Introduction
An abdominal aortic aneurysm (AAA) occurs if the
abdominal aorta becomes focally enlarged, resulting from a weakened
abdominal aortic wall. The prevalence of AAA has continued to
increase worldwide over the last four decades. Data from clinical
investigations involving different populations demonstrate that the
prevalence is 2.4–16.9% in male and 0.5–2.2% in female patients
older than 65 years of age (1). AAA
usually develops asymptomatically until the outbreak of acute
aneurysm rupture, which can cause severe internal bleeding and
accounts for a mortality of 85–90% (2). The pathogenesis of AAA initiation,
progression, and ultimate rupture has been rarely elucidated by
previous studies. Dilatation and weakening of the aorta is
accompanied by other phenotypic changes including local
inflammation, smooth muscle cell apoptosis, oxidation stress
increase and, in particularly, dramatic extracellular matrix (ECM)
degradation (3).
MicroRNAs (miRNAs) are a class of endogenous
non-coding RNAs encompassing 18–23 nucleotides that regulate the
expression of mRNAs by targeting their 3′-untranslated regions
(3′-UTRs) and inhibiting translation (4). Recently, miRNAs have been deemed as
vital modulators of diverse cell events and have been implicated in
a wide array of pathologies, such as cardiovascular disorders
(5). Expression alterations in
miRNAs have been revealed to impact the vascular angiogenesis,
inflammation, and remodeling (6,7). As a
member of the miR-29 family, microRNA-29b (miR-29b) has been
previously proven to be remarkably upregulated in patients with AAA
in comparison to the normal population (8). The miR-29 family can accelerate
fibrosis in various tissues via regulating downstream ECM genes
(9–11), including multiple collagens isoforms
(COL1A1, COL5A1, COL3A1) and components of aortic wall, such as and
elastin (ELN) and fibrillin-1 (FBN1). Dysregulation of ECM
homeostasis is responsible for several pathological conditions,
such as fibrosis and cancerous invasion. In addition, the
aforementioned collagens that are widely expressed in the aortic
wall play a vital role in aneurysm formation (12–14).
Also, matrix metalloproteinases (MMPs, such as MMP2 and MMP9) have
been identified as direct target genes of miR-29b and relate to AAA
onset and progression (15,16). These data in aggregate suggest that
miR-29b may participate in AAA development via altering ECM
microenvironment and suppressing ECM protein expression.
Prostaglandins (PGs) are essentially a category of
fatty acids that can result in inflammation, pain, redness and
swelling. There are four major bioactive PGs that are ubiquitously
generated in vivo and function as lipid mediators in
autocrine and paracrine manner. Among them, prostaglandin E2
(PGE2) is one of the most abundant PGs synthesized in
the human body and possesses versatile physiological and/or
pathological functions. While the pro-inflammatory property of
PGE2 during acute inflammatory response is profoundly
established, increasing studies have been launched with regard to
its role in multiple vascular pathological conditions. For example,
PGE2 induces augmentation of arterial dilatation and
enhances microvascular permeability, thereby increasing blood flow
into the inflamed tissues (17). On
the other hand, PGE2 restrains the aortic smooth muscle
cell (ASMC) proliferation and decreases cytokine secretion in
vitro (18).
Prior studies have also shown that PGE2
is abundantly produced in the aneurysm wall, which may exert
inhibitory effects on collagen synthesis (19,20). In
addition, PGE2 is significantly implicated in vascular
wall remodeling via the regulation of MMP activities in human AAA
(21). It has been demonstrated that
the miR-29 family members were obviously upregulated in trabecular
meshwork cells by exogenous PGE2-evoked stimuli
(22). Fortunately we found that the
expression of miR-29b in the ASMCs was elevated on PGE2
treatment in our tentative trial, justifying the assumption that
PGE2 improves miR-29b-mediated ECM remodeling in AAA
development.
Materials and methods
Cell culture
The Ethics Committee of the Provincial Hospital
Affiliated to Shandong University approved the study (Jinan,
China). Human ASMCs (passage no. 3) propagated in growth media
SmGM-2 were both purchased from Lonza (Walkersville, MD, USA)
supplemented with 5% fetal bovine serum (FBS) following the
manufacturer's instructions. PGE2 and indomethacin were
purchased from Cayman Chemical (Ann Arbor, MI, USA). Cells were
treated with 500 ng/ml PGE2 or 10 mmol/l indomethacin,
with DMSO employed as a control. Cell containing plates were
harvested for RNA or protein analysis at ~90% confluence.
