Introduction
Cardiovascular disease (CVD) is a class of diseases
that affect the blood vessels or heart (1). CVD causes 17.3 million death every year
worldwide and the death is expected to increase to 23.6 million by
the year 2030 (2). CVDs, including
diabetic cardiomyopathy, atherosclerosis, myocardial infarction
(MI) and heart failure, are the leading cause of death in diabetes
(3). CVD is commonly diagnosed among
Type 2 diabetics; however, the death risk of CVD in Type 1
diabetics is higher (4). Although
the death rate of CVD has been decreased by 40% in U.S. diabetic
adults (5), the burdens of patients
remain high and effective therapeutic strategies are still
lacking.
The endoplasmic reticulum (ER), the largest
membrane-bound organelle in eukaryotic cells, plays important roles
in protein synthesis, posttranslational modification and
processing, and folding, assembly and trafficking of secretory
protein and membrane (6,7). Excessive unfolded proteins lead to
occurrence of ER stress and then activate a protective signaling
pathway termed the unfolded protein response (UPR). The activation
of UPR causes reduction of protein synthesis, promotion of correct
protein folding, and degradation of misfolded proteins (8,9). It has
been reported that ER stress triggers apoptosis of β cells and
results in the onset of diabetes (10). Additionally, ER stress and myocardial
apoptosis are associated with acute MI (AMI) development (11). Activation of CCAAT/enhancer binding
protein (C/EBP)-homologous protein (CHOP) (a marker of ER stress)
plays an essential role in ER stress-mediated apoptosis (12). Takada et al (13) have found that ER stress is a leading
cause of left ventricular diastolic dysfunction in Type 2 diabetes.
In addition, Barr et al (14)
have found that ER stress is suppressed by hydrogen sulfide during
treatment of cardiovascular complications in diabetes. However, the
roles of ER stress and CHOP-induced myocardial injury in the
progression of MI in diabetes have not yet been fully explored.
In the present study, whether ER stress contributed
to the development of MI in diabetes in vivo was examined.
The effects of CHOP knockdown on cell viability, cell cycle
progression, apoptosis, ER stress and ER redox homeostasis were
then investigated in rat cardiomyocytes in an in vitro model
of diabetes/MI.
Materials and methods
Induction of diabetes and MI
All procedures were performed in accordance with the
Guide for the Care and Use of Laboratory Animals and approved by
the Institutional Animal Care and Use Committee of China Medical
University (Shenyang, China). The 8-week-old Sprague-Dawley rats
(Vital River Laboratories, Beijing, China) were fed with food and
water ad libitum (12-h light-dark cycle) and randomly
divided into 5 groups: i) Control group, ii) Diabetes group, iii)
Diabetes/Sham group, iv) MI group and v) Diabetes/MI group (n=12
rats in each group). At 12 h prior to animal experiments, the rats
were allowed access only to water. Diabetes was induced by 65 mg/kg
streptozotocin (STZ; in 0.01 M sodium citrate buffer, pH 4.4)
(Beijing Solarbio Science & Technology Co., Ltd., Beijing,
China) via intraperitoneal injection. One week after induction of
diabetes, blood glucose levels were measured and rats with glucose
levels greater than 396 mg/dl were included in this study. The
control rats received an injection of 0.01 M sodium citrate buffer.
MI was established by ligation of left anterior descending (LAD)
coronary artery. Briefly, after measurement of blood glucose
levels, anesthetization was performed and the rats were placed in
the supine position. The skin was cut off using surgical scissors
and the rats were ventilated with HX-300S animal respirator
(Chengdu Techman Software Co., Ltd., Chengdu, China; tidal volume,
6–8 ml/kg; rate, 80 breaths/min). Then, left thoracotomy was
performed and the LAD coronary artery (3–5 mm from the root of
aorta) was ligated using nylon suture for 1 week. The rats in the
Diabetes/Sham group were underwent left thoracotomy without LAD
coronary artery ligation after diabetes induction. After that, the
rats were anesthetized and hemodynamic parameters were measured.
Then, the blood was collected and the heart was removed. The rats
were euthanized by exsanguinations before tissue collection.
Electrocardiogram (ECG) and
hemodynamic parameters
ECG, heart rate, left ventricular systolic pressure
(LVSP) and LV end-diastolic pressure (LVEDP) were measured by
Biological Data Acquisition and Analysis System BL-420 (Chengdu
Techman Software Co., Ltd.).
ELISA
Serum cardiac enzymes cardiac troponin T (cTnT) and
creatine kinase-MB isoenzyme (CK-MB) were measured by ELISA kits
according to the manufacturer's instructions. CK-MB assay kit and
ELISA kit for cTnT were purchased from WHB Scientific (Shanghai,
China).
Triphenyltetrazolium chloride (TTC)
staining
Infarct size was evaluated by TTC staining. After
euthanasia, the heart was excised, incubated with 10% KCl solution
and frozen at −20°C. The heart was cut into slices and stained with
1% TTC solution (Beijing Solarbio Science & Technology Co.,
Ltd.) at 37°C. These slices stained with TTC were photographed. The
infarct area (white) and total area (white and red) were analyzed
by Image-Pro Plus software 6.0 (Media Cybernetics, Inc., Rockville,
MD, USA). Infarct size (%) was expressed as a percentage of infarct
area over total area.
H&E staining
The fixed heart tissues were dehydrated in ethanol
and embedded in paraffin. Then, the obtained 5-µm-thick sections
were dewaxed, rehydrated and stained with hematoxylin and eosin
dye. The pathological changes of H&E-stained sections were
observed under a microscope (Olympus, Tokyo, Japan; ×400
magnification).
TUNEL assay
Cell apoptosis was examined by in situ Cell
Death Detection kit (Roche, Mannheim, Germany) according to the
manufacturer's instructions. Briefly, the fixed tissues were
embedded in paraffin and sectioned into 5-µm-thick sections.
Immediately, the sections were dewaxed in xylene, rehydrated in
different concentrations of ethanol and permeabilized with 0.1%
Triton X-100 at room temperature for 8 min. After being washed with
PBS, the sections were blocked with 3% H2O2
solution and incubated with TUNEL reaction mixture (Roche) at 37°C
for 1 h in the dark prior to being counterstained with hematoxylin
(Solarbio). The stained sections were observed under an Olympus
microscope (Olympus; ×400 magnification).
Western blotting
Total proteins isolated from the heart and
cardiomyocytes were quantified by bicinchoninic acid (BCA)
(Beyotime Institute of Biotechnology, Haimen, China) and subjected
to Western blotting. Briefly, equal amounts of proteins were boiled
for 5 min with loading buffer and then separated by SDS-PAGE. The
proteins were transferred to PVDF membranes (EMD Millipore,
Bedford, MA, USA) and blocked with buffer (5% non-fat milk in TBST
buffer) for 1 h. The PVDF membranes were incubated with primary
antibody against glucose-regulated protein 78 (GRP78) (D151791;
Sangon Biotech, Shanghai, China; 1:500 dilution), CHOP (bs-20669R;
Bioss, Beijing, China; 1:500 dilution), B-cell lymphoma 2 (Bcl-2;
BA0412; Boster, Wuhan, China; 1:400 dilution), Bcl-2-associated X
protein (Bax; D120073; Sangon Biotech; 1:500 dilution), endoplasmic
reticulum oxidoreductase 1α (Ero1α; bs-10551M; Bioss; 1:500
dilution), Ero1β (bs-14627R; Bioss; 1:500 dilution) or protein
disulfide isomerase (PDI; bs-4250R; Bioss; 1:500 dilution). After
being washed with TBST buffer, the membranes were incubated with
corresponding secondary antibody (A0208 and A0216; Beyotime
Institute of Biotechnology; 1:5,000 dilution) at 37°C. After being
washed, the bands were developed using ECL regent (Beyotime
Institute of Biotechnology) and quantified by Media Cybernetics
Gel-Pro analyzer. β-actin was used as an internal control.
