evaluated the effect of GGA on the
levels of inflammatory mediators in APP/PS1 mice by western
blotting (Fig. 4). Relative to WT
mice, APP/PS1 mice exhibited significantly higher levels of TNF-α,
IL-1β and COX-2 (P<0.05). In turn, all doses of orally
administered GGA significantly reversed the elevated levels of
TNF-α, IL-1β and COX-2 in the brain of APP/PS1 mice (P<0.05). By
contrast, GGA administration had no effect on the levels of TNF-α,
IL-1β and COX-2 in WT mice.
Underlying mechanism of GGA effects on
AD-related phenotypes
As MAPK is considered to serve a key role in AD
pathophysiology (27), the current
study investigated whether GGA affects MAPK signaling in APP/PS1
mice. As presented in Fig. 5A, it
was observed by western blot analysis that the levels of
phosphorylated (p)c-Jun N-terminal kinase (JNK) did not differ
significantly between all groups. By contrast, the levels of p-p38
mitogen-activated protein kinase (p38 MAPK) and p-extracellular
signal-regulated kinases (ERK) were significantly higher in
Tg+GGA400 mice relative to Tg mice (P<0.05). In turn, p-p38 and
p-ERK levels were significantly decreased in Tg+GGA+quercetin mice,
compared with Tg+GGA400 mice (P<0.05; Fig. 5B and C). These data suggest a
potential role of ERK/p38 MAPK signaling in APP/PS1 mice
administered with GGA.
