Introduction
Lung cancer has been a common cause of
cancer-associated mortality for several decades (1) and ~85% of lung cancers are non-small
cell lung cancer (NSCLC) (1).
Current treatments for NSCLC include surgery, chemotherapy, target
therapy and novel immune checkpoint blockades (1,2).
Epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors
are recommended for patients with NSCLC who have enhanced EGFR
signal transduction (1). However,
resistance to EGFR tyrosine kinase inhibitors occurs in a large
proportion of patients with NSCLC, resulting in disease progression
(3,4).
Tumor necrosis factor (TNF)-α, a 17-kDa protein
produced primarily by macrophages, is currently used in the
regional treatment of locally advanced soft tissue sarcomas and
metastatic melanomas (5). TNF-α
functions via its receptors, TNF receptor (TNFR)-1 and TNFR-2
(5). TNFR-1 is widely expressed on
the cell surface of most cells and is essential for the induction
of cell apoptosis (6,7). TNFR-2 expression is limited to certain
neuronal, immune, hematopoietic and endothelial cells (8). Nuclear factor (NF)-κB has been
demonstrated to be an essential transcription factor activated by
TNF-α to prevent TNF-α and TNFR-1-mediated cell death (9). Inhibiting NF-κB increases the
sensitivity of NSCLC cells to apoptosis-inducing cancer therapies
(10).
Overexpression and mutation of EGFR may lead to the
constitutive activation of the EGF/EGFR signaling pathway and is
associated with increased tumor proliferation and chemotherapy
resistance (11). The EGF/EGFR
signaling pathway also induces NF-κB activation (12–14). The
results of these previous studies suggest that inhibiting the EGF
signaling pathway may enhance TNF-α-induced cell death in lung
cancer. In the present study, the sensitivity of a number of NSCLC
cell lines to TNF-α was investigated, as well as the effect of
nimotuzumab (Ni) on NSCLC cell sensitivity to TNF-α.
Materials and methods
Cell culture
Human NSCLC cell lines, H292 (Sigma-Aldrich; Merck
KGaA, Darmstadt, Germany), H1299, H1975 and H460 (all American Type
Culture Collection, Manassas, VA, USA) were cultured in RMPI-1640
medium supplemented with 10% fetal bovine serum, 100 µg/ml
streptomycin and 100 U/ml penicillin (all Thermo Fisher Scientific,
Inc., Waltham, MA, USA) in a humidified incubator with at 37°C in
an atmosphere containing 5% CO2. Subculture was
performed when the cells were ~90% confluent.
Cell transfection
NF-κB knockdown in H292 and H1975 cells was
performed using a NF-κB p65 short hairpin (sh) RNA lentivirus
vector (Santa Cruz Biotechnology, Inc., Dallas, TX, USA) according
to the manufacturer's protocol. Briefly, NF-κB p65 shRNA lentivirus
vector (500 ng) or the control vector (500 ng) was transfected into
105 293 cells by ViraPower™ Lentiviral Packaging Mix
reagent (Thermo Fisher Scientific, Inc.) to produce the lentivirus.
The harvested lentivirus particles were subsequently used to infect
H292 and H1975 cells. Western blotting confirmed knockdown of NF-κB
expression at 48 h following transfection.
Cell viability
An equal number of NSCLC cells (5×103
cells/well) was seeded in 96-well plates. Treatments, including
TNF-α (20, 40 or 80 ng/ml) and Ni (1 mM as the high dose or 0.5 mM
as the low dose) were administered at 37°C 24 h post seeding.
Single treatment and combination treatment were used. In the
combination treatment, 0.5 nM Ni was used together with 20, 40 or
80 ng/ml TNF-α to study the effects of Ni on TNF-α mediated cell
death. Following the specified treatment time (6, 12, 24, 36 or 48
h), cell viability was measured by using a Cell Counting kit-8
(CCK-8; Sigma-Aldrich; Merck KGaA). Briefly, 10 µl CCK-8 solution
was added to each well and incubated for 30 min at 37°C. Absorbance
was measured at 450 nm using an MRX® II microplate
reader (Dynex Technologies, Chantilly, VA, USA). The final cell
viability was calculated as: [Treatment optical density (OD)
value-blank OD value]/(control OD value-blank OD value). The cell
viability was normalized to the control group at 0 h of
treatment.
Western blotting
Western blotting was used to measure the expression
of NF-κB in NSCLC cell lines. Cells were harvested from cell
culture and lysed using radioimmunoprecipitation buffer with
proteinase inhibitor and phosphatase inhibitor (Thermo Fisher
Scientific, Inc.). A bicinchoninic acid (BCA) assay was used to
quantify proteins. Samples (30 µg total protein per lane) were
separated by 4–20% SDS-PAGE and transferred to polyvinylidene
difluoride membranes. Membranes were subsequently blocked in 5%
BSA-PBS buffer (at room temperature for 1 h) and incubated with
antibodies against anti-NF-κB (1:1,500) with β-actin (1:2,000) as
the internal control. Horseradish peroxidase (HRP)-conjugated
secondary antibody (1:5,000 dilution) and electrochemiluminescence
western blotting detection reagents (Pierce ECL Western Blotting
substrate; Thermo Fisher Scientific, Inc.) were used for signal
development. All antibodies were purchased from Abcam (Cambridge,
MA, USA).