Grouping and treatments
Rat cardiomyocyte H9c2 cells were cultured in DMEM
with 10% FBS at 37°C and divided into 6 groups: i) Control group,
normal H9c2 cells were cultured for 96 h; ii) High glucose (HG)
group, the cells were firstly maintained for 24 h under normal
conditions and then incubated for 72 h with DMEM containing 33 mM
glucose before analysis; iii) Hypoxia group, the cells were firstly
maintained for 72 h under normal conditions and then cultured under
a hypoxic environment (3% O2, 5% CO2 and 92%
N2) for 24 h prior to analysis; iv) HG/Hypoxia group,
after 24 h of normal culture, the cells were firstly subjected to
HG/Normoxia treatment for 48 h and then HG/Hypoxia treatment for 24
h prior to analysis; v) HG/Hypoxia/siNC group, 24 h after 30 pmol
siRNA (GenePharma, Shanghai, China) transfection using
Lipofectamine RNAiMAX (Invitrogen; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) according to the manufacturer's protocol, the
cells underwent 48 h of HG/Normoxia and 24 h of HG/Hypoxia
treatments; vi) HG/Hypoxia/siCHOP group, 24 h post-transfection,
siCHOP-transfected H9c2 cells were subjected to 48 h of HG/Normoxia
and 24 h of HG/Hypoxia treatments. Western blotting was performed
to confirm the successful knockdown of CHOP expression. The
sequences of siRNAs were shown below: siCHOP,
5′-GAACUAGGAAACGGAAACATT-3′; siNC, 5′-UUCUCCGAACGUGUCACGUTT-3′.
Cell Counting Kit-8 (CCK-8) assay
H9c2 cells were seeded in 96-well plates (at a
density of 3,000 cells/well) and cultured in a 5% CO2
incubator. Cell viability was examined by CCK-8 assay. The cells
were treated with 10 µl CCK-8 solution (Beyotime) at 37°C for 1 h.
The absorbance at 450 nm was measured by a microplate reader
(BioTek, Winooski, Vermont, USA).
Cell cycle and apoptosis analysis
For cell cycle analysis, H9c2 cells were washed with
PBS and fixed in 70% ethanol. The cells were incubated with 25 µl
propidium iodide (PI)/10 µl RNaseA in staining buffer (Beyotime
Institute of Biotechnology) at 37°C for 30 min in the dark. After
incubation, the cells were subjected to cytometric analysis. For
apoptosis detection, the cells were treated with 5 µl Annexin
V-FITC/5 µl propidium iodide (PI) in binding buffer (KeyGen,
Nanjing, China) for 15 min before analysis.
Hoechst 33258 staining
H9c2 cells were seeded onto coverslips in 12-well
plates at the density of 4×104 cells/coverslip. After
the aforementioned treatment, the cells were fixed with 4%
paraformaldehyde at room temperature for 20 min and stained with
Hoechst 33258 (Beyotime Institute of Biotechnology) for 5 min.
Subsequently, images were captured by a fluorescent microscope
(IX53; Olympus; ×400 magnification).
Statistical analysis
Statistical analyses for comparisons were performed
by ANOVA followed by Tukey's post-hoc test using GraphPad Prism
version 5.01 (GraphPad Software, Inc., San Diego, CA, USA). Data
are expressed as mean ± standard deviation. P<0.05 was
considered to indicate a statistically significant difference.
Results
Effects of diabetes and MI on ECG
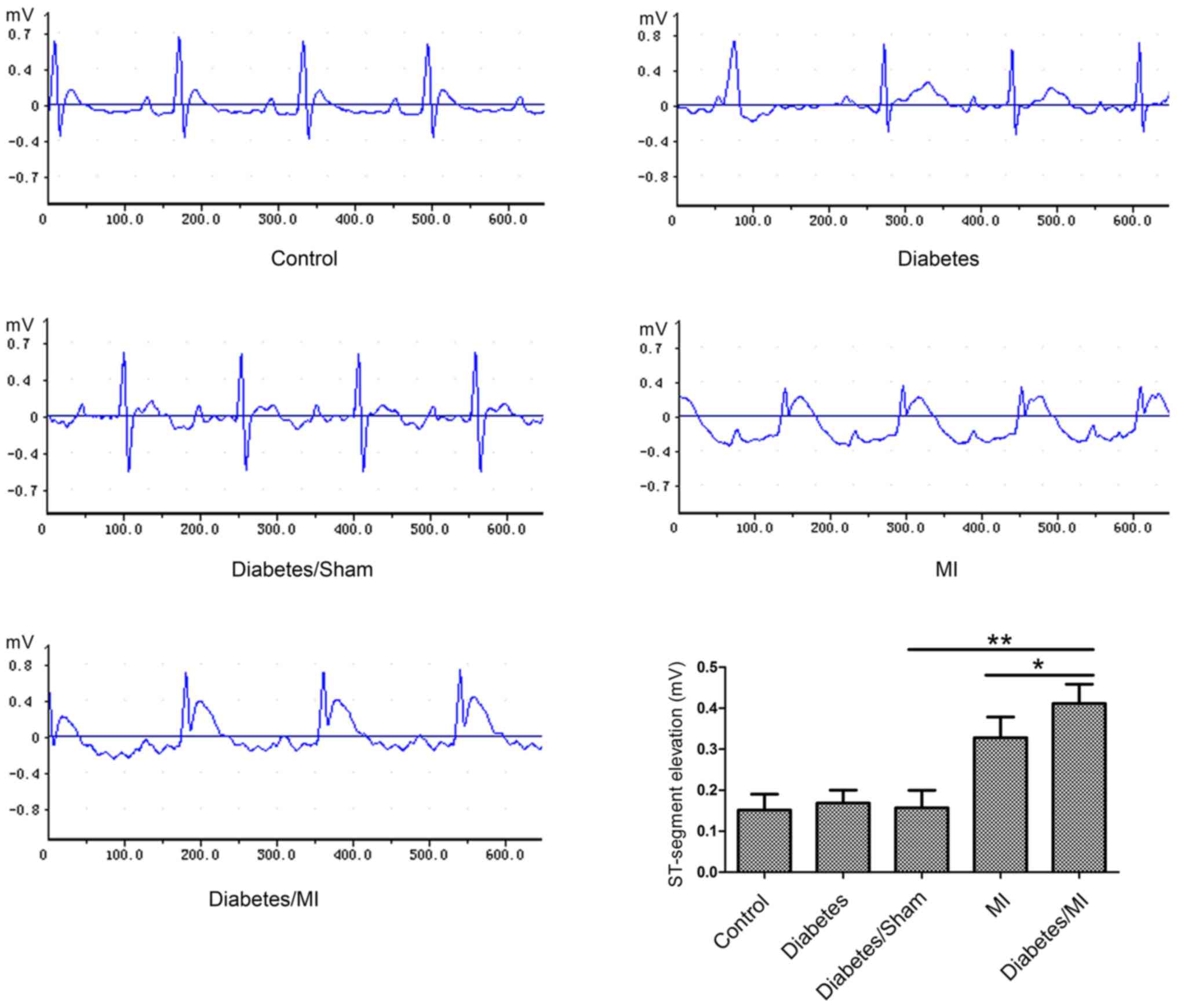
As shown in Fig. 1,
the rats in the Control group showed normal patterns of ECG.
ST-segments were markedly elevated in rats in the Diabetes/MI group
as compared with the Diabetes/Sham group or the MI group.
Diabetes aggravates heart dysfunction
in rat hearts after MI
The LVEDP, LVSP and heart rate were recorded to
assess heart function. As shown in Table
I, diabetes increased LVEDP, while decreased LVSP and heart
rate compared with the Control group. MI increased LVEDP and
decreased LVSP in diabetic rats compared with the Sham-operated
diabetic rats. The rats in the Diabetes/MI group exhibited
elevation in LVEDP and reductions in LVSP and heart rate compared
with the Diabetes/Sham group or MI group.
 | Table I.Heart function. |
Table I.
