Introduction
Liver cancer is a growing problem in less developed
countries and, according to the GLOBOCAN 2012 (1), it is the second most common cause of
cancer-associated mortality worldwide. The disease burden of liver
cancer is increasing markedly across the world (2). If liver cancer is diagnosed at an early
stage, patients are able to undergo effective treatment via liver
resection, transplantation or ablation, and the 5-year survival
rate of such patients is >50% (3). However, the prognosis of patients with
advanced liver cancer is very poor and the 5-year survival rate of
such patients is <5% (4).
Hepatoblastoma is a highly malignant liver cancer
that occurs most commonly in children (5). The 5-year overall survival rate of
patients with stage IV hepatoblastoma is ~40% (6). The standard method of treating patients
with hepatoblastoma is surgery combined with high dose,
non-targeted chemotherapy; however, such chemotherapy induces
severe side effects in patients, including ototoxicity and
cardiotoxicity (7,8). Therefore, novel therapeutic strategies
to treat patients with hepatoblastoma are required.
Due to advances in molecular biology, a number of
tumor-associated genes have been identified, including CTNNB1 and
β-Catenin in hepatoblastoma (9).
Current studies focusing on gene therapy aim to develop molecular
biological methods to inhibit the in vivo expression of such
genes in order to treat patients with cancer. Studies investigating
methods of treating cancer via gene therapy tend to focus on genes
that induce the apoptosis of cancer cells (10–12).
The abnormal activation of apoptosis pathways may
induce the onset and progression of numerous diseases, including
different types of cancer. The Fas/Fas ligand (FasL) system is one
such important apoptotic pathway (12). Ogasawara et al (13) demonstrated that the liver is more
sensitive to Fas-mediated apoptosis compared with other organs.
FasL, a type II membrane protein, belongs to the tumor necrosis
factor (TNF) superfamily (14). Fas,
which is a member of the TNF receptor (TNFR) family, is widely
expressed in normal cells, including liver, kidney and heart cells,
and binds to FasL (15). Fas is
involved in transferring the apoptotic signal, resulting in cell
apoptosis (16) and the Fas/FasL
system is a key physiological regulator of programmed cell death
(17). It has been demonstrated that
resistance to apoptosis due to loss of Fas function may serve an
important role in the pathogenesis of several malignancies
(18).
FasL contains two receptors, Fas and decoy receptor
3 (DcR3). DcR3, initially identified by Pitti et al
(19), does not contain a death
domain and so cannot transfer apoptotic signals (20). DcR3 competitively binds to FasL over
Fas, thus inhibiting FasL-induced apoptosis (21) and may contribute to tumor growth in
this manner (22). Furthermore, DcR3
binds to FasL and inhibits FasL-induced apoptosis. DcR3 is highly
expressed in many malignant tumors, including liver cancer
(23–25). It has been demonstrated that the
expression of DcR3 mRNA is 60.4% in liver cancer tissues; however,
it is not expressed in the adjacent normal tissues. Furthermore,
DcR3 expression is associated with tumor size, clinical stage,
tumor invasion and metastasis (23).
Yu et al (26) demonstrated
that decreasing DcR3 expression in SW480 colon cancer cells
inhibited cell growth and metastatic ability, induced apoptosis and
altered the cell cycle profile of these cells. However the exact
molecular mechanism underlying the oncogenic property of DcR3 in
liver cancer remains unclear.
Our previous study by the current study transfected
human liver cancer HepG2 cells with lentivirus-based short hairpin
RNA vector targeting DcR3 stably and indicated the that loss of
DcR3 impaired the growth and invasive ability of HepG2 (27). The present study also used the HepG2
cell line, which was established in 1979 and mistakenly reported as
a hepatocellular carcinoma cell line (28). However, in 2009, it was demonstrated
that HepG2 cells originated from hepatoblastoma, not hepatocellular
carcinoma (29). The results of the
current study are relevant to hepatoblastoma and liver cancer in
general, however, they may not be useful regarding hepatocellular
carcinoma.
Therefore, in the current study, HepG2 cells that
had undergone DcR3 knockdown by small interfering (si)RNA knockdown
were treated with FasL to assess the effect of FasL on HepG2 cell
activity and invasive capabilities.
Materials and methods
Reagents
Reagents and kits included: Dulbecco's modified
Eagle's medium (DMEM) and fetal bovine serum (FBS; Gibco; Thermo
Fisher Scientific, Inc., Waltham, MA, USA), TRIzol (Invitrogen;
Thermo Fisher Scientific, Inc.), a prime Script RT reagent kit and
SYBR Premix Ex Taq (Takara Bio, Inc., Otsu, Japan), Transwell
inserts (Axygen; Corning Inc., Corning, NY, USA), Matrigel (BD
Biosciences, San Jose, CA, USA), human recombinant FasL (R&D
Systems, Inc., Minneapolis, MN, USA), rabbit anti-human GAPDH
polyclonal antibody (cat. no. 5714; Cell Signaling Technology,
Inc., Danvers, MA, USA) and rabbit anti-human matrix
metallopeptidase 9 (MMP9) polyclonal antibody (cat. no. ab38898),
rabbit anti-human vascular endothelial growth factor C (VEGF-C)
monoclonal antibody (cat. no. ab191274), rabbit anti-human VEGF-D
polyclonal antibody (cat. no. ab155288) and horseradish peroxidase
(HRP)-conjugated goat anti-rabbit secondary antibodies (cat. no.
ab205718) (all Abcam, Cambridge, UK).
Cell culture
Hepatoblastoma HepG2 cells were purchased from the
Cell Bank at Peking Union Medical College Cancer Hospital (Beijing,
China). Cells were cultured at 37°C, in 5% CO2 and
saturated humidity in DMEM with 10% FBS, 100 U/ml ampicillin, and
100 µg/ml streptomycin.
Cell transfection
In our previous study (27), HepG2 cells were successfully
transfected with target lentivirus Lv-DcR3-EGFP-shRNA [the sequence
with a stem-loop structure was
CGCTGGTTTCTGCTTGGAGCA-CTCGAG-TGCTCCAAGCAGAAACCAGCG (Beijing
TransGen Biotech, Co., Ltd., Beijing, China)] and blank lentivirus
Lv-NC-EGFP-shRNA(Beijing TransGen Biotech, Co., Ltd.) using the
Lipofectamine 2000 reagent (Invitrogen; Thermo Fisher Scientific,
Inc.). The ratio of plasmid DNA (µg)/Lipofectamine 2000 reagent
(µl) was 1:3.5, and DcR3 siRNA knockdown in HepG2 cells (KD) was
achieved and DcR3 blank plasmid negative control HepG2 cells were
utilized (negative control, NC). The interval between transfection
and subsequent experimentation was 4 weeks. Wild-type HepG2 cells
(WT) were used as a normal control.
Detection of apoptosis
HepG2 WT cells and KD cells were seeded into 6-well
plates at 5×105 cells/well. Following 24 h incubation,
KD cells were incubated with dimethyl sulfoxide (DMSO) containing
FasL (10 ng/ml; KD + Fas L). KD and WT cells incubated with DMSO
alone acted as controls. Following 48 h incubation, cells were
digested with trypsin and centrifuged at 1,000 × g for 5 min at
room temperature. Cells were then washed with PBS and centrifuged
at 1,000 × g for 5 min at room temperature. Cells were resuspended
in PBS, 10×104 cells were transferred into a centrifuge
tube and centrifuged at 1,000 × g for 5 min at room temperature.