NF-κB activity assay
NF-κB activity was measured on H460, H1299, H292 and
H1975 cells using the NF-κB p65 Transcription Factor Assay kit
(ab133112; Abcam). Nuclear extraction was performed using the
Nuclear Extraction kit (Abcam) according to the manufacturer's
protocol. Briefly, the nuclear extracts containing activated
transcription factor were added to the wells of the assay plate and
incubated at room temperature for 1 h. The transcription factor
NFκB (p65) primary antibodies (from the kit; ab133112) were then
added and incubated at room temperature for 1 h. The HRP conjugated
secondary antibodies (from the kit; ab133112) were subsequently
added and incubated at room temperature for 1 h. The substrate was
then added and the plate was read at OD 450 nm. The result was
presented as the fold change vs. H460 cells.
Measurement of cleaved caspase-3 and
poly ADP ribose polymerase (PARP)
Levels of cleaved caspase-3 and cleaved PARP in the
NSCLC cell lines were measured using ELISA. The human cleaved PARP1
ELISA kit (cat. no. ab174441; Abcam) and cleaved caspase-3 human
ELISA kit (cat. no. KHO1091; Thermo Fisher Scientific, Inc.) were
used according to the manufacturer's protocol. The total protein of
each sample was quantified using a BCA protein assay.
Statistical analysis
All data are presented as the mean ± standard
deviation. Statistical analyses were performed using GraphPad Prism
version 7 software (GraphPad Software, Inc., La Jolla, CA, USA).
Statistical differences between the groups were analyzed by
two-sample t-test (for two groups) or by one-way analysis of