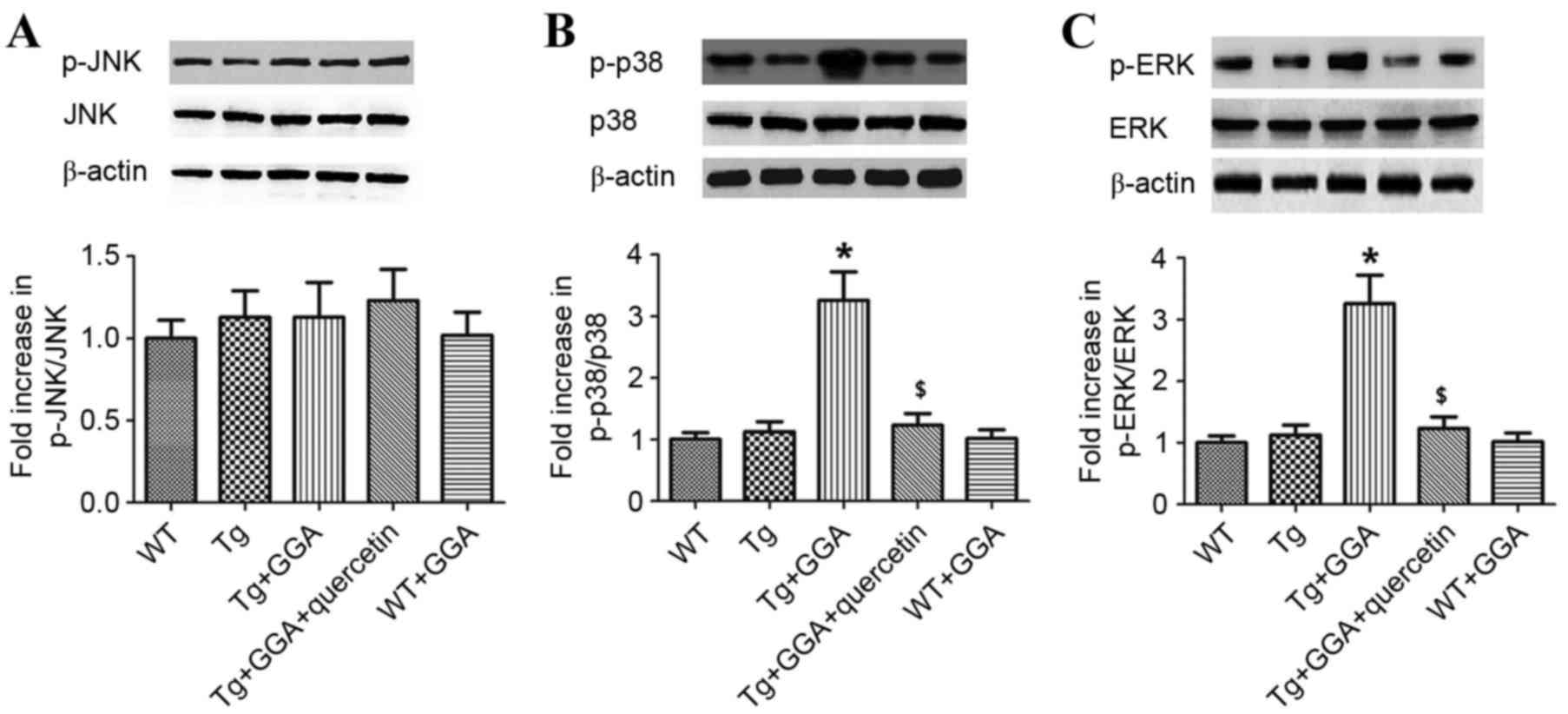 | Figure 5.Effects of orally administered GGA on
ERK/p38 MAPK signaling in APP/PS1 mice. Whole cell extracts were
prepared from the brains of GGA-treated WT and APP/PS1 mice prior
to western blot analysis to measure the levels of (A) p-JNK, (B)
p-p38 and (C) p-ERK. β-actin was used as an internal standard.
*P<0.05 vs. untreated Tg mice, $P<0.05 vs. Tg+GGA
mice. GGA, geranylgeranylacetone; ERK, extracellular
signal-regulated protein kinases; p38 MAPK, p38 mitogen-activated
protein kinase; APP/PS1, transgenic mouse strain; WT, wild type;
Tg, transgenic; JNK, c-Jun N-terminal kinase; p-,
phosphorylated. |
To verify a potential link between ERK/p38 MAPK
signaling and the alleviation of AD-related phenotypes in APP/PS1
mice administered with GGA, the effects of ERK/p38 MAPK inhibition
on levels of soluble Aβ and LRP-1 were evaluated. In accordance
with aforementioned results (Fig.
2), Aβ40 and Aβ42 levels were significantly higher in APP/PS1
mice, relative to WT mice (P<0.05), and oral administration of
GGA significantly reversed the elevated levels of Aβ40 and Aβ42 in
APP/PS1 mice (P<0.05; Fig. 6A and
B). In addition, APP/PS1 mice orally administered with GGA
exhibited significant increases in the levels of Aβ40 and Aβ42
following treatment with quercetin, SB-203580 or PD98059
(P<0.05; Fig. 6A and B).