Heart function.
| Group | LVEDP (mm Hg) | LVSP (mm Hg) | Heart rate
(beats/min) |
|---|
| Control |
5.133±0.819 |
110.000±8.690 |
400.167±15.651 |
| Diabetes |
6.750±1.048 |
99.650±9.764 |
361.167±15.562a |
| Diabetes/Sham |
6.217±0.662 |
95.217±9.251 |
368.667±17.397 |
| MI |
8.567±0.792 |
86.300±6.165 |
376.667±16.318 |
| Diabetes/MI |
10.433±2.455b |
81.317±8.259 |
321.000±15.388b,c |
Diabetes aggravates myocardial injury
in rat hearts after MI
To examine whether diabetes aggravates myocardial
injury in rat hearts after MI, serum levels of CK-MB and cTnT were
measured by ELISA. The results showed that STZ-induced diabetes had
no significant effect on CK-MB and cTnT (Table II). Serum CK-MB and cTnT levels were
higher in the Diabetes/MI group than those in the Diabetes/Sham
group or the MI group.
| Table II.CK-MB and cTnT levels in serum. |
Table II.
CK-MB and cTnT levels in serum.
| Group | CK-MB (ng/ml) | cTnT (ng/l) |
|---|
| Control |
4.102±0.838 |
49.778±13.641 |
| Diabetes |
3.864±0.890 |
56.656±11.491 |
| Diabetes/Sham |
4.554±1.050 |
55.806±15.380 |
| MI |
7.337±1.455 |
146.272±27.636 |
| Diabetes/MI |
9.016±1.456a |
167.183±31.626a |
Diabetes enlarges myocardial infarct
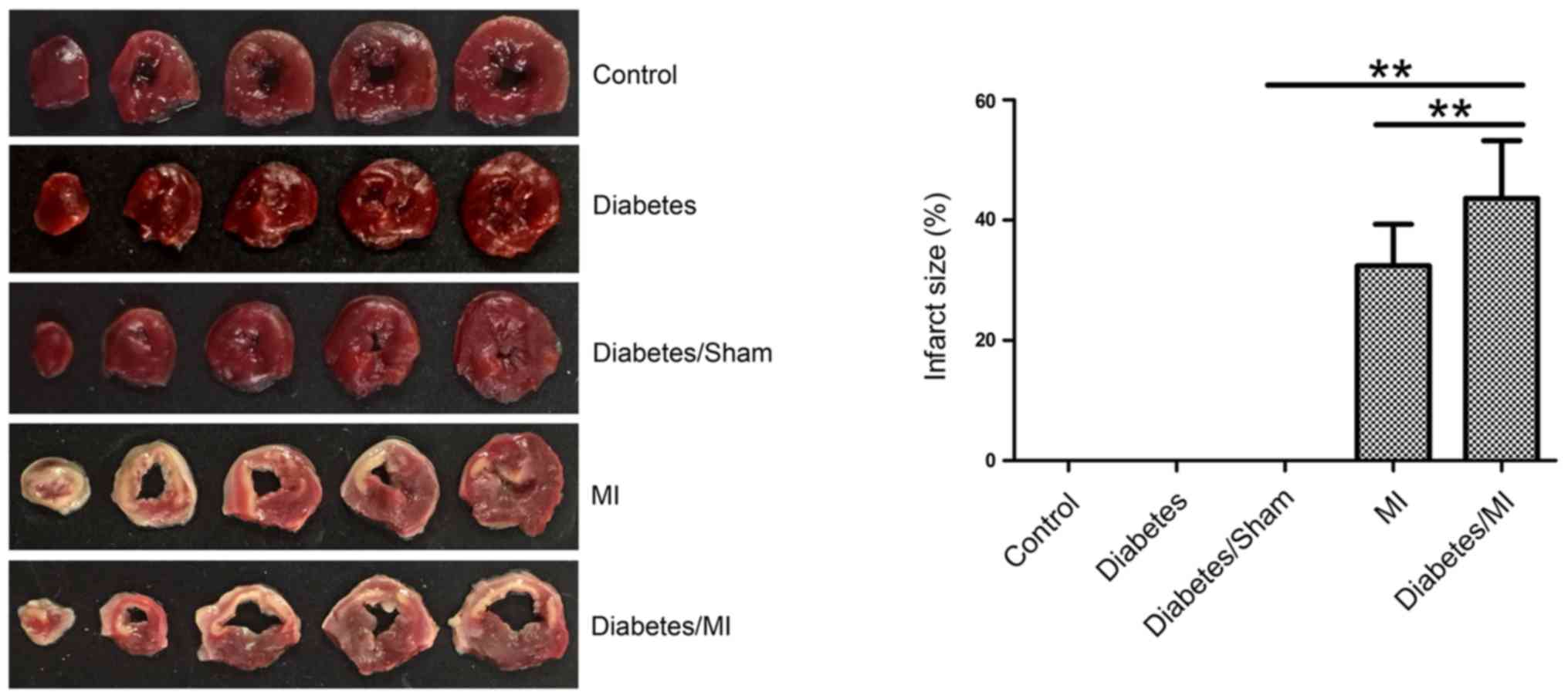
size in rats after MI
Myocardial infarct size was measured by TTC
staining. As shown in Fig. 2, there
was no MI in the rats of the Control, Diabetes or Diabetes/Sham
group. The infarct sizes in diabetic rats after MI (43.614±9.615)%
were larger than those in both the Diabetes/Sham group (0%) and the
MI group (32.430±6.890)%.
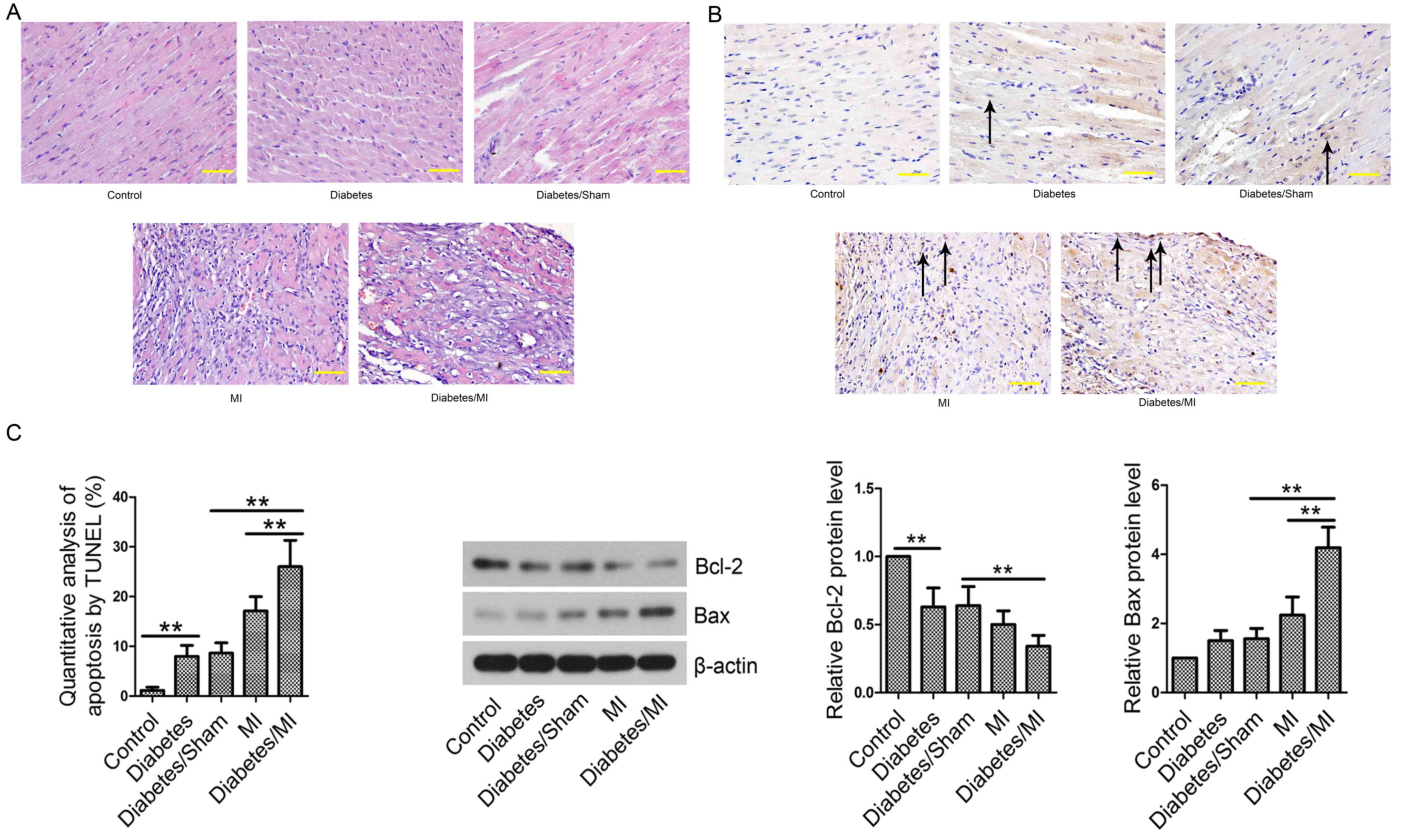
Diabetes induces pathological changes
in rat hearts after MI
In the Control group, the myocardial cells were
regularly arranged and did not show any pathological changes
(Fig. 3A). The myocardial tissues of
the Diabetes group, Diabetes/Sham group, MI group and Diabetes/MI
group exhibited pathological changes in infarcted border zone,
including disordered arrangement and rupture of myocardial fibers,
nuclear condensation/fragmentation, myocardial interstitial edema,
and inflammatory cell infiltration.