variance (for more than two groups) according to the group number.
Tukey's multiple comparisons test was used to evaluate the
difference of each group with every other group. P<0.05 was
considered to indicate a statistically significant difference.
Two-tailed P-values were used to determine the statistical
significance.
Results
TNF-α resistance varies between NSCLC
cell lines
To explore the effect of TNF-α on NSCLC cells, four
NSCLC cell lines were treated with 20, 40 or 80 ng/ml TNF-α for
6–48 h. No significant differences in cell viability were observed
in H292 or H1975 cells treated with different concentrations of
TNF-α (Fig. 1A and B). However,
treatment with 40 or 80 ng/ml TNF-α significantly decreased the
viability of H460 (Fig. 1C) and
H1299 (Fig. 1D) cells (Fig. 1C and D). Treatment with 80 ng/ml
TNF-α for 24 or 48 h caused a significant increase in the level of
cleaved caspase-3 in H460 and H1299 cells, which suggests that
TNF-α has an apoptosis-inducing effect (Fig. 1E and F). These results indicate the
heterogeneous responses of NSCLC cells to TNF-α.
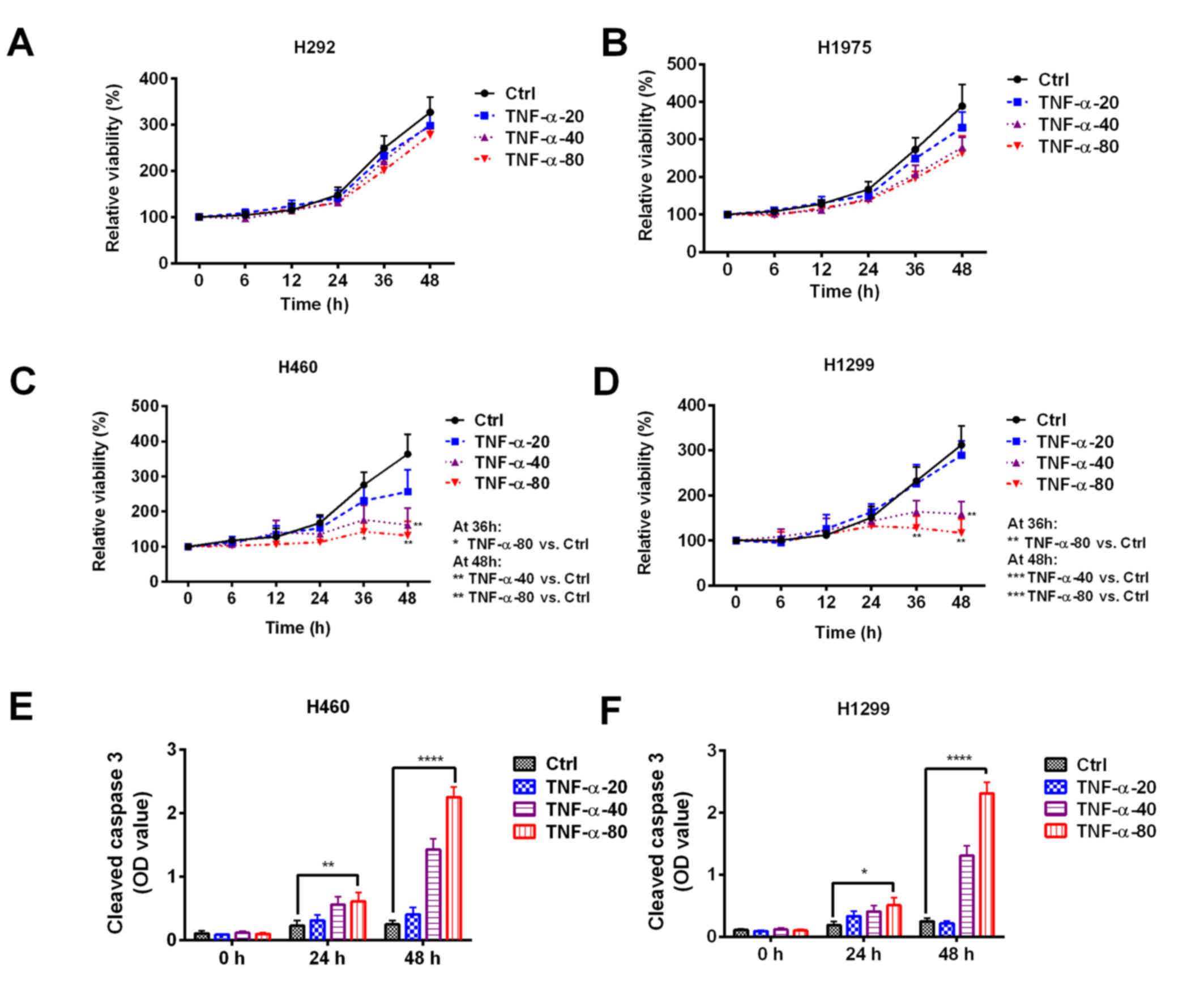 | Figure 1.Sensitivity of non-small cell lung
cancer cell lines to TNF-α treatment. (A) H292, (B) H1975, (C)
H460, and (D) H1299 cell lines were treated with 20, 40, or 80
ng/ml TNF-α and cell viability was assessed. The cell viability was
normalized to the control group at 0 h. An increased level of
cleaved caspase-3 was observed in (E) H460 and (F) H1299 cells
following TNF-α treatment. *P<0.05, **P<0.01, ***P<0.001
and ****P<0.0001 vs. Ctrl. Ctrl, control; NSCLC, non-small cell