Apoptosis was detected using an Annexin V-fluorescein
isothiocyanate (FITC) cell apoptosis assay kit (Beyotime Institute
of Biotechnology, Haimen, China). Cells were resuspended in 195 µl
Annexin V-FITC; then 5 µl Annexin V-FITC was added and mixed well.
Following 10 min incubation at 20–25°C, cells were centrifuged at
1,000 × g for 5 min and the supernatant was discarded. Cells were
then resuspended in 190 µl Annexin V-FITC and 10 µl propidium
iodide (PI) was added and mixed well. PI staining was performed at
room temperature for 20 min in the dark. A flow cytometer was used
to assess the results; cells stained with Annexin V-FITC exhibited
green fluorescence and those stained with PI exhibited red
fluorescence. Flow cytometry data were analyzed using CellQuest Pro
5.10 (BD Biosciences, Franklin Lakes, NJ, USA). The cells were
properly gated and a dual-parameter dot plot of FL1-H (x-axis;
Annexin V fluorescence) vs. FL2-H (y-axis; PI fluorescence) was
demonstrated to be logarithmic in fluorescence intensity.
Cell cycle analysis
HepG2 WT cells and KD cells were seeded into 6-well
platesat 5×105 cells/well. Following 24 h incubation, KD
+ Fas L cells were incubated with DMSO containingn Fas (10 ng/ml).
KD and WT cells incubated with DMSO alone acted as controls.
Following 48 h, cells were washed with PBS and digested with
trypsin. Cells were then transferred to a centrifuge tube and
centrifuged at 1,000 × g for 5 min at room temperature; the
supernatant was then discarded. The cells were resuspended with
0.9% NaCl and then the supernatant was discarded. Subsequently,
cells were fixed with 1 ml 75% ethanol overnight at −20°C.
Following fixation, cells were centrifuged at 1,000 × g at 4°C for
5 min and the supernatant was discarded. The cells were washed
twice with NaCl 0.9% with 1% bovine serum albumin (Sigma-Aldrich;
Merck KGaA, Darmstadt, Germany) added. Following the addition of 50
µl RNAse (Sigma-Aldrich; Merck KGaA) and 50 µl PI for 20 min, cells
were left in the dark at 4°C. Cells were mixed gently on ice in the
dark and then flow cytometry was performed. Flow cytometry data
were analysed using Cell Quest Pro 5.10(BD Biosciences). The DNA
content (x-axis, PI fluorescence) vs. counts (y-axis) was plotted
as a histogram.
MTS cell proliferation assay
Cells in the WT, NC, and KD groups were seeded into
96-well plates at 2,000 cells/well. Following 24 h incubation, each
group of cells (WT, NC and KD) were incubated either with DMEM
containing FasL (10 ng/ml) or DMEM alone. Cells were incubated at
37°C in 5% CO2 for 8 days. Subsequently, 20 µl CellTiter
96® AQueous One Solution Reagent (Promega Corporation,
Madison, WI, USA) was added to each well and incubated at 37°C for
3 h in 5% CO2. Optical density was measured at 490 nm
everyday using Plate reader spectrophotometer (Bio-Rad
Laboratories, Inc., Hercules, CA, USA).
Clonogenic assay
For each experimental group (WT, NC and KD cells),
800 cells/well were seeded into a 6-well plate. Following 24 h
incubation, each group of cells (WT, NC and KD) were incubated
either with DMEM containing FasL (10 ng/ml) or DMEM alone. When
clones became visible, the supernatant was discarded and clones
were washed with PBS for 3 times. Cells were fixed in 4%
paraformaldehyde for 15 min at room temperature. Following three
washes in PBS, cells were stained with 1% crystal violet solution
at 37°C for 10–30 min. Plates were then air-dried and the numbers
of clones were calculated. The colony formation rate (%) was
calculated using the following formula:(number of clones/number of
inoculated cells) ×100.
Wound healing assay
WT, NC and KD cells were seeded at
1×105/ml into 6-well plates and cultured until cells
grew to filled the plates. A 200 µl pipet tip was used to evenly
scratch across the well (at intervals of 0.5–1.0 cm) 5 times, with
the tip kept vertical. Removed cells were washed with PBS (3 times)
and each group of cells (WT, NC and KD) was incubated either with
DMEM containing FasL (10 ng/ml) or DMEM alone. Plates were
incubated at 37°C with 5% CO2 for 48 h, and samples were
photographed at 0, 24 and 48 h.
Cell invasion assay
The apical side of the membrane of the Transwell
inserts was coated with 50 mg/l Matrigel at a dilution of 1:8 and
was then air-dried at 4°C. A total of 3×103 cells from
each group in 200 µl serum-free medium was added to the upper
chamber of the Transwell assay. DMEM containing FasL (10 ng/ml) and
1% FBS (Gibco; Thermo Fisher Scientific, Inc.) was added to the
chamber below the insert, DMEM without FasL was used as the
control. Following incubation at 37°C for 24 h, the inserts were
taken out and washed twice with PBS. A cotton swab was used to
remove the membrane matrix and the cells on the apical side of the
insert. Cells on the basal side of the insert were fixed using
formaldehyde for 30 min at room temperature and subsequently
stained with crystal violet for 20 min at room temperature. Cells
were washed with water 3–5 times. The mean cell count of five
randomly selected microscopic fields using a light microscope was
used for each sample. For each group, 3 repeats were carried out.
The relative invasion rate (%) was calculated using the following
formula: (The number of invasive cells in the treated sample/the
number of invasive cells in the control) ×100%.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
WT, NC and KD cells were inoculated in 6-well plates
at 5×105 cells/well. Following 24 h incubation, each
group of cells (WT, NC and KD) were incubated either with DMEM
containing FasL (10 ng/ml) or DMEM alone. After 48 h, total RNA was
extracted from each group using TRIzol and reverse transcribed into
cDNA using the Prime Script RT reagent kit following the
manufacturers' protocol. qPCR was performed using the SYBR Premix
Ex Taq (Takara Bio, Inc.). The primer sequences used were as
follows: DcR3, forward, 5′-GGTACCAGGAGCTGAGGAGTGT'-3 and reverse,
5′-CCTTGGTGTCGGACCCCA-3′; MMP 9, forward,
5′-TGTACCGCTATGGTTACACTCG-3′ and reverse,
5′-GGCAGGGACAGTTGCTTCT-3′; VEGF-C, forward,
5′-GGCTGGCAACATAACAGAGAA-3′ and reverse,
5′-CCCCACATCTATACACACCTCC-3′; VEGF-D, forward,
5′-TCCCATCGGTCCACTAGGTTT-3′ and reverse,
5′-AGGGCTGCACTGAGTTCTTTG-3′; GAPDH (the internal control), forward,
5′-GTCGGAGTCAACGGATTTGG-3′ and reverse,
5′-AAAAGCAGCCCTGGTGACC-3′.
The qPCR conditions were: Initial denaturation at
94°C 2 min, 30 cycles of denaturation at 94°C for 30 sec, annealing
at 55°C for 30 sec and elongation at 72°C for 60 sec; and final
extension at 72°C for 10 min. Relative mRNA expression was
calculated using the 2−ΔΔCq method (30).