Diabetes promotes myocardial cell
apoptosis in rat hearts after MI
Cell apoptosis was then evaluated by TUNEL assay. As
shown in Fig. 3B, few apoptotic
cells were observed in rat hearts from the Control group.
TUNEL-positive cell rate in the Diabetes group was significantly
increased as compared with that in the Control group. Diabetes
complicated by MI further led to increased percentage of apoptotic
cells compared with the Diabetes/Sham or MI group. Next, the levels
of apoptosis-related proteins Bcl-2 and Bax were examined by
Western blotting. The results showed that the Bcl-2 level was lower
and Bax was higher in the Diabetes group than those in the Control
group (Fig. 3C). MI further
decreased Bcl-2 expression and increased Bax expression in diabetic
rats compared with the Diabetes/Sham or MI group. Diabetes
aggravated MI by inducing cell apoptosis.
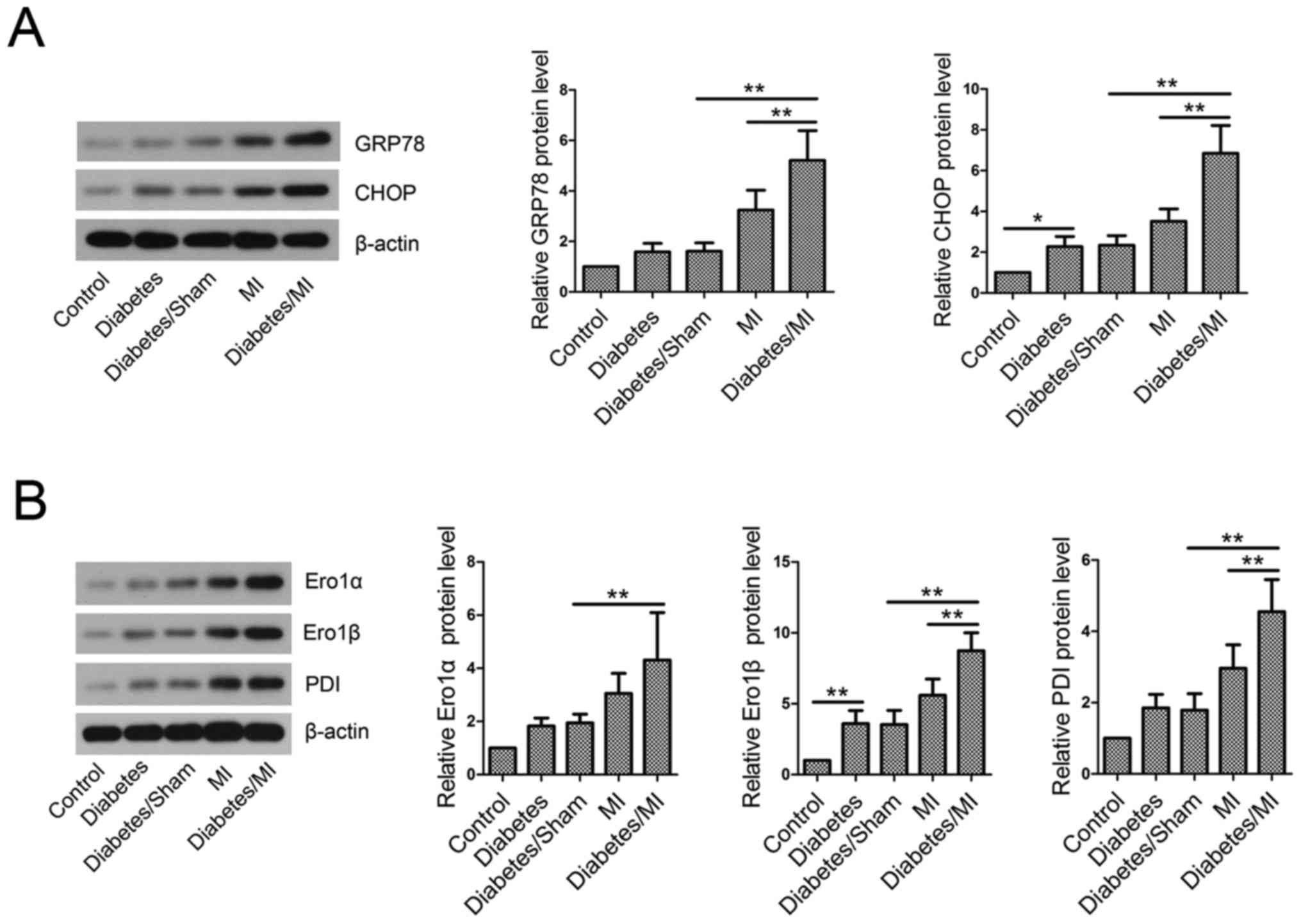
Diabetes activates ER stress in rat
hearts after MI
To investigate the effect of diabetes on ER stress
in MI rats, GRP78 and CHOP levels in myocardial tissues were
examined by western blotting. The results showed that STZ-induced
diabetes elevated GRP78 and CHOP levels compared with the control
rats (Fig. 4A). Additionally,
STZ-induced diabetes further activated ER stress in MI rats, as
evidenced by GRP78 and CHOP elevation.
Disruption of ER redox homeostasis in
diabetic rats with MI
The results showed that diabetes upregulated Ero1α,
Ero1β and PDI protein levels as compared with the Control group
(Fig. 4B). The levels of Ero1α,
Ero1β and PDI were greatly increased in diabetic rats with MI
compared with the Diabetes/Sham group or MI group.