lung cancer; TNF, tumor necrosis factor; OD, optical density. |
Ni enhances TNF-α sensitivity in NSCLC
cell lines
As TNF-α resistance was observed in certain NSCLC
cell lines, the possible mechanisms of the resistance were
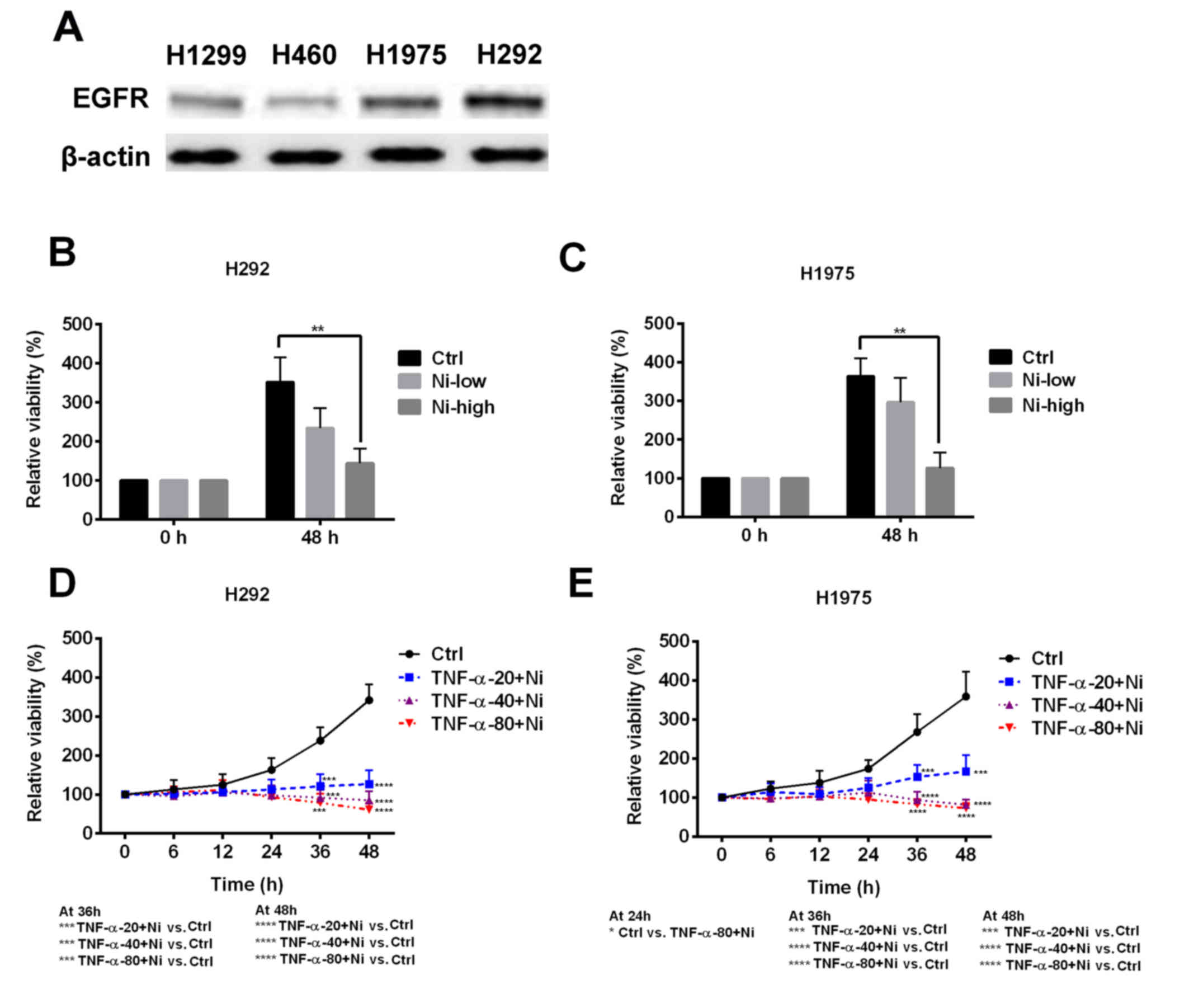
explored. H292 and H1975 cells expressed a notably higher level of
EGFR compared with H1299 and H460 cells (Fig. 2A). In H292 and H1975 cells, low dose
Ni treatment slightly inhibited cell viability and high dose Ni
significantly inhibited cell viability (Fig. 2B and C). Low dose Ni combined with
TNF-α was used to treat the H292 and H1975 cells. Low dose Ni
ameliorated TNF-α resistance in H292 and H1975 cells, resulting in
decreased cell viability at all TNF-α concentrations (Fig. 2D and E). These results suggest that
the EGFR signaling pathway is one of the mechanisms responsible for
TNF-α resistance in H292 and H1975 cells.
Combined treatment with TNF-α and Ni
stimulates apoptosis in NSCLC cell lines
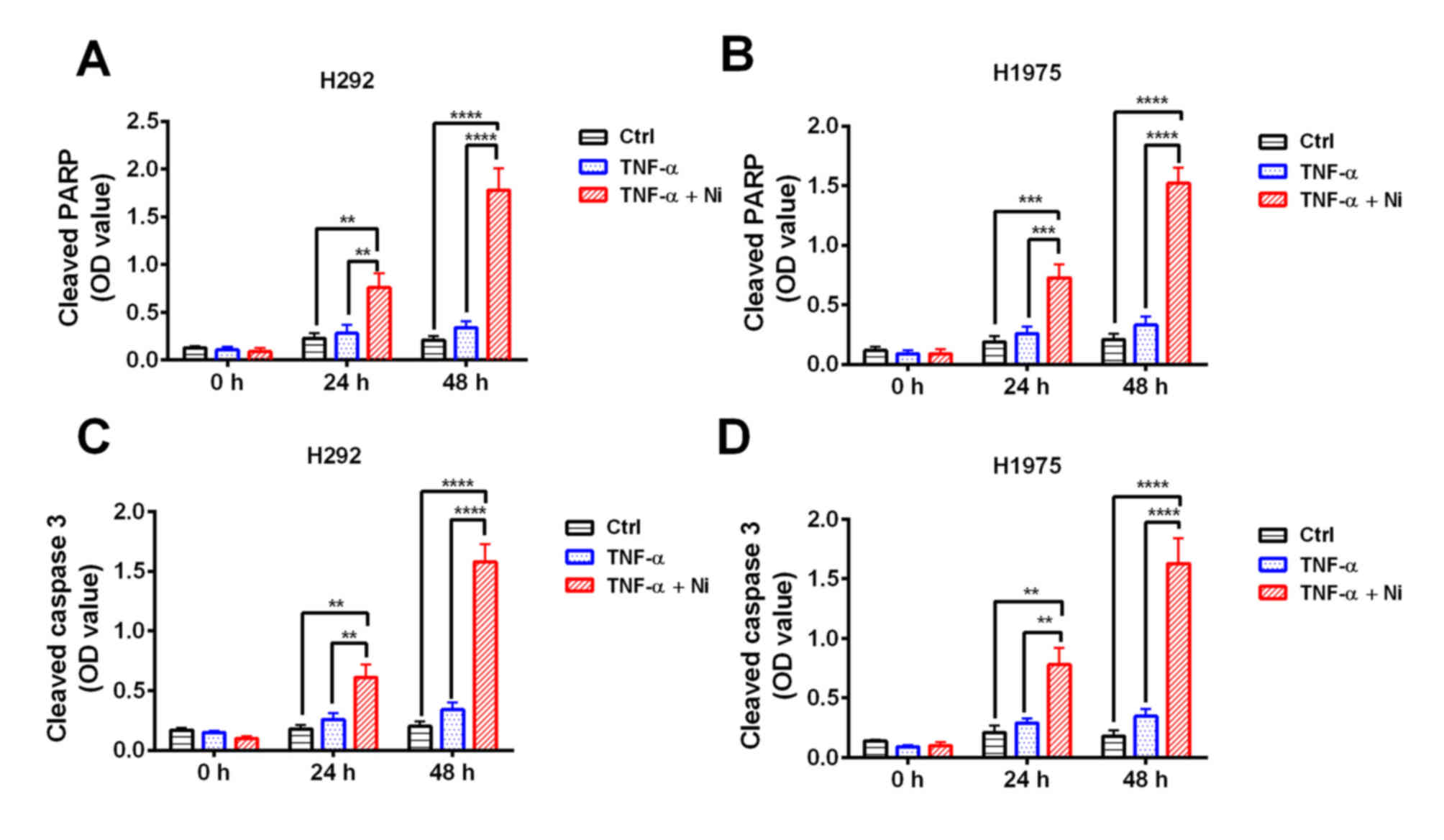
To confirm the effect of TNF-α and low dose Ni
combination treatment cleaved PARP and cleaved caspase-3 were
measured in H292 and H1975 cells. The results revealed that TNF-α
treatment caused no significant increase in cleaved PARP or
caspase-3 (Fig. 3) However, combined
treatment with TNF-α and low dose Ni significantly increased the
levels of cleaved PARP and cleaved caspase-3 in H292 and H1975
cells at 48 and 24 h compared with the TNF-α only and control
groups (Fig. 3). These results
suggest that TNF-α and low dose Ni treatment work synergistically
to induce apoptosis in H292 and H1975 cells.