Western blot analysis
Each group of cells was washed in PBS at 4°C and
then radioimmuniprecipitation assay lysis buffer (Nanjing KeyGen
Biotech Co., Ltd., Nanjing, China) containing phenylmethylsulfonyl
fluoride was added for 5 min. Lysates were centrifuged at 12,000 ×
g for 5 min at 4°C. The protein concentration in the supernatant
was quantified using a BCA kit. Protein samples (30 µg) were
separated using 12% SDS-PAGE and then transferred to nitrocellulose
membranes. Following blocking with 5% skimmed milk for 1 h at room
temperature, rabbit anti-human MMP9 polyclonal antibody (1:1,000),
rabbit anti-human VEGF-C polyclonal antibody (1:1,000), rabbit
anti-human VEGF-D monoclonal antibody (1:1,000), rabbit anti-human
GAPDH antibody (1:5,000) were added and membranes were incubated at
4°C overnight. Following washing with TBST, horseradish
peroxidase-labeled goat anti-rabbit secondary antibody (1:5,000)
was added and incubated at room temperature for 1 h. Enhanced
chemiluminescent agent (EMD Millipore, Billerica, MA, USA) was
added following washing with TBST. A Bio-Rad gel imaging system
(BioradGelDoc XR; Bio-Rad Laboratories, Inc.) was used to capture
the images, and GAPDH was used as the internal control for the
analysis of DcR3, MMP9, VEGF-C, and VEGF-D.
Statistical analysis
Values are expressed as the mean ± standard
deviation. All statistical analyses were performed using GraphPad
Prism software version 6.0 (GraphPad Software, Inc., La Jolla, CA,
USA). The statistical significance among groups was determined by
two-tailed analysis of variance followed by Dunnett's multiple
comparisons test. P<0.05 was considered to indicate a
significant difference. All experiments were performed in
triplicate and repeated at least three times.
Results
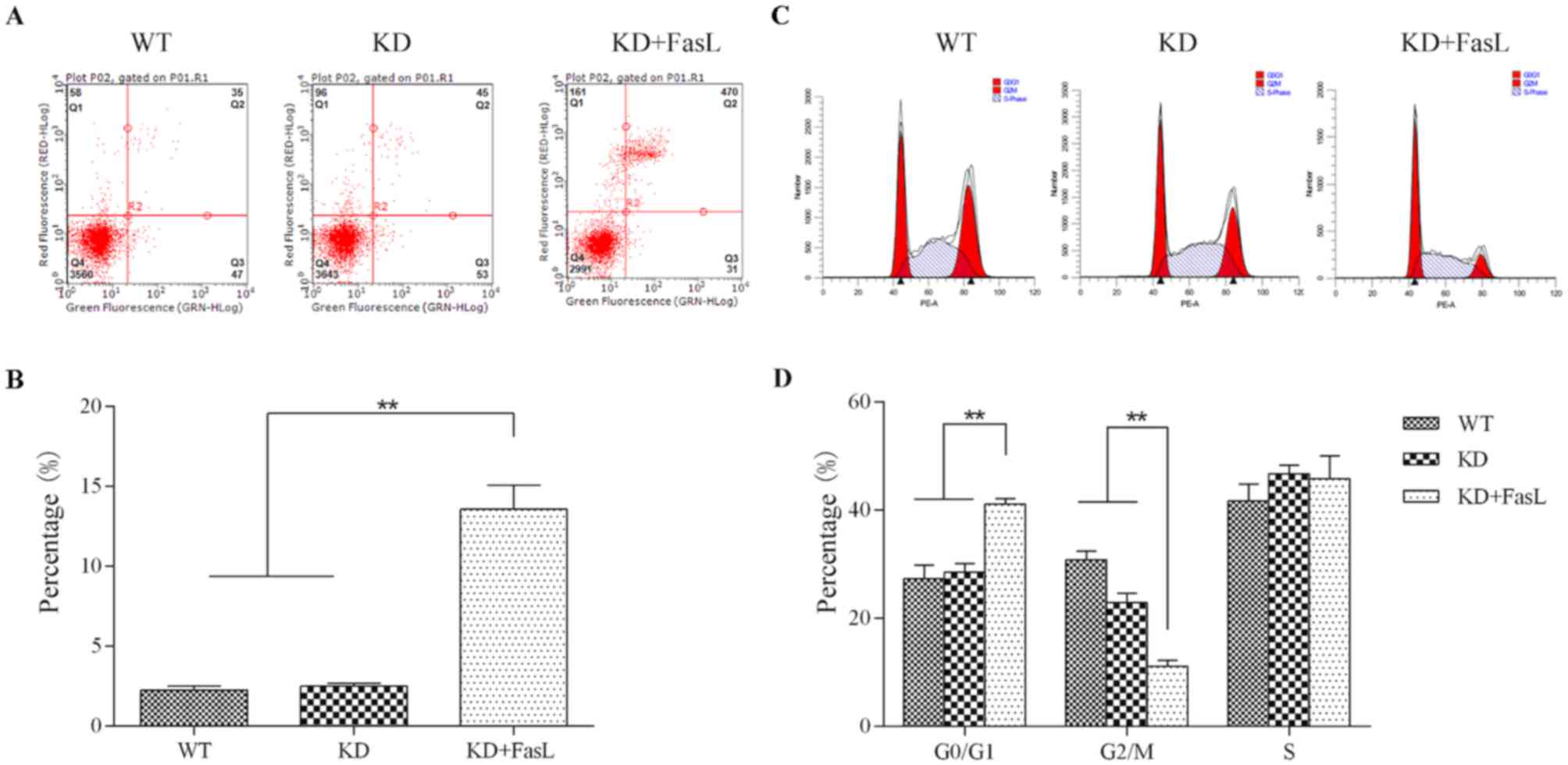
Following DcR3 knockdown, HepG2 cells
became more sensitive to FasL-induced apoptosis
Flow cytometric analysis indicated that, following
treatment with FasL, the proportion of KD cells exhibiting signs of
early apoptosis was 12.29±1.12%, which was significantly higher
than that of the KD (1.12±0.10%) and WT (0.99±0.10%) groups
(P<0.01; Fig. 1A and B).
Cell cycle analysis by flow cytometry indicated that
the proportion of cells in the KD + FasL group in the
G0/G1 phase (41.12±1.02%) was significantly
greater than that of the KD (28.59±1.51%) and WT (27.34±2.52%)
groups (P<0.01; Fig. 1C and D).
Furthermore, the proportion of cells in the G2/M phase in the KD +
FasL group (11.11±1.10%) was significantly lower than that of the
KD (22.98±1.71%) and the WT (30.84 ± 1.61%)groups (P<0.01).
Following DcR3 knockdown, FasL
decreases the proliferation, migration and invasion of HepG2
cells
The results of the MTS assay demonstrated that,
compared with the WT and NC groups, the proliferation rates of
cells in the KD, WT + FasL, NC + FasL and WT + FasL groups from day
3 onwards were significantly decreased compared with the WT group
(P<0.05 or P<0.01; Fig. 2A).
The proliferation rate of cells in the KD + FasL group was
significantly lower. These results indicate that FasL decreases
cell proliferation and that this impact is enhanced following DcR3
knockdown.