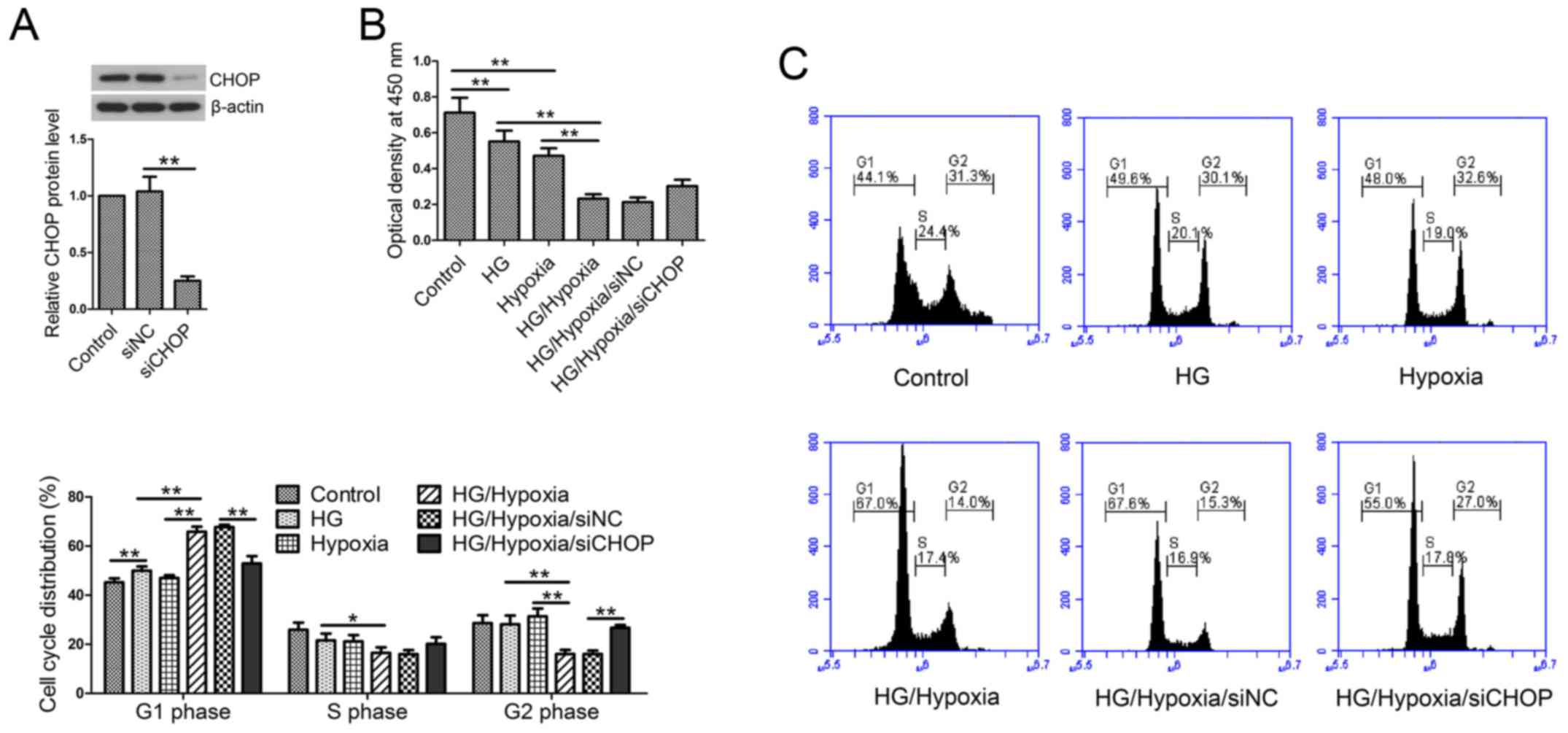
CHOP knockdown reverses the reduced
cell viability and cell cycle arrest
Firstly, in our in vitro studies, CHOP
expression was evaluated by western blotting post-siRNA
transfection. The results showed that CHOP siRNA transfection
significantly reduced CHOP expression in H9c2 cells (Fig. 5A). Cell viability was measured by
CCK-8 assay. Treatment with HG or hypoxia resulted in significant
decreases in cell viability as compared with the control cells
(Fig. 5B). Additionally, H9c2 cells
exposed to both HG and hypoxic treatments showed lower cell
viability than the single treatment group. However, siCHOP
transfection partially restored the reduced cell viability induced
by HG and hypoxia compared with the siNC-transfected cells. Then,
cell cycle distribution was analyzed by flow cytometry. The results
showed that HG followed by hypoxic treatment significantly arrested
the cell cycle at the G1 phase, whereas CHOP knockdown reversed the
cell cycle arrest (Fig. 5C).
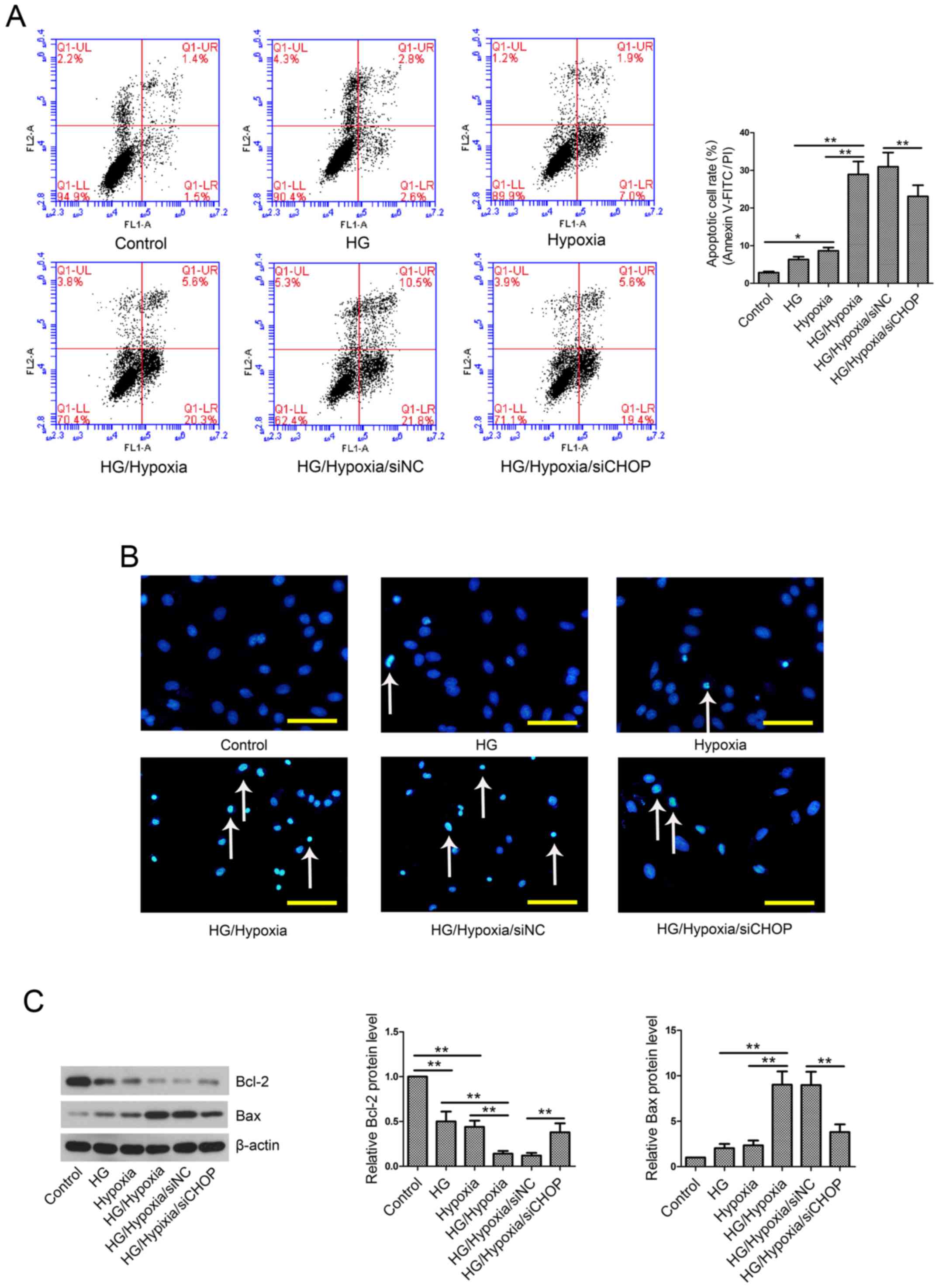
CHOP knockdown reduces cell apoptosis
rate in an in vitro model
Cell apoptosis was evaluated by Annexin V-FITC/PI
assay and Hoechst staining. The results showed that exposure of
H9c2 cells to HG and/or hypoxia resulted in a remarkable increase
in cell apoptotic rate (Fig. 6A and
B) as well as upregulation of Bax and downregulation of Bcl-2
(Fig. 6C). However, this
pro-apoptotic effect was partially abolished by CHOP knockdown.
CHOP knockdown inhibits ER stress and
maintains ER redox homeostasis in H9c2 cells
Then, the effects of CHOP knockdown on ER stress and
ER redox homeostasis were investigated by western blotting. HG
incubation followed by hypoxic treatment induced ER stress and
resulted in an imbalance in ER redox homeostasis, as evidenced by
upregulated levels of GRP78, CHOP, Ero1α, Ero1β and PDI (Fig. 7). However, CHOP knockdown partly
restored the elevated levels of these key proteins.