Ni treatment inhibits NF-κB expression
in NSCLC cell lines
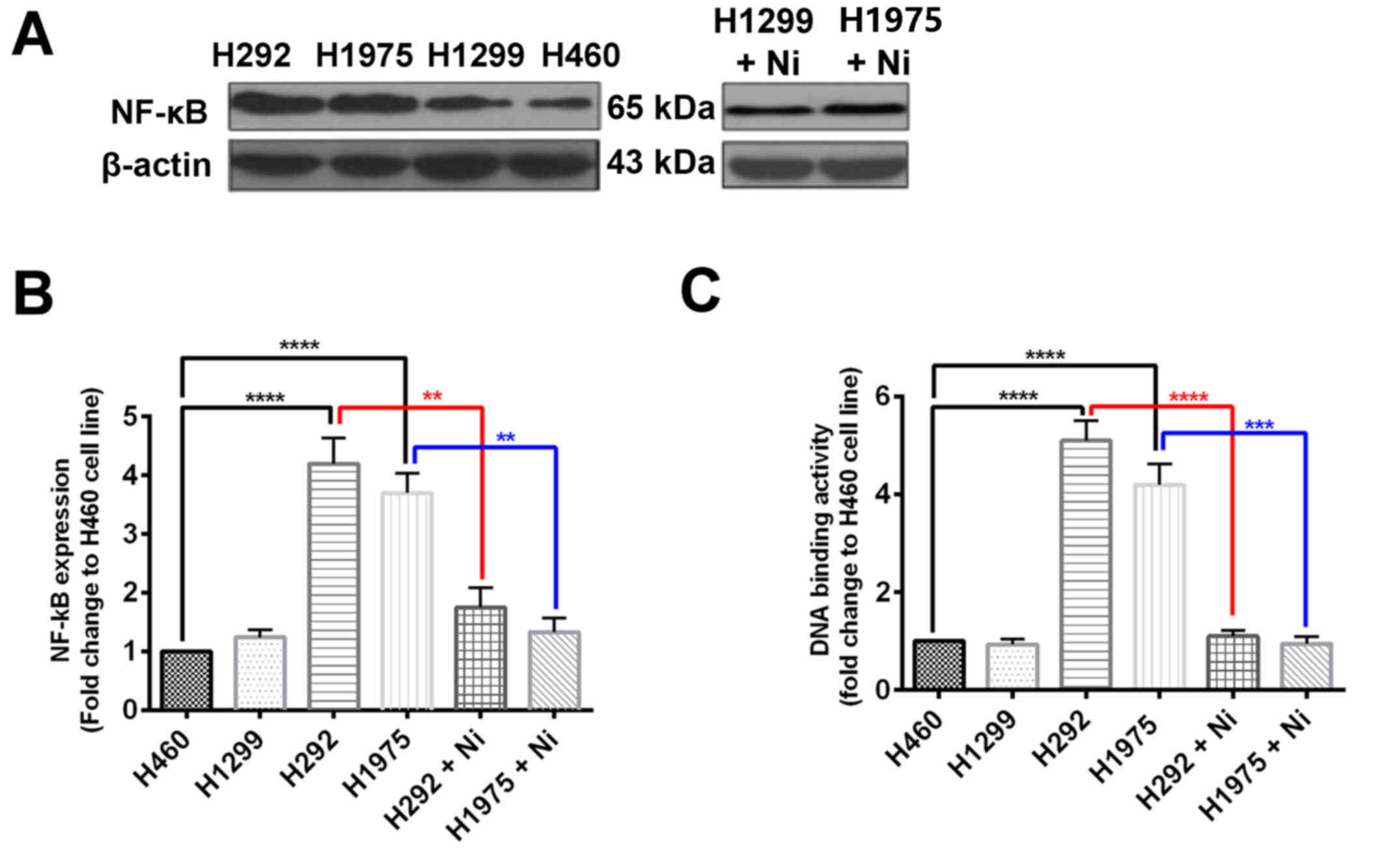
To explore the mechanism by which Ni increases the
efficacy of TNF-α treatment in H292 and H1975 cells, NF-κB protein
expression was measured using western blotting and NF-κB activity
was also determined. It was revealed that H460 and H1299 cells had
comparable NF-κB protein expression, whereas NF-κB protein
expression was significantly increased in H292 and H1975 cells
compared with H460 cells (Fig. 4A and
B). When subjected to Ni treatment, NF-κB protein expression
was signifaicntly reduced in H292 and H1975 cells (Fig. 4A and B). The DNA binding activity of
NF-κB was significantly higher in H292 and H1975 cells compared
with H460 cells (Fig. 4C). The Ni
treatment significantly inhibited the DNA binding activity of NF-κB
in H292 and H1975 cells (Fig. 4C).