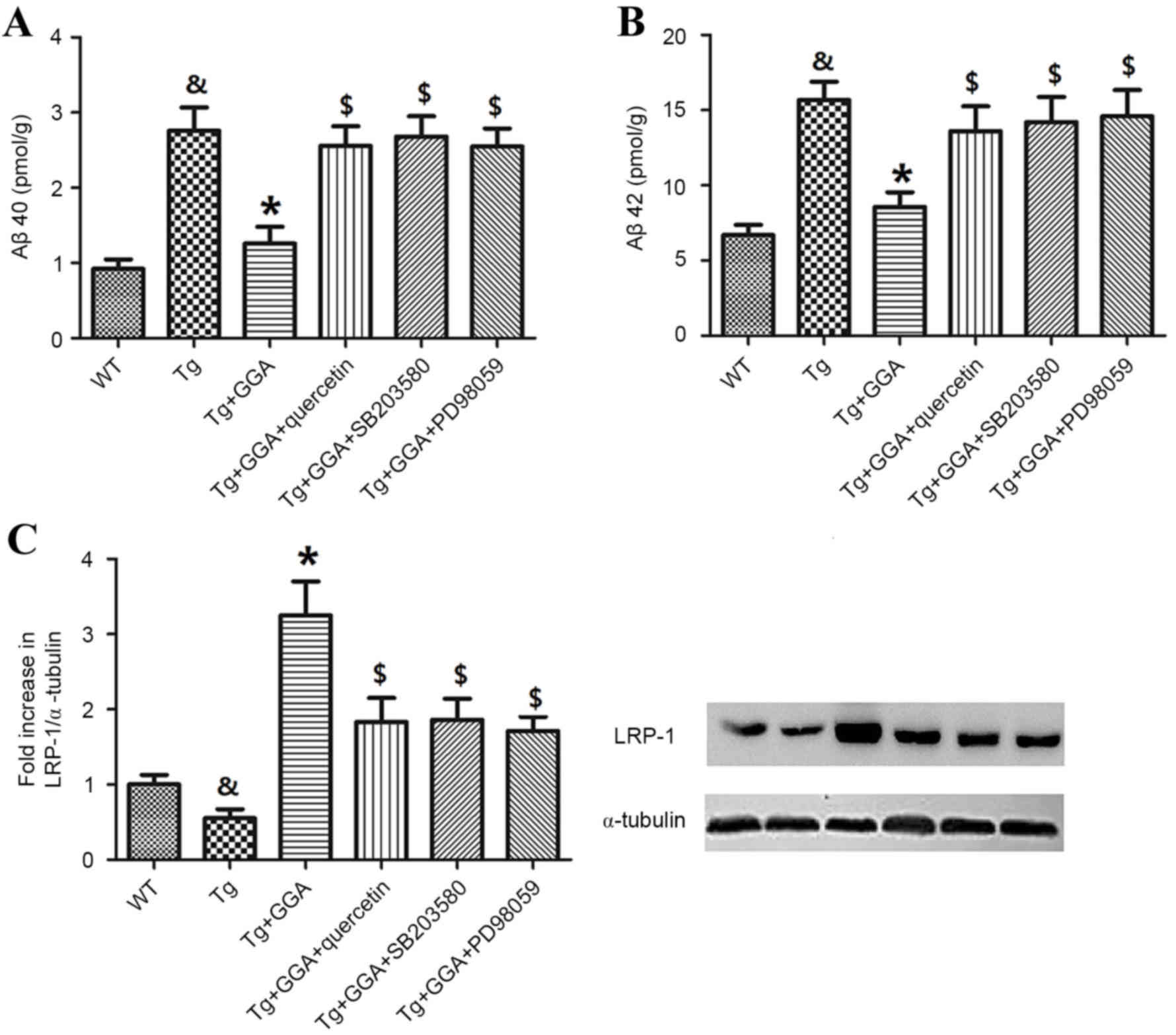 | Figure 6.Effects of ERK/p38 MAPK inhibition on
pathological phenotypes in APP/PS1 mice. Following oral
administration with GGA, WT and APP/PS1 mice were treated with
quercetin, SB-203580 or PD98059 to evaluate the involvement of
ERK/p38 MAPK signaling in the alleviation of pathological
phenotypes in GGA-treated APP/PS1 mice. Levels of soluble (A) Aβ40
and (B) Aβ42 in the brain of WT and APP/PS1 mice were measured by
sandwich ELISA. (C) The level of LRP-1 expression was measured by
western blotting. &P<0.05 vs. untreated WT mice,
*P<0.05 vs. untreated Tg mice, $P<0.05 vs. Tg+GGA
mice. ERK, extracellular signal-regulated protein kinases; p38
MAPK, p38 mitogen-activated protein kinase; APP/PS1, transgenic
mouse strain; GGA, geranylgeranylacetone; SB-203580, p38 inhibitor;
PD98059, ERK inhibitor; Aβ, amyloid-β; LRP-1, lipoprotein
receptor-related protein 1; WT, wild type; Tg, transgenic. |
Furthermore, analogous to previous results (Fig. 3A), the level of LRP-1 was
significantly lower in the brain of APP/PS1 mice relative to WT
mice (P<0.05), and orally administered GGA significantly
increased the lower levels of LRP-1 in APP/PS1 mice (P<0.05). As
depicted in Fig. 6C, it was also
observed that treatment with quercetin, SB-203580 or PD98059
following GGA administration in APP/PS1 mice led to a significant
decrease in the level of LRP-1, relative to Tg+GGA mice
(P<0.05).