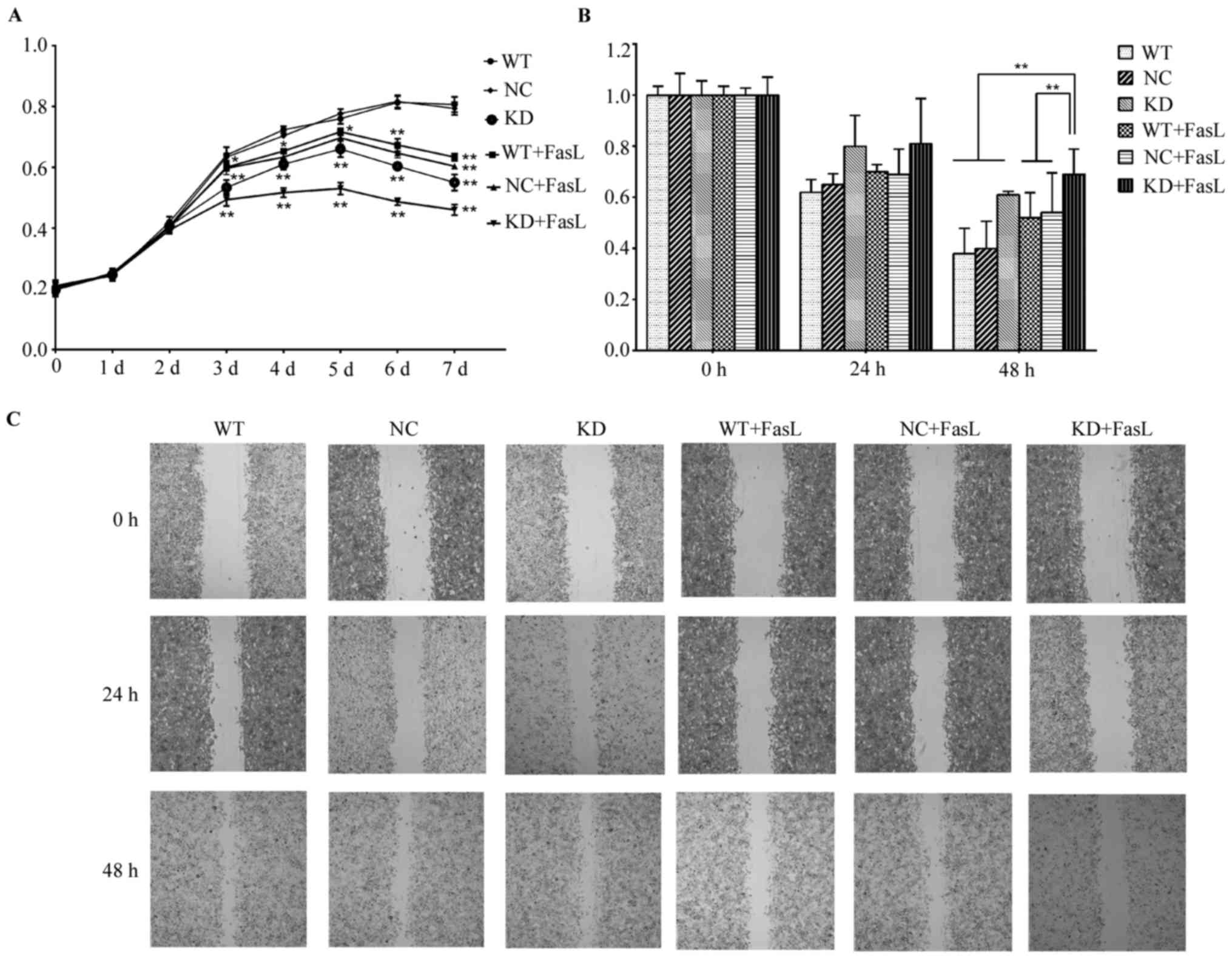 | Figure 2.Effects of FasL on the activity and
migration of HepG2 cells. (A) The results of the MTS assay. From
Day 3, the OD values of the KD, WT + FasL, NC + FasL and KD + FasL
groups were lower than the WT group, respectively. The OD value in
the KD + FasL group was the lowest. (B and C) The results of the
wound healing assay indicated that following the addition of FasL,
cell migration was reduced in all treatment groups; the decrease in
the KD cells was the most significant. *P<0.05, **P<0.01. OD,
optical density; FasL, Fas ligand; KD, DcR3 knockdown HepG2 cells;
WT, wild-type HepG2 cells; NC, negative control HepG2 cells; DcR3,
Fas and decoy receptor 3. |
The results of the wound healing assay indicated
that FasL reduced cell migration, with the most significant
decrease identified in the KD + FasL group. Cell migration was
significantly decreased in the KD + FasL group compared with all
other groups at 48 h (P<0.01; Fig. 2B
and C).
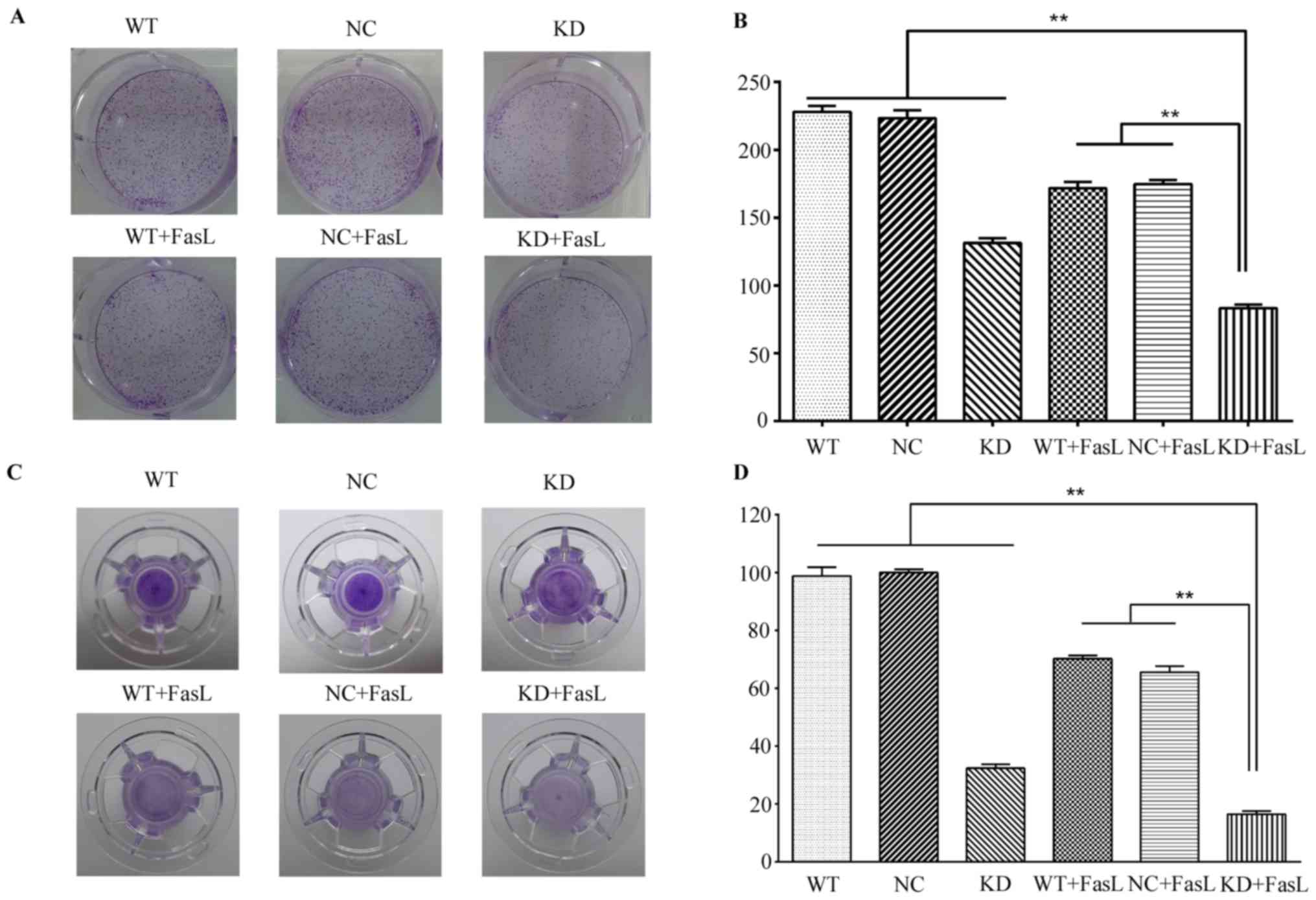
The results of the clonogenic assay demonstrated
that compared with the WT group, the number of clones in the KD, WT
+ FasL, NC + FasL and KD + FasL groups all decreased (Fig. 3A and B). The greatest decrease was
found in the KD + FasL group; the number of clones was
significantly decreased in this group compared with all other
groups, (P<0.01). These results indicate following DcR3
silencing, treatment with FasL further decreases HepG2 cell
proliferation.