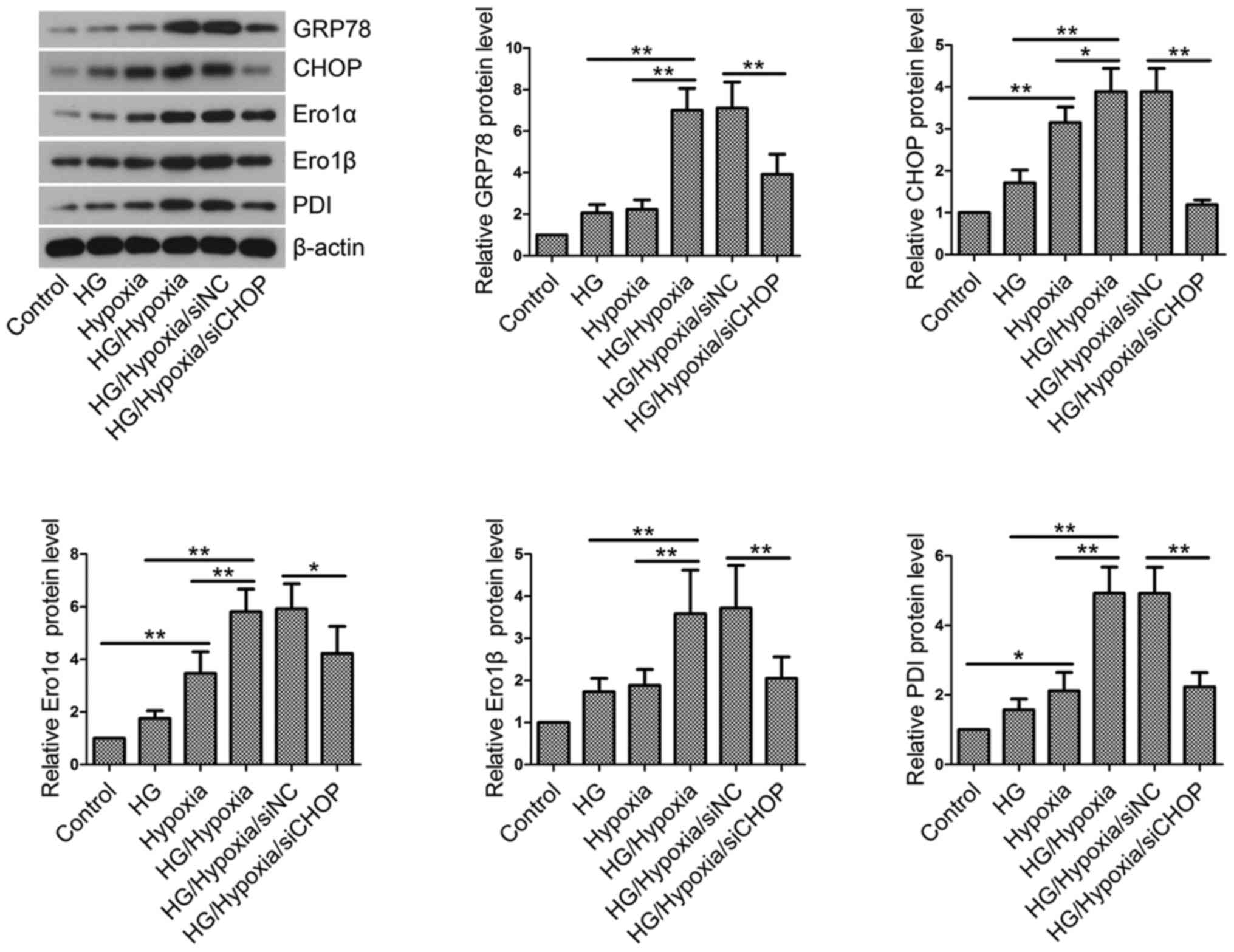 | Figure 7.Endoplasmic reticulum stress and
redox homeostasis in vitro. Total proteins were isolated
from H9c2 cells in each group and western blotting was performed to
measure GRP78, CHOP, Ero1α, Ero1β and PDI levels. β-actin served as
an internal control. Values are presented as the mean ± standard
deviation. *P<0.05 and **P<0.01, as indicated. GRP78,
glucose-regulated protein 78; CHOP, CCAAT/enhancer binding
protein-homologous protein; Ero1, endoplasmic reticulum
oxidoreductase 1; PDI, protein disulfide isomerase; HG, high
glucose; NC, negative control; si/siRNA, small interfering RNA. |
Discussion
Clinical studies have revealed that CVD is the major
cause of morbidity and mortality in diabetics (15). Patients with diabetes are more
susceptible to I/R injury than non-diabetic patients (16). Therefore, effective therapeutic
targets are urgently needed to alleviate MI in diabetes.
In the present study, diabetic SD rats induced by a
single injection of STZ were subjected to LAD coronary artery
ligation. Severity of myocardial injury is associated with the
levels of serum enzymes, such as lactate dehydrogenase (LDH), CK-MB
and cTnT (17). Moreover, cTnT is
more specific and sensitive than CK-MB in the diagnosis of AMI
(18). Zhang et al (19) have found that cardiac enzymes,
including CK-MB and cTnT, are higher in the MI group than the
Control group. Therefore, serum levels of CK-MB and cTnT were
measured and the pathological changes were evaluated in diabetic
rats following MI. The results showed that diabetes significantly
increased CK-MB and cTnT levels and induced pathological changes in
MI rats, which are consistent with previous findings (20). These data suggest that diabetes may
aggravate MI-induced myocardial injury.
LVEDP and LVSP are indicators of heart function
(21,22). LVEDP reflects the cardiac diastolic
function, while LVSP indicates the cardiac contractile function
(23). In the present study, the
results showed that LVEDP was increased and LVSP was decreased in
STZ-induced diabetic rats with MI as compared with the
Diabetes/Sham group or MI group, indicating that diabetes may
impair heart function in rats with MI by altering hemodynamic
parameters. The obtained results are in line with previous findings
(24).
Previous evidences have revealed that myocardial
apoptosis also plays a vital role in the progression of MI in
humans and animal models (25). In
the present study, myocardial apoptosis was investigated in
diabetes complicated by MI in animal models. Myocardial I/R injury
or MI can lead to myocardial apoptosis, which is correlated with
infarct size and ventricular dysfunction (26). Infarct size is commonly used to
evaluate the severity of I/R injury (27). The results showed that diabetes
significantly increased infarct size and induced myocardial
apoptosis in rats with MI, suggesting that diabetes may aggravate
MI by enhancing infarct size enlargement and myocardial
apoptosis.
GRP78 is a glucose-regulated protein and a major
molecular chaperone in ER (28).
GRP78 is a marker of ER stress (29). It has been reported that GRP78 is
associated with folding of newly synthesized protein, translocation
across the ER membrane and regulation of Ca2+
homeostasis (28). GRP78 forms a
protein complex with PERK, IRE1 (inositol-requiring transmembrane
kinase/endonuclease) and ATF6 under physiological conditions
(30). When excessive misfolded
proteins are accumulated in ER or under ER stress, these proteins
are released from the protein complex and activated. Finally, the
folding capacities of ER and CHOP expression are enhanced (30). CHOP is ubiquitously expressed,
including normal myocardial tissues, at low levels in the cytosol
under physiological conditions; while it accumulates in the nucleus
when activated (31). Previous
evidences have demonstrated that ER stress is associated with the
development of cerebral and myocardial ischemia/reperfusion injury
(32,33). ER stress can lead to cell apoptosis
(34). ER stress-induced apoptosis
has been shown to be associated with transcription factors, caspase
family proteins, Bcl-2 family members and JNK signaling pathway
(35). CHOP is the first identified
regulator that participates in ER stress-induced apoptosis
(31). It has been reported that
apoptosis is attenuated in CHOP-deficient mice in response to ER
stress (36). In the present study,
the results showed that the protein levels of GRP78 and CHOP were
greatly higher in the Diabetes/MI group than those in the MI and
Diabetes/Sham group. The results indicate that diabetes may
aggravate MI by modulating ER stress and CHOP may be a key mediator
in ER stress-induced apoptosis in an animal model of diabetes and
MI.