Discussion
Overexpression of HSP70 alleviates symptoms in a
number of neurodegenerative diseases, including Alzheimer's
disease, Parkinson's disease, Huntington's disease and spinal
cerebellar ataxias (19,28), and thus HSP70 inducers may be
potential therapeutics in the treatment of neurodegeneration. It
has been demonstrated in a mouse model of AD that oral
administration of GGA alleviates AD-related phenotypes (29), and in the present study, the effects
of GGA on the phenotypes displayed by a transgenic APP/PS1 mouse
model of AD were evaluated. It has also been previously documented
that GGA exerts antiulcer effects on gastric ulcers following its
absorption through the intestinal mucosa (30), and that GGA is able to pass through
the BBB (31), suggesting that HSP70
expression may be influenced by orally administered GGA. In the
present study, it was observed that oral administration of GGA
rescued cognitive impairment and reversed the pathological
phenotypes (decreased levels of Aβ40 and Aβ42) in APP/PS1 mice.
These results are analogous to those observed in APP23 mice
administered with GGA (29),
suggesting that GGA has an alleviative effect on AD-related
phenotypes in the brain of APP/PS1 mice.
In addition, it was demonstrated that orally
administered GGA did not affect the levels of RAGE and p-gp in the
brain of APP/PS1 mice, while levels of LRP-1 were significantly
increased and levels of TNF-α, IL-1β and COX-2 were significantly
decreased. Previous studies have demonstrated associations between
AD and LRP-1, RAGE and p-gp (32,33).
However, the current study observed that levels of RAGE and p-gp in
APP/PS1 mice were not affected by orally administered GGA, possibly
due to a direct effect of HSP70 upregulation on LRP-1 in APP/PS1
mice. Previous studies have also investigated the role of
inflammation in the pathogenesis of AD. Inflammation typically
occurs in regions that are pathologically susceptible in AD brains
and is accompanied by elevated levels of proinflammatory cytokines,
including IL-1β, IL-1 and IL-6 (34–36).
Considering that LRP-1 serves a key role in Aβ elimination from the
brain (37) and that HSPs have
protective effects in inflammation (38), oral administration of GGA may have
inhibited inflammation and increased the levels of LRP-1, thus
stimulating Aβ clearance, through upregulation of HSP70 in APP/PS1
mice.
In addition to AD-related phenotypes, the present
study found that ERK/p38 MAPK signaling was also affected by GGA
treatment, with significant increases in levels of p-p38 and p-ERK
observed in the brain of GGA-treated APP/PS1 mice. In a previous
study by Colombo et al (39),
it was demonstrated in an AD model that JNK regulated the
phosphorylation and degradation of amyloid precursor protein (APP),
thus causing significant decreases in the levels of APP, Aβ
oligomers and Aβ fragments, thus suggesting a link between the JNK
pathway and APP metabolism. By contrast, the present study found
that the levels of p-JNK did not differ significantly in
GGA-treated APP/PS1 mice relative to WT and Tg mice. This may be
due to differences between the current mouse model and the
H4-APPsw cell line employed by Colombo et al
(39).