Furthermore, the results of the Transwell cell
migration assay demonstrated that the number of cells in the KD +
FasL group passing through the Matrigel membrane was significantly
decreased compared with all other groups (P<0.01; Fig. 3C and D). These results suggest that
FasL decreases the invasiveness of HepG2 cells.
Following DcR3 knockdown, the
expression of VEGF-C, VEGF-D and MMP9 decreases following treatment
of HepG2 cells with FasL
The results of RT-qPCR demonstrated that, compared
with the other groups, the expression of VEGF-C, VEGF-D and MMP 9
mRNA in the KD + FasL group was significantly decreased (P<0.01;
Fig. 4). The same trend was observed
regarding the expression of VEGF-C, VEGF-D and MMP9 protein
following western blotting.
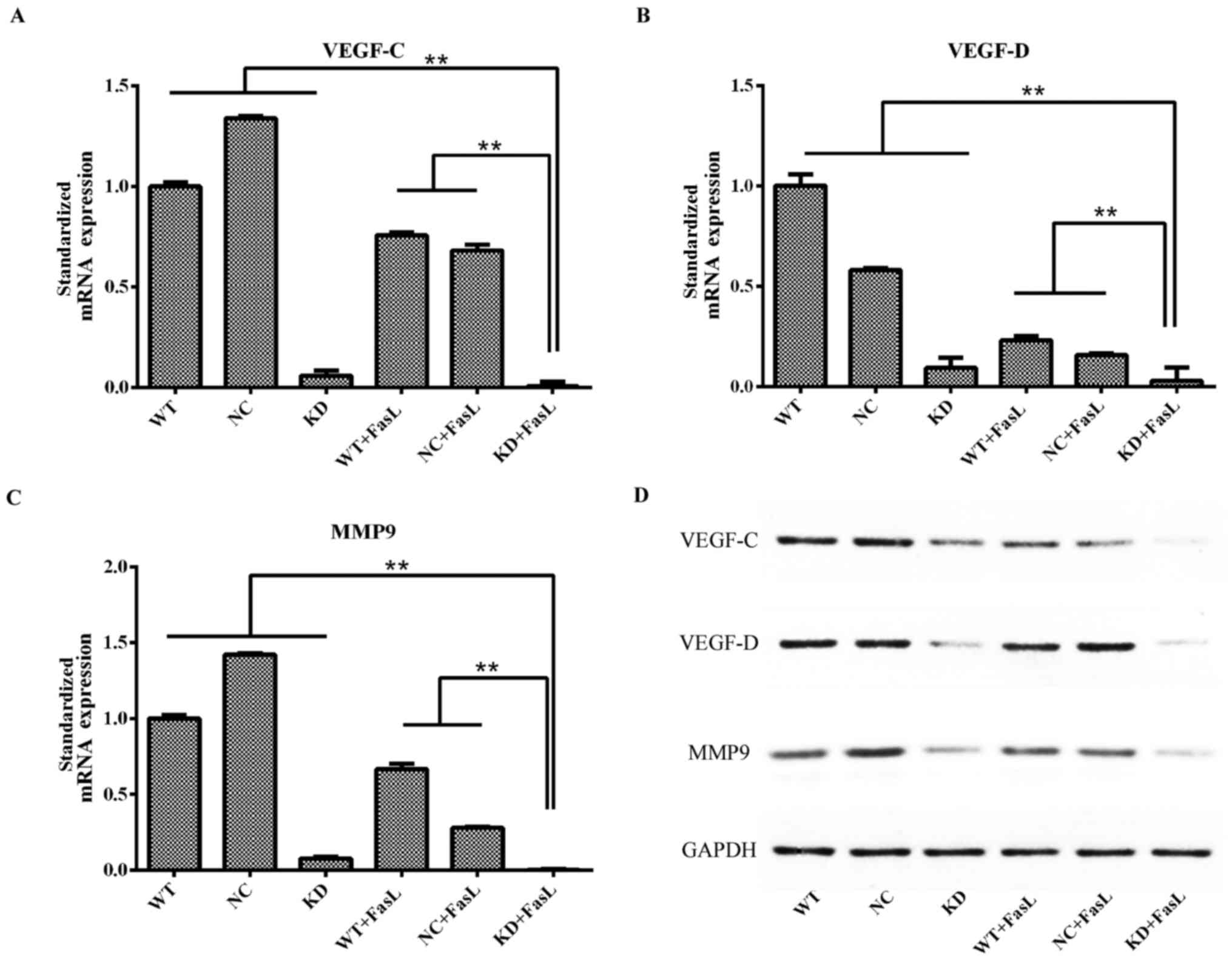 | Figure 4.Changes in the expression of (A)
VEGF-C, (B) VEGF-D and (C) MMP9 mRNA in HepG2 cells following
exposure to FasL. **P<0.01. (D) Results of western blotting
presenting the expression of VEGF-C, VEGF-D and MMP9 protein in
each of the groups. MMP9, matrix metallopeptidase 9, VEGF-C,
vascular endothelial growth factor C, VEGF-D, vascular endothelial
growth factor D; FasL, Fas ligand; WT, wild-type HepG2 cells; KD,
DcR3 knockdown HepG2 cells; NC, negative control HepG2 cells; DcR3,
Fas and decoy receptor 3. |
Discussion
The Fas/FasL system is an apoptotic pathway that
serves an important role in maintaining immune homeostasis
(31) and tumorigenesis (32). Under normal circumstances, Fas is
highly expressed in various cells and the development of tumors is
often associated with a lack of Fas expression or function, and the
increased expression of FasL in tumor cells (33). It has been established that Fas/FasL
not only induces apoptosis, but also triggers multiple signaling
pathways to inhibit apoptosis; furthermore, its abnormal expression
is closely associated with tumorigenesis (34). It may stimulate tumor cell
proliferation and promote tumor cell migration and invasion via the
nuclear factor-κB, mitogen-activated protein kinase and
phosphatidylinositol 3-kinase pathways (35). FasL contains two receptors, Fas and
DcR3. DcR3 competitively binds to FasL over Fas, thereby inhibiting
FasL-induced apoptosis.