These results suggest that the EGFR signaling pathway regulates
NF-κB in NSCLCs, which is a key transcriptional factor in the TNF-α
response.
NF-κB deficiency reduces the NSCLC
cell line resistance to TNF-α
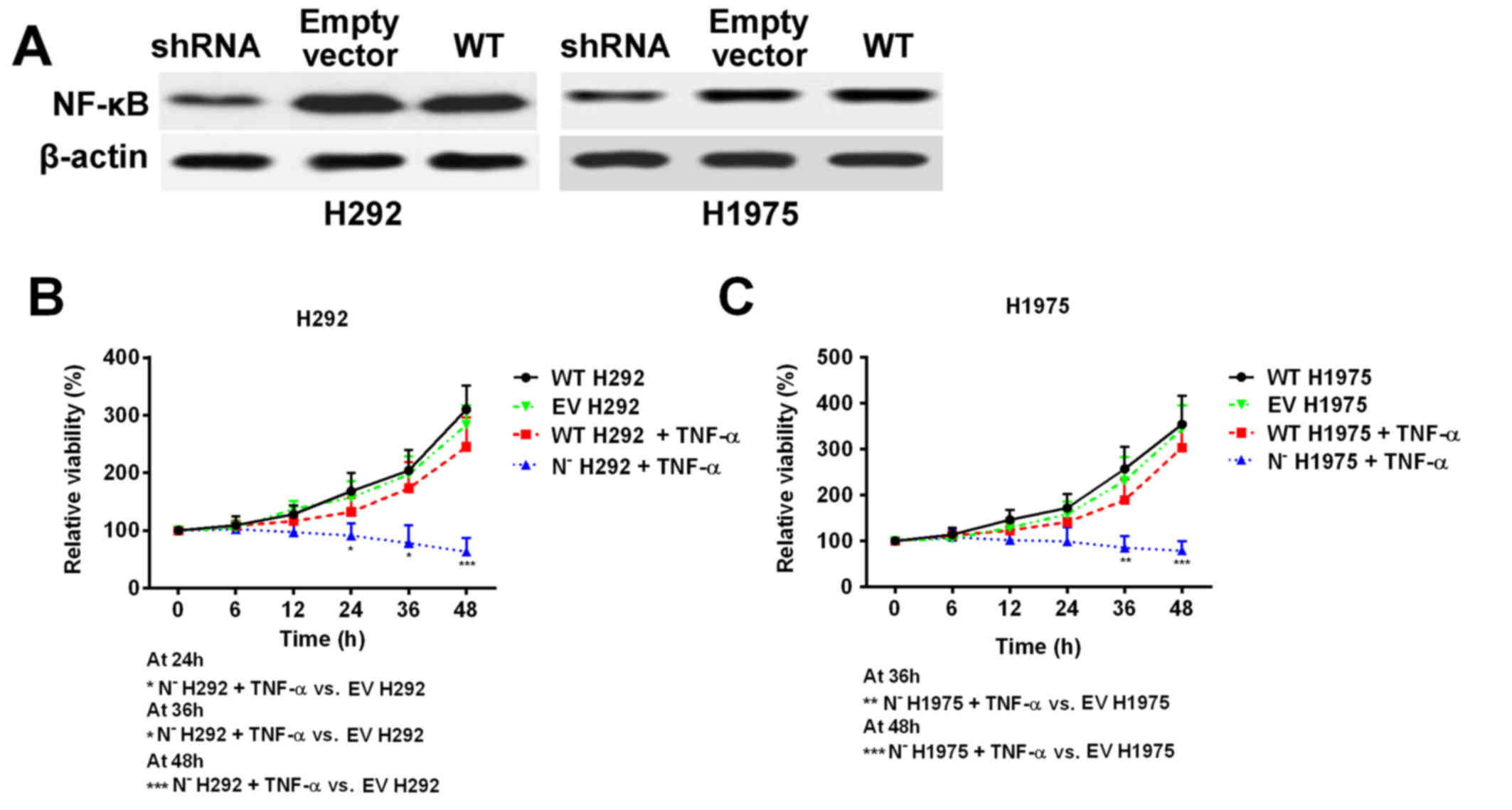
As Ni treatment was demonstrated to suppress NF-κB
protein expression and activity, shRNA was used to knock down the
expression of NF-κB in H292 and H1975 cell lines to mimic Ni
treatment (Fig. 5A). The cell
response to TNF-α treatment was subsequently investigated. Cell
viability was significantly decreased in NF-κB deficient cells
following TNF-α treatment, whereas the wild type cell lines
remained resistant to TNF-α treatment (Fig. 5B and C). These results, together with
the previous data, indicate that overexpression of NF-κB in NSCLC
cells with high EGFR signaling pathway activity is an important
factor that contributes to TNF-α resistance.