The ERK/p38 MAPK pathway also regulates
neuroinflammation, neuronal differentiation and synaptic plasticity
(40–42), and p38 MAPK is considered to be a
novel therapeutic target in the treatment of AD (43). Furthermore, a study by Khan and Alkon
(44) identified AD-specific
alterations in the ratio of p-Erk1/Erk2. In the current study, the
role of ERK/p38 MAPK signaling in GGA-treated APP/PS1 mice was
evaluated by subsequent treatment with either SB-203580 (p38
inhibitor) or PD98059 (ERK inhibitor). Significantly increased
levels of soluble Aβ40 and Aβ42 in GGA-treated APP/PS1 mice were
detected following SB-203580 or PD98059 administration. A
significant decrease in the level of LRP-1 was also observed in
GGA-treated APP/PS1 mice following SB-203580 or PD98059
administration. These results indicate that orally administered GGA
in APP/PS1 mice suppresses AD-related phenotypes through regulation
of the ERK/p38 MAPK signaling pathway. In conclusion, results of
the present study suggest a novel molecular mechanism for the
suppression of AD-related phenotypes in APP/PS1 mice orally
administered with GGA. However, the direct association between
LRP-1 and Aβ remains unclear, and requires further study.
References
|
1
|
Katzman R: The prevalence and malignancy
of Alzheimer disease: A Major Killer. Arch Neurol. 33:217–218.
2008. View Article : Google Scholar
|
|
2
|
Brookmeyer R, Johnson E, Ziegler-Graham K
and Arrighi HM: Forecasting the global burden of Alzheimer's
disease. Alzheimers Dement. 3:186–191. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
McKhann G, Drachman D, Folstein M, Katzman
R, Price D and Stadlan EM: Clinical diagnosis of Alzheimer's
disease: Report of the NINCDS-ADRDA Work Group under the auspices
of Department of Health and Human Services Task Force on
Alzheimer's Disease. Neurology. 34:939–944. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hardy J and Selkoe DJ: The amyloid
hypothesis of Alzheimer's disease: Progress and problems on the
road to therapeutics. Science. 297:353–356. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Whitwell JL, Dickson DW, Murray ME,
Weigand SD, Tosakulwong N, Senjem ML, Knopman DS, Boeve BF, Parisi
JE, Petersen RC, et al: Neuroimaging correlates of pathologically
defined subtypes of Alzheimer's disease: A case-control study.
Lancet Neurol. 11:868–877. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kurz A and Perneczky R: Amyloid clearance
as a treatment target against Alzheimer's disease. J Alzheimers
Dis. 24 Suppl 2:S61–S73. 2011.
|
|
7
|
Haass C and Selkoe DJ: Soluble protein
oligomers in neurodegeneration: Lessons from the Alzheimer's
amyloid beta-peptide. Nat Rev Mol Cell Biol. 8:101–112. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tanzi RE, Moir RD and Wagner SL: Clearance
of Alzheimer's Aβ Peptide: The Many Roads to Perdition. Neuron.
43:605–608. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yoon SS and Jo SA: Mechanisms of amyloid-β
peptide clearance: Potential therapeutic targets for Alzheimer's
disease. Biomol Ther (Seoul). 20:245–255. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zlokovic BV: Clearing amyloid through the
blood-brain barrier. J Neurochem. 89:807–811. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tuppo EE and Arias HR: The role of
inflammation in Alzheimer's disease. Int J Biochem Cell Biol.
37:289–305. 2015. View Article : Google Scholar
|
|
12
|
Selkoe DJ: Cell biology of protein
misfolding: The examples of Alzheimer's and Parkinson's diseases.
Nat Cell Biol. 6:1054–1061. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Forloni G, Terreni L, Fogliarino S,
Invernizzi R, Assini A, Ribizzi G, Negro A, Calabrese E, Volonté
MA, Mariani C, et al: Protein misfolding in Alzheimer's and
Parkinson's disease: Genetics and molecular mechanisms. Neurobiol
Aging. 23:957–976. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liberek K, Lewandowska A and Zietkiewicz
S: Chaperones in control of protein disaggregation. EMBO J.