The DcR3 gene was identified in 1998 by Pitti et
al (19) and is closely
associated with tumorigenesis. In the majority of tissues, the
expression of DcR3 is negligible; however, it is highly expressed
in many different types of cancer, including esophageal (19), gastric (19), colon (19), rectal (19), pancreatic (23), liver (23–25),
lung (19,23), kidney (36), breast (23), ovarian (37) and skin cancer (38).
Connolly et al (21) intravenously injected FasL to Fas+
mice and demonstrated that the mice succumbed to acute fatal
fulminant hepatic necrosis in ≤2 h; however, this did not occur in
mice pre-treated with DcR3. It was therefore hypothesized that DcR3
reduces Fas-mediated liver cell necrosis. Hayashi et al
(39) demonstrated that
downregulating DcR3 expression using siRNA may increase
Fas-mediated apoptosis and using TNF-α to increase the expression
of DcR3 could inhibit Fas-mediated apoptosis in rheumatoid
arthritis synovial fibroblasts. Li et al (40) indicated that DcR3 expression in the
human gastric cancer BGC823 and breast cancer MCF-7 cell lines were
35.3 and 21.6%, whereas the apoptosis induced by FasL was 15.6 and
58.2%, respectively. Therefore, it was suggested that FasL-mediated
apoptosis was directly associated with levels of DcR3 expression
(40). The results of the current
study demonstrated that FasL-induced apoptosis was increased when
DcR3 in HepG2 cells was silenced, which is in accordance with the
aforementioned experimental results. Furthermore, FasL
significantly decreased the proliferation and invasiveness of KD
cells. These results indicate that FasL may impair the
proliferation and invasiveness of HepG2 cells more effectively when
DcR3 expression is decreased.
Tumor invasion and metastasis is a complex
multi-step process. When leaving the primary tumor and moving in
and out of blood vessels, cancer cells must initially break through
the extracellular matrix and basement membrane barrier to
infiltrate the adjacent fibrous connective tissue before moving to
more distant sites (41). The
degradation of the extracellular matrix and the basement membrane
is generally conductedby MMPs, hyaluronidase, elastase, cathepsin
and large numbers of macrophages (42). MMPs are proteolytic enzymes that are
closely associated with tumor invasion and metastasis (43). They are directly involved in tissue
damage, induce VEGF and stimulate endothelial cells to synthesize
proteases that lead to endothelial cell proliferation and migration
(44). MMPs are considered to be
important inducers and regulators of angiogenesis (45). MMP9 is a type IV collagenase that
acts as a protease in tumor invasion and metastasis primarily by
degrading collagen type III and IV in the extracellular matrix and
the basement membrane of blood vessel walls (46). MMP9 is also involved in the formation
of blood vessels in tumors (47).
The increased expression of MMP9 is associated with tumor
malignancy and prognosis (48,49) and
it has been demonstrated that disease-free and 5-year survival
rates are significantly decreased in patients with gliomas that are
MMP9 positive compared with those that are MMP9 negative. In the
present study, the results of RT-qPCR and western blotting
demonstrated that the expression of MMP9 in DcR3-knockdown HepG2
cells treated with FasL was significantly lower than in normal and
FasL-treated normal HepG2 cells, indicating that FasL may reduce
the expression of MMP9 and inhibit tumor angiogenesis invasion and
metastasis in HepG2 cells following DcR3 silencing.
There have been few studies investigating tumor
lymphangiogenesis; attention has focused on angiogenesis, due to
its importance during the tumorigenesis of solid tumors (50–52). In
fact, due to their structure and function, lymphatic vessels are
not only involved in maintaining tumor growth, but are also the
major cause of metastasis indifferent types of cancer and are key
factors determining cancer stage and patient prognosis (53). Lymph node metastasis is commonly used
as a guide for treatment selection (54). The liver is an organ containing
abundant blood vessels and liver cancer tissues exhibit a large
amount of lymphangiogenesis that is closely associated with liver
cancer cell migration and invasion (55).
Tumor lymphangiogenesis is extremely complex. Tumor
cells, stromal cells, macrophages and activated platelets all can
secrete a variety of lymphatic growth factors that induce the
formation of new tumor lymphatic vessels and promote tumor
lymphatic metastasis (56). The
dimeric glycoproteins VEGF-C and VEGF-D, are the most well-studied
lymphatic endothelial growth factors. VEGF-C was the first
lymphatic endothelial growth factor identified to serve a role in
promoting lymphangiogenesis (57).
VEGF-D was identified in 1998 and was determined to have a similar
structure, function and common specific receptor to VEGF-C
(58). It has been demonstrated that
VEGF receptor 3 (VEGFR3) has the highest affinity in binding to
VEGF-C/D (59). VEGFR3 is primarily
expressed in embryonic and adult lymphatic endothelial cells and is
an important signal that mediates lymphangiogenesis. Following the
binding of VEGFR-3 to VEGF-C/D, lymphatic endothelial cell
proliferation and migration is stimulated, tumor lymphangiogenesis
is promoted, tumor cells move into lymphatic vessels and cancer
metastasis occurs via the lymphatic system (60–62). It
has been demonstrated that VEGF-C exhibits the strongest promoting
effect on lymphangiogenesis (63).
It is highly expressed in lung, breast, stomach and colon cancer
tumors and is closely associated with tumor lymphoid metastasis
(64). Tumor cells are able to
secrete large amount of growth factors to promote angiogenesis,
providing essential nutrients to the tumor tissue, promoting tumor
lymphangiogenesis and mediating tumor invasion. It has been
reported that tumor angiogenesis is associated with VEGF-A/VEGFR-2
and that lymphangiogenesis is primarily regulated by VEGF-C, VEGF-D
and VEGFR-3 (65). Under normal
physiological conditions, VEGF-C and VEGF-D are able to mediate
endothelial cell mitosis, cell migration and apoptosis (66). However, when cells are in abnormal
states of proliferation and activation (i.e., tumorigenesis), tumor
cells stimulate the surrounding tissues and induce
lymphangiogenesis via autocrine and paracrine signaling. Growth of
the newly formed lymphatic vessels is promoted, as is tumor
lymphatic metastasis (67). The
results of the current study demonstrated that the expression of
VEGF-C and VEGF-D in DcR3-silenced cells following treatment with
FasL was significantly decreased compared with normal and
FasL-treated normal cells, indicating that FasL reduces the
expression of VEGF-C and VEGF-D following the downregulation of
DcR3 in vitro, thus impairing the ability of
lymphangiogenesis and lymph node metastasis of HepG2 cells.
In conclusion, the results of the current study
indicate that FasL increases the apoptosis of HepG2 cells and
significantly decreases the proliferation and invasion of HepG2
cells following DcR3 silencing. This may be due to the
downregulation of MMP9, VEGF-C, VEGF-Dexpression; however, its
exact mechanism of action remains unclear and further studies are
required. The results of the present study suggest that DcR3
silencing combined with FasL treatment may be a novel method of
treating patients with liver cancer, particularly those with
hepatoblastoma.
Acknowledgements
Not applicable.