Bcl-2 family members are involved in apoptosis,
including pro-apoptotic protein Bax and anti-apoptotic protein
Bcl-2 (37). CHOP overexpression
results in a reduction of Bcl-2 and promotes Bax translocation to
mitochondria, thereby regulating the mitochondrial apoptosis
pathway; whereas, Bcl-2 overexpression reverses CHOP-induced
apoptosis (38). The present study
investigated whether Bax and Bcl-2 participated in CHOP-induced
apoptosis in diabetes complicated by MI. The results showed that
the rats in the Diabetes/MI group exhibited higher Bax and lower
Bcl-2 than those in the MI group or Diabetes/Sham group. These
results suggest that Bax and Bcl-2 may be the downstream regulators
in CHOP-induced apoptosis in this animal model.
There are two isoforms of Ero1 in human cells,
namely Ero1α and Ero1β. Ero1α is identified in almost all cell
types, while Ero1β is only expressed in select tissues (39). Ero1α and Ero1β expression levels can
be elevated by hypoxia and UPR (40). PDI (also named PDIA1) belongs to PDI
family, which contains 21 family members. The family members have
at least one thioredoxin-like βαβαβαββα domain (41,42).
Both Ero1α and Ero1β can catalyze the reoxidation of PDI, in which
oxygen was consumed to produce hydrogen peroxide (43). Previous evidence has revealed that
the calcium signaling pathway is a mechanism of CHOP-induced
apoptosis (44). Ero1α, a target
gene of CHOP, promotes ER calcium channel IP3R1-induced calcium
release and thereby induces the activation of calcium-sensing
enzyme CaMKII, eventually leading to apoptosis (44). In this study, Ero1α, Ero1β and PDI
levels were examined by western blotting. The results showed that
diabetes complicated with MI significantly elevated Ero1α, Ero1β
and PDI levels compared with the MI group or Diabetes/Sham group.
The results suggest that diabetes may exacerbate MI by disrupting
ER redox homeostasis.
Consistent with previous reports (45,46), our
in vivo studies demonstrated that ER stress was activated in
the development of ischemic injury in diabetes. Based on the in
vivo findings in diabetic rats with MI, an in vitro
model was established using H9c2 cells. In the present study,
siCHOP-transfected H9c2 cells were incubated with HG and subjected
to hypoxic treatment to investigate the role of CHOP.
Woo et al (47) have reported that delivery of cyclin
A2 activates cell cycle in cardiomyocytes and improves heart
function in a rat model of ischemic heart failure. Pasumarthi et
al (48) have found that cyclin
D2 overexpression promotes DNA synthesis in cardiomyocytes and
infarct regression in transgenic mice. The aforementioned evidences
suggest that cardiomyocyte proliferation is critical for
maintaining heart function in injured heart. Thus, in the present
study, cell proliferation was assessed by CCK-8 assay and flow
cytometry. The results showed that HG combined with hypoxia reduced
cell viability and caused cell cycle arrest at G1 phase. ER
stress-induced apoptosis contributes to progression of acute MI and
anti-apoptotic treatment can reduce infarct size in rats with MI
(11). Our in vitro studies
showed that HG combined with hypoxia induced cell apoptosis,
activated ER stress and dysregulated ER redox homeostasis, which
were consistent with our in vivo results. However, CHOP
knockdown by specific siRNA transfection partly reversed the effect
of HG and hypoxia.
In summary, CHOP knockdown alleviated MI in diabetes
via the GRP78/CHOP and Ero1/PDI signaling pathways. CHOP is
expected to be a promising target for the treatment of diabetes
complicated by MI.
References
|
1
|
Bai Y, Wang X, Zhao S, Ma C, Cui J and
Zheng Y: Sulforaphane protects against cardiovascular disease via
Nrf2 activation. Oxid Med Cell Longev. 2015:4075802015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yang ZH, Emma-Okon B and Remaley AT:
Dietary marine-derived long-chain monounsaturated fatty acids and
cardiovascular disease risk: A mini review. Lipids Health Dis.
15:2012016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bernardi S, Michelli A, Zuolo G, Candido R
and Fabris B: Update on RAAS modulation for the treatment of
diabetic cardiovascular disease. J Diabetes Res. 2016:89175782016.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lipes MA and Galderisi A: Cardiac
autoimmunity as a novel biomarker, mediator and therapeutic target
of heart disease in type 1 diabetes. Curr Diab Rep. 15:302015.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gregg EW, Cheng YJ, Saydah S, Cowie C,
Garfield S, Geiss L and Barker L: Trends in death rates among U.S.
adults with and without diabetes between 1997 and 2006: Findings
from the National Health Interview Survey. Diabetes Care.
35:1252–1257. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lee JM: Nuclear receptors resolve
endoplasmic reticulum stress to improve hepatic insulin resistance.
Diabetes Metab J. 41:10–19. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Phillips MJ and Voeltz GK: Structure and
function of ER membrane contact sites with other organelles. Nat
Rev Mol Cell Biol. 17:69–82. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chen Y, Tsai YH and Tseng SH: HDAC
inhibitors and RECK modulate endoplasmic reticulum stress in tumor
cells. Int J Mol Sci. 18:pii: E258. 2017.
|
|
9
|
Lee YT, Lin HY, Chan YW, Li KH, To OT, Yan
BP, Liu T, Li G, Wong WT, Keung W and Tse G: Mouse models of
atherosclerosis: A historical perspective and recent advances.
Lipids Health Dis. 16:122017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Oyadomari S, Koizumi A, Takeda K, Gotoh T,
Akira S, Araki E and Mori M: Targeted disruption of the Chop gene
delays endoplasmic reticulum stress-mediated diabetes. J Clin
Invest. 109:525–532. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Shi ZY, Liu Y, Dong L, Zhang B, Zhao M,
Liu WX, Zhang X and Yin XH: Cortistatin improves cardiac function
after acute myocardial infarction in rats by suppressing myocardial
apoptosis and endoplasmic reticulum stress. J Cardiovasc Pharmacol
Ther: pii: 1074248416644988. 2016.
|
|
12
|
Rao J, Zhang C, Wang P, Lu L, Qian X, Qin
J, Pan X, Li G, Wang X and Zhang F: C/EBP homologous protein (CHOP)
contributes to hepatocyte death via the promotion of ERO1α
signalling in acute liver failure. Biochem J. 466:369–378. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Takada A, Miki T, Kuno A, Kouzu H, Sunaga
D, Itoh T, Tanno M, Yano T, Sato T, Ishikawa S and Miura T: Role of
ER stress in ventricular contractile dysfunction in type 2
diabetes. PLoS One. 7:e398932012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Barr LA, Shimizu Y, Lambert JP, Nicholson
CK and Calvert JW: Hydrogen sulfide attenuates high fat
diet-induced cardiac dysfunction via the suppression of endoplasmic
reticulum stress. Nitric Oxide. 46:145–156. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Rawal S, Manning P and Katare R:
Cardiovascular microRNAs: As modulators and diagnostic biomarkers
of diabetic heart disease. Cardiovasc Diabetol. 13:442014.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kuusisto J and Laakso M: Update on type 2
diabetes as a cardiovascular disease risk equivalent. Curr Cardiol
Rep. 15:3312013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lu SF, Huang Y, Wang N, Shen WX, Fu SP, Li
Q, Yu ML, Liu WX, Chen X, Jing XY and Zhu BM: Cardioprotective
effect of electroacupuncture pretreatment on myocardial
ischemia/reperfusion injury via antiapoptotic signaling. Evid Based
Complement Alternat Med. 2016:46097842016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tiwari RP, Jain A, Khan Z, Kohli V,
Bharmal RN, Kartikeyan S and Bisen PS: Cardiac troponins I and T:
Molecular markers for early diagnosis, prognosis and accurate
triaging of patients with acute myocardial infarction. Mol Diagn
Ther. 16:371–381. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang J, Yu P, Chen M, Peng Q, Wang Z and
Dong N: Remote ischaemic preconditioning and sevoflurane
postconditioning synergistically protect rats from myocardial
injury induced by ischemia and reperfusion partly via inhibition
TLR4/MyD88/NF-κB signaling pathway. Cell Physiol Biochem. 41:22–32.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ding M, Lei J, Han H, Li W, Qu Y, Fu E, Fu
F and Wang X: SIRT1 protects against myocardial
ischemia-reperfusion injury via activating eNOS in diabetic rats.