27:328–335. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Meriin AB and Sherman MY: Role of
molecular chaperones in neurodegenerative disorders. Int J
Hyperthermia. 21:403–419. 2009. View Article : Google Scholar
|
|
16
|
Wyttenbach A and Arrigo AP: The role of
heat shock proteins during neurodegeneration in Alzheimer's,
Parkinson's and Huntington's Disease. Heat Shock Proteins in Neural
Cells. 81–99. 2009. View Article : Google Scholar
|
|
17
|
Bobkova NV, Garbuz DG, Nesterova I,
Medvinskaya N, Samokhin A, Alexandrova I, Yashin V, Karpov V,
Kukharsky MS, Ninkina NN, et al: Therapeutic effect of exogenous
hsp70 in mouse models of Alzheimer's disease. J Alzheimers Dis.
38:425–435. 2014.PubMed/NCBI
|
|
18
|
Sinadinos C, Quraishe S, Sealey M, Samson
PB, Mudher A and Wyttenbach A: Low endogenous and chemical induced
heat shock protein induction in a 0N3Rtau-expressing drosophila
larval model of Alzheimer's disease. J Alzheimers Dis.
33:1117–1133. 2013.PubMed/NCBI
|
|
19
|
Hoshino T, Murao N, Namba T, Takehara M,
Adachi H, Katsuno M, Sobue G, Matsushima T, Suzuki T and Mizushima
T: Suppression of Alzheimer's disease-related phenotypes by
expression of heat shock protein 70 in mice. J Neurosci.
31:5225–5234. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mikuriya T, Sugahara K, Takemoto T, Tanaka
K, Takeno K, Shimogori H, Nakai A and Yamashita H:
Geranylgeranylacetone, a heat shock protein inducer, prevents
acoustic injury in the guinea pig. Brain Res. 1065:107–114. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Jankowsky JL, Fadale DJ, Anderson J, Xu
GM, Gonzales V, Jenkins NA, Copeland NG, Lee MK, Younkin LH, Wagner
SL, et al: Mutant presenilins specifically elevate the levels of
the 42 residue beta-amyloid peptide in vivo: Evidence for
augmentation of a 42-specific gamma secretase. Hum Mol Genet.
13:159–170. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Katsuno M, Sang CH, Adachi H, Minamiyama
M, Waza M, Tanaka F, Doyu M and Sobue G: Pharmacological induction
of heat-shock proteins alleviates polyglutamine-mediated motor
neuron disease. Proc Natl Acad Sci USA. 102:pp. 16801–16806. 2005,
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bromley-Brits K, Deng Y and Song W: Morris
water maze test for learning and memory deficits in Alzheimer's
disease model mice. J Vis Exp: pii. e29202011.
|
|
24
|
Hughes RN: The value of spontaneous
alternation behavior (SAB) as a test of retention in
pharmacological investigations of memory. Neurosci Biobehav Rev.
28:497–505. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Broadbent NJ, Gaskin S, Squire LR and
Clark RE: Object recognition memory and the rodent hippocampus.
Learn Mem. 17:5–11. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Iwata N, Mizukami H, Shirotani K, Takaki
Y, Muramatsu S, Lu B, Gerard NP, Gerard C, Ozawa K and Saido TC:
Presynaptic localization of neprilysin contributes to efficient
clearance of amyloid-beta peptide in mouse brain. J Neurosci.
24:991–998. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Munoz L and Ammit AJ: Targeting p38 MAPK
pathway for the treatment of Alzheimer's disease.