Funding
The present study was funded by the National Natural
Science Foundation of China (Grant no. 81550033).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
TZ designed the study, performed experiments,
analyzed the data and wrote the paper. YX, SR and XZ performed
experiments. CL analyzed data. JW initiated, organized and designed
the study, and wrote the paper
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ferlay J, Soerjomataram I, Dikshit R, Eser
S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F: Cancer
incidence and mortality worldwide: Sources, methods and major
patterns in GLOBOCAN 2012. Int J Cancer. 136:E359–E386. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Altekruse S, McGlynn K and Reichman M:
Hepatocellular carcinoma incidence, mortality, and survival trends
in the United States from 1975 to 2005. J Clin Oncol. 27:1485–1491.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Forner A, Llovet J and Bruix J:
Hepatocellular carcinoma. Lancet. 379:1245–1255. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Xu X, Chen W, Miao R, Zhou YY, Wang ZX,
Zhang LQ, Qu K, Pang Q, Wang RT and Liu C: Survival analysis of
hepatocellular carcinoma: A comparison between young patients and
aged patients. Chin Med J (Engl). 128:1793–1800. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Darbari A, Sabin KM, Shapiro CN and
Schwarz KB: Epidemiology of primary hepatic malignancies in U.S.
children. Hepatology. 38:560–566. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Meyers RL, Rowland JR, Krailo M, Chen Z,
Katzenstein HM and Malogolowkin MH: Predictive power of
pretreatment prognostic factors in children with hepatoblastoma: A
report from the Children's Oncology Group. Pediatr Blood Cancer.
53:1016–1022. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Watanabe K: Current chemotherapeutic
approaches for hepatoblastoma. Int J Clin Oncol. 18:955–961. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zsiros J, Brugieres L, Brock P, Roebuck D,
Maibach R, Zimmermann A, Childs M, Pariente D, Laithier V, Otte JB,
et al: Dose-dense cisplatin-based chemotherapy and surgery for
children with high-risk hepatoblastoma (SIOPEL-4): A prospective,
single-arm, feasibility study. Lancet Oncol. 14:834–842. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bell D, Ranganathan S, Tao J and Monga SP:
Novel advances in understanding of molecular pathogenesis of
hepatoblastoma: A Wnt/β-catenin perspective. Gene Expr. 17:141–154.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ganju A, Khan S, Hafeez BB, Behrman SW,
Yallapu MM, Chauhan SC and Jaggi M: miRNA nanotherapeutics for
cancer. Drug Discov Today. 22:424–432. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang Z, Wang N, Liu P and Xie X: AMPK and
cancer. EXS. 107:203–226. 2016.PubMed/NCBI
|
|
12
|
De la Rosa AJ, Gomez MA, Morales S,
Padillo FJ and Muntane J: CD95 signaling in cancer treatment. Curr
Pharm Des. 20:2809–2818. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ogasawara J, Watanabe-Fukunaga R, Adachi
M, Matsuzawa A, Kasugai T, Kitamura Y, Itoh N, Suda T and Nagata S:
Lethal effect of the anti-Fas antibody in mice. Nature.
364:806–809. 1993. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Suda T, Takahashi T, Golstein P and Nagata
S: Molecular cloning and expression of the Fas ligand, a novel
member of the tumor necrosis factor family. Cell. 75:1169–1178.
1993. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mor G, Gutierrez L, Eliza M, Kahyaoglu F
and Arici A: Fas-fas ligand system-induced apoptosis in human
placenta and gestational trophoblastic disease. Am J Reprod
Immunol. 40:89–94. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Müschen M, Warskulat U and Beckmann M:
Defining CD95 as a tumor suppressor gene. J Mol Med (Berl).
78:312–325. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kim SK, Yoon YD, Park YS, Seo JT and Kim
JH: Involvement of the Fas-Fas ligand system and active caspase-3
in abnormal apoptosis in human testes with maturation arrest and
Sertoli cell-only syndrome. Fertil Steril. 87:547–553. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lau HT, Yu M, Fontana A and Stoeckert C
Jr: Prevention of islet allograft rejection with engineered
myoblasts expressing FasL in mice. Science. 273:109–112. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Pitti RM, Marsters SA, Lawrence DA, Roy M,
Kischkel FC, Dowd P, Huang A, Donahue CJ, Sherwood SW, Baldwin DT,
et al: Genomic amplification of a decoy receptor for Fas ligand in
lung and colon cancer. Nature. 396:699–703. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liu YQ, Mu ZQ, You S, Tashiro S, Onodera S
and Ikejima T: Fas/FasL signaling allows extracelluar-signal
regulated kinase to regulate cytochrome c release in
oridonin-induced apoptotic U937 cells. Biol Pharm Bull.
29:1873–1879. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Connolly K, Cho YH, Duan R, Fikes J,
Gregorio T, LaFleur DW, Okoye Z, Salcedo TW, Santiago G, Ullrich S,
et al: In vivo inhibition of Fas ligand-mediated killing by TR6, a
Fas ligand decoy receptor. J Pharmacol Exp Ther. 298:25–33.
2001.PubMed/NCBI
|
|
22
|
Ge Z, Sanders AJ, Ye L and Jiang WG:
Aberrant expression and function of death receptor-3 and death
decoy receptor-3 in human cancer. Exp Ther Med. 2:167–172. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wu Y, Han B, Sheng H, Lin M, Moore PA,
Zhang J and Wu J: Clinical significance of detecting elevated serum
DcR3/TR6/M68 in malignant tumor patients. Int J Cancer.
105:724–732. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Shen HW, Gao SL, Wu YL and Peng SY:
Overexpression of decoy receptor 3 in hepatocellular carcinoma and
its association with resistance to Fas ligand-mediated apoptosis.
World J Gastroenterol. 11:5926–5930. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chen G and Luo D: Expression of decoy
receptor 3 in liver tissue microarrays. Nat Med J India.
21:275–278. 2008.
|
|
26
|
Yu W, Xu YC, Tao Y, He P, Li Y, Wu T, Zhu
YP, Li J, Wu JX and Dai J: DcR3 regulates the growth and metastatic
potential of SW480 colon cancer cells. Oncol Rep. 30:2741–2748.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhou XN, Li GM, Xu YC, Zhao TJ and Wu JX:
Knockdown of decoy receptor 3 impairs growth and invasiveness of
hepatocellular carcinoma cell line of HepG2. Chin Med J (Engl).
129:2623–2629. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Aden DP, Fogel A, Plotkin S, Damjanov I
and Knowles BB: Controlled synthesis of HBsAg in a differentiated
human liver carcinoma-derived cell line. Nature. 282:615–616. 1979.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
López-Terrada D, Cheung SW, Finegold MJ
and Knowles BB: Hep G2 is a hepatoblastoma-derived cell line. Hum
Pathol. 40:1512–1515. 2009. View Article : Google Scholar
|
|
30
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Strasser A, Jost PJ and Nagata S: The many
roles of FAS receptor signaling in the immune system. Immunity.
30:180–192. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chen L, Park SM, Tumanov AV, Hau A, Sawada
K, Feig C, Turner JR, Fu YX, Romero IL, Lengyel E and Peter ME:
CD95 promotes tumour growth. Nature. 465:492–496. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Barnhart BC, Legembre P, Pietras E, Bubici
C, Franzoso G and Peter ME: CD95 ligand induces motility and
invasiveness of apoptosis-resistant tumor cells. EMBO J.