Cardiovasc Diabetol. 14:1432015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Liu J, Wang N, Xie F, Sun LH, Chen QX, Ye
JH, Cai BZ, Yang BF and Ai J: Blockage of peripheral NPY Y1 and Y2
receptors modulates barorefex sensitivity of diabetic rats. Cell
Physiol Biochem. 31:421–431. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chang R, Li Y, Yang X, Yue Y, Dou L, Wang
Y, Zhang W and Li X: Protective role of deoxyschizandrin and
schisantherin A against myocardial ischemia-reperfusion injury in
rats. PLoS One. 8:e615902013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Xie F, Sun L, Su X, Wang Y, Liu J, Zhang
R, Wang N, Zhao J, Ban T, Niu H and Ai J: Neuropeptide Y reverses
chronic stress-induced baroreflex hypersensitivity in rats. Cell
Physiol Biochem. 29:463–474. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Agrawal YO, Sharma PK, Shrivastava B, Arya
DS and Goyal SN: Hesperidin blunts streptozotocin-isoproternol
induced myocardial toxicity in rats by altering of PPAR-γ receptor.
Chem Biol Interact. 219:211–220. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bussani R, Abbate A, Biondi-Zoccai GG,
Dobrina A, Leone AM, Camilot D, Di Marino MP, Baldi F, Silvestri F,
Biasucci LM and Baldi A: Right ventricular dilatation after left
ventricular acute myocardial infarction is predictive of extremely
high peri-infarctual apoptosis at postmortem examination in humans.
J Clin Pathol. 56:672–676. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang X, Wang Q, Guo W and Zhu YZ: Hydrogen
sulfide attenuates cardiac dysfunction in a rat model of heart
failure: A mechanism through cardiac mitochondrial protection.
Biosci Rep. 31:87–98. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Han Q, Zhang HY, Zhong BL, Zhang B and
Chen H: Antiapoptotic effect of recombinant HMGB1 A-box protein via
regulation of microRNA-21 in myocardial ischemia-reperfusion injury
model in rats. DNA Cell Biol. 35:192–202. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Li J, Ni M, Lee B, Barron E, Hinton DR and
Lee AS: The unfolded protein response regulator GRP78/BiP is
required for endoplasmic reticulum integrity and stress-induced
autophagy in mammalian cells. Cell Death Differ. 15:1460–1471.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yu H, Zhen J, Yang Y, Gu J, Wu S and Liu
Q: Ginsenoside Rg1 ameliorates diabetic cardiomyopathy by
inhibiting endoplasmic reticulum stress-induced apoptosis in a
streptozotocin-induced diabetes rat model. J Cell Mol Med.
20:623–631. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bennett HL, Fleming JT, O'Prey J, Ryan KM
and Leung HY: Androgens modulate autophagy and cell death via
regulation of the endoplasmic reticulum chaperone glucose-regulated
protein 78/BiP in prostate cancer cells. Cell Death Dis. 1:e722010.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Oyadomari S and Mori M: Roles of
CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ.
11:381–389. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
DeGracia DJ and Montie HL: Cerebral
ischemia and the unfolded protein response. J Neurochem. 91:1–8.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhang M, Yu LM, Zhao H, Zhou XX, Yang Q,
Song F, Yan L, Zhai ME, Li BY, Zhang B, et al:
2,3,5,4′-Tetrahydroxystilbene-2-O-β-D-glucoside protects murine
hearts against ischemia/reperfusion injury by activating
Notch1/Hes1 signaling and attenuating endoplasmic reticulum stress.
Acta Pharmacol Sin. 38:317–330. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Nakagawa T, Zhu H, Morishima N, Li E, Xu
J, Yankner BA and Yuan J: Caspase-12 mediates
endoplasmic-reticulum-specific apoptosis and cytotoxicity by
amyloid-beta. Nature. 403:98–103. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kadowaki H, Nishitoh H and Ichijo H:
Survival and apoptosis signals in ER stress: The role of protein
kinases. J Chem Neuroanat. 28:93–100. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zinszner H, Kuroda M, Wang X, Batchvarova
N, Lightfoot RT, Remotti H, Stevens JL and Ron D: CHOP is
implicated in programmed cell death in response to impaired
function of the endoplasmic reticulum. Genes Dev. 12:982–995. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Gross A: BCL-2 family proteins as
regulators of mitochondria metabolism. Biochim Biophys Acta.
1857:1243–1246. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Fu HY, Okada K, Liao Y, Tsukamoto O,
Isomura T, Asai M, Sawada T, Okuda K, Asano Y, Sanada S, et al:
Ablation of C/EBP homologous protein attenuates endoplasmic
reticulum-mediated apoptosis and cardiac dysfunction induced by
pressure overload. Circulation. 122:361–369. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Inaba K, Masui S, Iida H, Vavassori S,
Sitia R and Suzuki M: Crystal structures of human Ero1alpha reveal
the mechanisms of regulated and targeted oxidation of PDI. EMBO J.
29:3330–3343. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Dias-Gunasekara S, Gubbens J, van Lith M,
Dunne C, Williams JA, Kataky R, Scoones D, Lapthorn A, Bulleid NJ,
Benham AM, et al: Tissue-specific expression and dimerization of
the endoplasmic reticulum oxidoreductase Ero1beta. J Biol Chem.
280:33066–33075. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kozlov G, Määttänen P, Thomas DY and
Gehring K: A structural overview of the PDI family of proteins.
FEBS J. 277:3924–3936. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Andreu CI, Woehlbier U, Torres M and Hetz
C: Protein disulfide isomerases in neurodegeneration: From disease
mechanisms to biomedical applications. FEBS Lett. 586:2826–2834.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Swiatkowska M, Padula G, Michalec L,
Stasiak M, Skurzynski S and Cierniewski CS: Ero1alpha is expressed
on blood platelets in association with protein-disulfide isomerase
and contributes to redox-controlled remodeling of alphaIIbbeta3. J
Biol Chem. 285:29874–29883. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Scull CM and Tabas I: Mechanisms of ER
stress-induced apoptosis in atherosclerosis. Arterioscler Thromb
Vasc Biol. 31:2792–2797. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Su D, Ma J, Yang J, Kang Y, Lv M and Li Y:
Monosialotetrahexosy-1 ganglioside attenuates diabetes-associated
cerebral ischemia/reperfusion injury through suppression of the
endoplasmic reticulum stress-induced apoptosis. J Clin Neurosci.
41:54–59. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Yu L, Li S, Tang X, Li Z, Zhang J, Xue X,
Han J, Liu Y, Zhang Y, Zhang Y, et al: Diallyl trisulfide
ameliorates myocardial ischemia-reperfusion injury by reducing
oxidative stress and endoplasmic reticulum stress-mediated
apoptosis in type 1 diabetic rats: Role of SIRT1 activation.
Apoptosis. 22:942–954. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Woo YJ, Panlilio CM, Cheng RK, Liao GP,
Atluri P, Hsu VM, Cohen JE and Chaudhry HW: Therapeutic delivery of
cyclin A2 induces myocardial regeneration and enhances cardiac
function in ischemic heart failure. Circulation. 114 1
Suppl:I206–I213. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Pasumarthi KB, Nakajima H, Nakajima HO,
Soonpaa MH and Field LJ: Targeted expression of cyclin D2 results
in cardiomyocyte DNA synthesis and infarct regression in transgenic
mice. Circ Res. 96:110–118. 2005. View Article : Google Scholar : PubMed/NCBI
|