Neuropharmacology. 58:561–568. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Adachi H, Katsuno M, Waz M, Minamiyam M,
Tanak F and Sobue G: Heat shock proteins in neurodegenerative
diseases: Pathogenic roles and therapeutic implications. Int J
Hyperthermia. 25:647–654. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hoshino T, Suzuki K, Matsushima T,
Yamakawa N, Suzuki T and Mizushima T: Suppression of Alzheimer's
disease-related phenotypes by geranylgeranylacetone in mice. PLoS
One. 8:e763062013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Seno K, Joh T, Yokoyama Y and Itoh M: Role
of mucus in gastric mucosal injury induced by local
ischemia/reperfusion. J Lab Clin Med. 126:287–293. 1995.PubMed/NCBI
|
|
31
|
Murakami M, Oketani K, Fujisaki H,
Wakabayashi T and Ohgo T: Antiulcer effect of
geranylgeranylacetone, a new acyclic polyisoprenoid on
experimentally induced gastric and duodenal ulcers in rats.
Arzneimittelforschung. 31:799–804. 1981.PubMed/NCBI
|
|
32
|
Donahue JE, Flaherty SL, Johanson CE,
Duncan JA III, Silverberg GD, Miller MC, Tavares R, Yang W, Wu Q,
Sabo E, et al: RAGE, LRP-1, and amyloid-beta protein in Alzheimer's
disease. Acta Neuropathol. 112:405–415. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Weller RO, Subash M, Preston SD, Mazanti I
and Carare RO: SYMPOSIUM: Clearance of Aβ from the brain in
Alzheimer's disease: Perivascular drainage of amyloid-β peptides
from the brain and its failure in cerebral amyloid angiopathy and
Alzheimer's disease. Brain Pathol. 18:253–266. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Finch CE and Morgan TE: Systemic
inflammation, infection, ApoE alleles, and Alzheimer disease: A
position paper. Curr Alzheimer Res. 4:185–189. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Griffin WS and Mrak RE: Interleukin-1 in
the genesis and progression of and risk for development of neuronal
degeneration in Alzheimer's disease. J Leukoc Biol. 72:233–238.
2002.PubMed/NCBI
|
|
36
|
Bona DD, Plaia A, Vasto S, Cavallone L,
Lescai F, Franceschi C, Licastro F, Colonna-Romano G, Lio D,
Candore G and Caruso C: Association between the interleukin-1beta
polymorphisms and Alzheimer's disease: A systematic review and
meta-analysis. Brain Res Rev. 59:155–163. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Deane R, Sagare A and Zlokovic BV: The
role of the cell Surface LRP and Soluble LRP in blood-brain barrier
abeta clearance in Alzheimer's disease. Curr Pharm Des.
14:1601–1605. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Jacquier-Sarlin MR, Fuller K, Dinh-Xuan
AT, Richard MJ and Polla BS: Protective effects of the hsp70 in
inflammation. Experimentia. 50:1031–1038. 1994. View Article : Google Scholar
|
|
39
|
Colombo A, Bastone A, Ploia C, Sclip A,
Salmona M, Forloni G and Borsello T: JNK regulates APP cleavage and
degradation in a model of Alzheimer's disease. Neurobiol Dis.
33:518–525. 2013. View Article : Google Scholar
|
|
40
|
Webber KM, Smith MA, Lee HG, Harris PL,
Moreira P, Perry G and Zhu X: Mitogen- and stress-activated protein
kinase 1: Convergence of the ERK and p38 pathways in Alzheimer's
disease. J Neurosci Res. 79:554–560. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Roux PP and Blenis J: ERK and p38
MAPK-activated protein kinases: A family of protein kinases with
diverse biological functions. Microbiol Mol Biol Rev. 68:320–344.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Sarina, Yagi Y, Nakano O, Hashimoto T,
Kimura K, Asakawa Y, Zhong M, Narimatsu S and Gohda E: Induction of
neurite outgrowth in PC12 cells by artemisinin through activation
of ERK and p38 MAPK signaling pathways. Brain Res. 1490:61–71.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Lenka M and Alaina JA: Targeting p38 MAPK
pathway for the treatment of Alzheimer's disease.
Neuropharmacology. 58:561–568. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Khan TK and Alkon DL: Alzheimer's
disease-specific alterations of the Erk1/Erk2 phosphorylation
ratio. Journal. 2009.
|