23:3175–3185. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
O'Reilly LA, Tai L, Lee L, Kruse EA,
Grabow S, Fairlie WD, Haynes NM, Tarlinton DM, Zhang JG, Belz GT,
et al: Membrane-bound Fas ligand only is essential for Fas-induced
apoptosis. Nature. 461:659–663. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wisniewski P, Ellert-Miklaszewska A,
Kwiatkowska A and Kaminska B: Non-apoptotic Fas signaling regulates
invasiveness of glioma cells and modulates MMP-2 activity via
NFkappaB-TIMP-2 pathway. Cell Signal. 22:212–220. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Macher-Goeppinger S, Aulmann S, Wagener N,
Funke B, Tagscherer KE, Haferkamp A, Hohenfellner M, Kim S,
Autschbach F, Schirmacher P and Roth W: Decoy receptor 3 is a
prognostic factor in renal cell cancer. Neoplasia. 10:1049–1056.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Connor JP and Felder M: Ascites from
epithelial ovarian cancer contain high levels of functional decoy
receptor 3 (DcR3) and is associated with platinum resistance.
Gynecol Oncol. 111:330–335. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Maeda T, Hao C and Tron VA: Ultraviolet
light (UV) regulation of the TNF family decoy receptors DcR2 and
DcR3 in human keratinocytes. J Cutan Med Surg. 5:294–298. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Hayashi S, Miura Y, Nishiyama T, Mitani M,
Tateishi K, Sakai Y, Hashiramoto A, Kurosaka M, Shiozawa S and
Doita M: Decoy receptor 3 expressed in rheumatoid synovial
fibroblasts protects the cells against fas-induced apoptosis.
Arthritis Rheum. 56:1067–1075. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Li W, Zhang C, Chen C and Zhuang G:
Correlation between expression of DcR3 on tumor cells and
sensitivity to FasL. Cell Mol Immunol. 4:455–460. 2007.PubMed/NCBI
|
|
41
|
Clark AG and Vignjevic DM: Modes of cancer
cell invasion and the role of the microenvironment. Curr Opin Cell
Biol. 36:13–22. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Laurenzana A, Fibbi G, Margheri F,
Biagioni A, Luciani C, Del Rosso M and Chillà A: Endothelial
progenitor cells in sprouting angiogenesis: Proteases pave the way.
Curr Mol Med. 15:606–620. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Jacob A and Prekeris R: The regulation of
MMP targeting to invadopodia during cancer metastasis. Front Cell
Dev Biol. 3:42015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Kessenbrock K, Wang CY and Werb Z: Matrix
metalloproteinases in stem cell regulation and cancer. Matrix Biol.
44–46:184–190. 2015. View Article : Google Scholar
|
|
45
|
DeClerck YA, Mercurio AM, Stack MS,
Chapman HA, Zutter MM, Muschel RJ, Raz A, Matrisian LM, Sloane BF,
Noel A, et al: Proteases, extracellular matrix, and cancer: A
workshop of the path B study section. Am J Pathol. 164:1131–1139.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Banday MZ, Sameer AS, Mir AH, Mokhdomi TA,
Chowdri NA and Haq E: Matrix metalloproteinase (MMP) −2, −7 and −9
promoter polymorphisms in colorectal cancer in ethnic Kashmiri
population-A case-control study and a mini review. Gene. 589:81–89.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Verma S, Kesh K, Gupta A and Swarnakar S:
An overview of matrix metalloproteinase 9 polymorphism and gastric
cancer risk. Asian Pac J Cancer Prev. 16:7393–7400. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Komatsu K, Nakanishi Y, Nemoto N, Hori T,
Sawada T and Kobayashi M: Expression and quantitative analysis of
matrix metalloproteinase-2 and-9 in human gliomas. Brain Tumor
Pathol. 21:105–112. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Visse R and Nagase H: Matrix
metalloproteinases and tissue inhibitors of metalloproteinases:
Structure, function, and biochemistry. Circ Res. 92:827–839. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Ji RC: Lymph nodes and cancer metastasis:
New perspectives on the role of intranodal lymphatic sinuses. Int J
Mol Sci. 18:E512016. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Paduch R: The role of lymphangiogenesis
and angiogenesis in tumor metastasis. Cell Oncol (Dordr).
39:397–410. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Dieterich LC and Detmar M: Tumor
lymphangiogenesis and new drug development. Adv Drug Deliv Rev.
99:148–160. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Fink DM, Steele MM and Hollingsworth MA:
The lymphatic system and pancreatic cancer. Cancer Lett.
381:217–236. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Christiansen A and Detmar M:
Lymphangiogenesis and cancer. Genes Cancer. 2:1146–1158. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Tanaka M and Iwakiri Y: The hepatic
lymphatic vascular system: Structure, function, markers, and
lymphangiogenesis. Cell Mol Gastroenterol Hepatol. 2:733–749. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Li S and Li Q: Cancer stem cells,
lymphangiogenesis, and lymphatic metastasis. Cancer Lett.
357:438–447. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Stacker SA and Achen MG: Emerging roles
for VEGF-D in human disease. Biomolecules. 8:E12018. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Heckman CA, Holopainen T, Wirzenius M,
Keskitalo S, Jeltsch M, Ylä-Herttuala S, Wedge SR, Jürgensmeier JM
and Alitalo K: The tyrosine kinase inhibitor cediranib blocks
ligand-induced vascular endothelial growth factor receptor-3
activity and lymphangiogenesis. Cancer Res. 68:4754–4762. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Wang P and Cheng Y: Gene expression
profile of lymphatic endothelial cells. Cell Biol Int.
35:1177–1187. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Hu X, Xing L, Wei X, Liu X, Pang R, Qi L
and Song S: Nonangiogenic function of VEGF and enhanced
radiosensitivity of HeLa cells by inhibition of VEGF expression.
Oncol Res. 20:93–101. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Gadducci A, Guerrieri ME and Greco C:
Tissue biomarkers as prognostic variables of cervical cancer. Crit
Rev Oncol Hematol. 86:104–129. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Watanabe S, Kato M, Kotani I, Ryoke K and
Hayashi K: Lymphatic vessel density and vascular endothelial growth
factor expression in squamous cell carcinomas of lip and oral
cavity: A clinicopathological analysis with immunohistochemistry
using antibodies to D2-40, VEGF-C and VEGF-D. Yonago Acta Med.
56:29–37. 2013.PubMed/NCBI
|
|
63
|
Biedka M, Makarewicz R, Kopczyńska E,
Marszałek A, Goralewska A and Kardymowicz H: Angiogenesis and
lymphangiogenesis as prognostic factors after therapy in patients
with cervical cancer. Contemp Oncol (Pozn). 16:6–11.
2012.PubMed/NCBI
|
|
64
|
Wang J, Huang Y, Zhang J, Wei Y, Mahoud S,
Bakheet AM, Wang L, Zhou S and Tang J: Pathway-related molecules of
VEGFC/D-VEGFR3/NRP2 axis in tumor lymphangiogenesis and lymphatic
metastasis. Clin Chim Acta. 461:165–171. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Zhang S, Yu H and Zhang L: Role of
vascular endothelial growth factor receptor-3/Flt-4 in early-stage
cervical cancer. Oncol Lett. 1:453–456. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Bernaudin JF, Kambouchner M and Lacave R:
Lymphatic vascular system, development and lymph formation. Review.
Rev Pneumol Clin. 69:93–101. 2013.(In French). View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Zhu X, Er K, Mao C, Yan Q, Xu H, Zhang Y,
Zhu J, Cui F, Zhao W and Shi H: miR-203 suppresses tumor growth and
angiogenesis by targeting VEGFA in cervical cancer. Cell Physiol
Biochem. 32:64–73. 2013. View Article : Google Scholar : PubMed/NCBI
|