Discussion
TNF-α is a multifunctional cytokine that is
associated with a number of essential cellular processes, including
regulating cell survival, apoptosis, inflammation and immune
activity (2). The role of TNF-α in
different types of cancer is varied and context dependent (2,5,7,15). TNF-α
may induce tumor cell death and has been used as a local treatment
for advanced soft tissue sarcoma, melanoma and other unresectable
tumors (5). Conversely,
TNF-α-mediated inflammation promotes tumor development and
inhibition of TNF-α reduces tumor progression (15). Previous studies have suggested that
NF-κB activation may be associated with resistance to TNF-α-induced
apoptosis in cancer cells (16,17). The
EGFR signaling pathway is critical in regulating NF-κB activation
(18,19). In the present study it was
hypothesized that inhibiting the EGFR signaling pathway may
sensitize NSCLC cells to TNF-α treatment by downregulating NF-κB
activation.
In the present study it was demonstrated that NSCLC
H460 and H1299 cell lines were sensitive to TNF-α treatment,
whereas H292 and H1975 cell lines were resistant to TNF-α
treatment, which suggests that TNF-α resistance is cell type
dependent. It was observed that H292 and H1975 cells had increased
levels of EGFR protein compared with H460 and H1299 cells. When
combined with Ni, the TNF-α-induced apoptosis of H292 and H1975
cells was increased. These results suggest that TNF-α resistance in
NSCLC cells is associated with activation of the EGFR signaling
pathway. A previous study demonstrated that TNF-α and EGF had
similar effects on the activation of NF-κB (12). NF-κB is a key transcription factor
that prevents TNF-α mediated cell death (9,20), which
may explain why Ni increased the sensitivity of NSCLC cells to
TNF-α-induced apoptosis.
To confirm that the EGFR signaling-induced
activation of NF-κB is a major factor leading to TNF-α resistance,
the effects of Ni on NF-κB activation were evaluated. NF-κB was
highly expressed in H292 and H1975 cells, which also highly
expressed EGFR. Ni treatment notably downregulated NF-κB expression
in these cells. In the NF-κB knockdown H292 and H1975 cells, TNF-α
had significantly enhanced anti-tumor effects compared with the
NF-κB wild type cells. The results of the present study suggest
that overactivation of NF-κB via EGFR overexpression is an
important factor that mediates TNF-α resistance in NSCLC cells.
These conclusions are in agreement with the results of previous
studies, in which the activation of NF-κB enhanced NSCLC cell
apoptosis (21).
The enhanced expression of growth factors or
overexpression of their receptors results in autonomously
constitutive proliferation in a number of tumor types (22–25).
Approximately 30% of patients with NSCLC exhibit increased EGFR
mRNA expression (23). Targeting the
EGFR signaling pathway using inhibitors, including Ni, has been
demonstrated to significantly improve patient survival rates
(26,27). However, the endogenous tumor
inhibiting mechanisms, including TNF-α and its associated signaling
pathways are also critical in controlling tumor development
(2,5). The present study demonstrated the
potential of using EGFR inhibitors to sensitize NSCLC cells to
TNF-α, which is an endogenous anti-tumor factor found in the human
body.
In conclusion, the results herein demonstrate that
Ni increases the sensitivity of NSCLC cells to TNF-α induced cell
death via inhibiting NF-κB expression and activation. The present
study also revealed a novel mechanism by which Ni suppresses NSCLC
development.
Acknowledgements
The authors would like to thank the Cancer Hospital
of Jilin Province (Changchun, China) for their funding and
support.
References
|
1
|
Reck M, Popat S, Reinmuth N, De Ruysscher
D, Kerr KM and Peters S; ESMO Guidelines Working Group, :
Metastatic non-small-cell lung cancer (NSCLC): ESMO clinical
practice guidelines for diagnosis, treatment and follow-up. Ann
Oncol. 25 Suppl 3:iii27–iii39. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wallach D: Cell death induction by TNF: A
matter of self control. Trends Biochem Sci. 22:107–109. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jackman D, Pao W, Riely GJ, Engelman JA,
Kris MG, Jänne PA, Lynch T, Johnson BE and Miller VA: Clinical
definition of acquired resistance to epidermal growth factor
receptor tyrosine kinase inhibitors in non-small-cell lung cancer.
J Clin Oncol. 28:357–360. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Massarelli E, Varella-Garcia M, Tang X,
Xavier AC, Ozburn NC, Liu DD, Bekele BN, Herbst RS and Wistuba II:
KRAS mutation is an important predictor of resistance to therapy
with epidermal growth factor receptor tyrosine kinase inhibitors in
non-small-cell lung cancer. Clin Cancer Res. 13:2890–2896. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
van Horssen R, Ten Hagen TL and Eggermont
AM: TNF-alpha in cancer treatment: Molecular insights, antitumor
effects, and clinical utility. Oncologist. 11:397–408. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chen G and Goeddel DV: TNF-R1 signaling: A
beautiful pathway. Science. 296:1634–1635. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Arnott CH, Scott KA, Moore RJ, Robinson
SC, Thompson RG and Balkwill FR: Expression of both TNF-αlpha
receptor subtypes is essential for optimal skin tumour development.
Oncogene. 23:1902–1910. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
McCoy MK and Tansey MG: TNF signaling
inhibition in the CNS: Implications for normal brain function and
neurodegenerative disease. J Neuroinflammation. 5:452008.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Beg AA and Baltimore D: An essential role
for NF-kappaB in preventing TNF-alpha-induced cell death. Science.
274:782–784. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Jones DR, Broad RM, Madrid LV, Baldwin AS
Jr and Mayo MW: Inhibition of NF-kappaB sensitizes non-small cell
lung cancer cells to chemotherapy-induced apoptosis. Ann Thorac
Surg. 70:930–937. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mendelsohn J and Baselga J: The EGF
receptor family as targets for cancer therapy. Oncogene.
19:6550–6565. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sethi G, Ahn KS, Chaturvedi MM and
Aggarwal BB: Epidermal growth factor (EGF) activates nuclear
factor-κB through IκBα kinase-independent but EGF receptor-kinase
dependent tyrosine 42 phosphorylation of IκBα. Oncogene.
34:54072015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Biswas DK, Cruz AP, Gansberger E and
Pardee AB: Epidermal growth factor-induced nuclear factor kappa B
activation: A major pathway of cell-cycle progression in
estrogen-receptor negative breast cancer cells. Proc Natl Acad Sci
USA. 97:pp. 8542–8547. 2000; View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sun L and Carpenter G: Epidermal growth
factor activation of NF-kappaB is mediated through IkappaBalpha
degradation and intracellular free calcium. Oncogene. 16:2095–2102.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhao X, Fan W, Xu Z, Chen H, He Y, Yang G,
Yang G, Hu H, Tang S, Wang P, et al: Inhibiting tumor necrosis
factor-alpha diminishes desmoplasia and inflammation to overcome
chemoresistance in pancreatic ductal adenocarcinoma. Oncotarget.
7:81110–81122. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang CY, Mayo MW and Baldwin AS Jr: TNF-
and cancer therapy-induced apoptosis: Potentiation by inhibition of
NF-kappaB. Science. 274:784–787. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kim JY, Lee S, Hwangbo B, Lee CT, Kim YW,
Han SK, Shim YS and Yoo CG: NF-kappaB activation is related to the
resistance of lung cancer cells to TNF-alpha-induced apoptosis.
Biochem Biophys Res Commun. 273:140–146. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tanaka K, Babic I, Nathanson D, Akhavan D,
Guo D, Gini B, Dang J, Zhu S, Yang H, De Jesus J, et al: Oncogenic
EGFR signaling activates an mTORC2-NF-κB pathway that promotes
chemotherapy resistance. Cancer Discov. 1:524–538. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lin Y, Bai L, Chen W and Xu S: The
NF-kappaB activation pathways, emerging molecular targets for
cancer prevention and therapy. Expert Opin Ther Targets. 14:45–55.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Delhalle S, Deregowski V, Benoit V,
Merville MP and Bours V: NF-kappaB-dependent MnSOD expression
protects adenocarcinoma cells from TNF-alpha-induced apoptosis.
Oncogene. 21:3917–3924. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Song L, Xiong H, Li J, Liao W, Wang L, Wu
J and Li M: Sphingosine kinase-1 enhances resistance to apoptosis
through activation of PI3K/Akt/NF-κB pathway in human non-small
cell lung cancer. Clin Cancer Res. 17:1839–1849. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Nicholson RI, Gee JM and Harper ME: EGFR
and cancer prognosis. Eur J Cancer. 37 Suppl 4:S9–S15. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Brabender J, Danenberg KD, Metzger R,
Schneider PM, Park J, Salonga D, Hölscher AH and Danenberg PV:
Epidermal growth factor receptor and HER2-neu mRNA expression in
non-small cell lung cancer is correlated with survival. Clin Cancer
Res. 7:1850–1855. 2001.PubMed/NCBI
|
|
24
|
Repetto L, Gianni W, Aglianò AM and
Gazzaniga P: Impact of EGFR expression on colorectal cancer patient
prognosis and survival: A response. Ann Oncol. 16:15572005.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bhargava R, Gerald WL, Li AR, Pan Q, Lal
P, Ladanyi M and Chen B: EGFR gene amplification in breast cancer:
Correlation with epidermal growth factor receptor mRNA and protein
expression and HER-2 status and absence of EGFR-activating
mutations. Mod Pathol. 18:1027–1033. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Reddy BK, Lokesh V, Vidyasagar MS, Shenoy
K, Babu KG, Shenoy A, Naveen T, Joseph B, Bonanthaya R,
Nanjundappa, et al: Nimotuzumab provides survival benefit to
patients with inoperable advanced squamous cell carcinoma of the
head and neck: A randomized, open-label, phase IIb, 5-year study in
Indian patients. Oral Oncol. 50:498–505. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Cabanas R, Saurez G, Rios M, Alert J,
Reyes A, Valdes J, Gonzalez MC, Pedrayes JL, Avila M, Herrera R, et
al: Treatment of children with high grade glioma with nimotuzumab:
A 5-year institutional experience. MAbs. 5:202–207. 2013.
View Article : Google Scholar : PubMed/NCBI
|