In particular, indomethacin solution was first
prepared by dropwise addition of 1 mol/l
Na2CO3 to the drug powder until dissolved,
and afterwards DMSO was added to make the solution concentration of
10.0 mmol/l, followed by sterile filtering.
Transfection of cultured cells
The ASMCs were transfected with miRNA-29b mimic,
inhibitor or Scr-miR (Dharmacon, Chicago, IL, USA) using
Lipofectamine 2000 (Invitrogen, Burlington, ON, Canada). miRNA
transfection efficiency was confirmed by RT-qPCR. Two hours after
transfection, cells were treated with PGE2 or
indomethacin for 24 h before they were harvested.
miRNA extraction and
quantification
miRNAs were extracted from cells using the mirVana
miRNA isolation kit (Ambion, Austin, TX, USA). Briefly, the cell
samples were collected and washed two times using PBS, prior to the
addition of miRNA additive (1:10) on ice for 15 min. The cell
lysate was added with equal volumes of acid-phenol:chloroform,
before centrifugation and removal of the aqueous phase, and then
the mixture was added 1.25-fold to 100% ethanol. The mixture was
passed through the filter cartridge and eluted. RT-qPCR was carried
out with a final reaction volume of 20 ml containing 10 ml TaqMan
Universal PCR Master Mix (Applied Biosystems; Thermo Fisher
Scientific Inc., Waltham, MA, USA), 8 ml DEPC-treated water, 1 ml
TaqMan microRNA assay (Applied Biosystems; Thermo Fisher Scientific
Inc.), and 1 ml RT product. The data were normalized to RNU6B small
nuclear RNA to calculate fold-changes using the method of ∆∆Cq.
Dual-luciferase reporter assay
Two online databases, miRBase and TargetScan, were
used to predict the potential binding sites for miR-29b. For
dual-luciferase reporter assays, the full-length 3′-UTR of COL1A1
containing three miR-29b binding sites was cloned into the
downstream of a pMIR-Report (Ambion) to generate pMir-COL1A1
3′-UTR, which was co-transfected with miR-29b mimics or Scr-miR
into ASMCs. The pRL-SV40 vector (Promega, Madison, WI, USA)
carrying the Renilla luciferase gene was used as an internal
control to normalize the transfection efficiency. Luciferase
activities were determined by using the Dual-Luciferase Reporter
assay system (Promega) following the manufacturer's instructions.
All the reactions were performed in triplicate.
mRNA quantification by RT-qPCR
Total RNA was isolated using TRIzol reagent
(Invitrogen, Carlsbad, CA, USA) following the manufacturer's
instructions. The total RNA 40 µg was used as template and then
reverse transcribed with M-MLV Reverse Transcriptase kit (Promega
Biotech Co., Ltd, Beijing, China) to synthesize the cDNA.
Expression levels of target genes were normalized to β-actin, the
internal positive control. RT-qPCR was carried out in a LightCycler
(Roche Diagnostics, Laval, QC, Canada) machine with the SYBR-Green
probe (SYBR Premix Ex Taq™ II; Takara, Dalian, China).
Melting-curve analysis was used to determine the melting
temperature (Tm) of specific amplification products and primer
dimers, which were used for the signal acquisition step (2–3°C
below Tm) for each gene. The comparative Cq (2-ΔΔCq) method was
introduced to account for the relative fold-changes of the amount
of template differences.
Protein extraction and western
blotting
The harvested cells were pelleted and resuspended in
RIPA lysis buffer, followed by incubating on ice for further lysing
and centrifuged at 15,000 × g for 5 min at 4°C. After
centrifugation, protein supernatant was kept at −80°C for future
analysis.
For immunoblotting, 30 µg of total protein was
separated on 10% SDS-PAGE and transferred to PVDF membrane at 250
mA for 1 h. The membrane was blocked with 5% silk milk and then
incubated overnight at 4°C with mouse monoclonal primary antibody
mouse monoclonal anti-COL1A1 (sc-293182; 1:1,000; Santa Cruz
Biotechnology, Inc., Santa Cruz, CA, USA), followed by subsequent
incubation with HRP-conjugated secondary goat anti-mouse polyclonal
IgG (SA132; 1:2,000; Beijing Solarbio Science and Technology Co.,
Ltd., Beijing, China) at room temperature for 1 h. Then, the
membrane was washed with PBST three times (5 min/time) before
photographed using ECL reagents (Millipore, Billerica, MA,
USA).
Soluble collagen assay
The total soluble collagen secreted from ASMCs was
evaluated following the manufacturer's instructions (QuickZyme
Biosciences, Leiden, Netherlands). In brief, after treatment with
PGE2 or indomethacin for 24 h, companied with
transfection with miR-29b mimics, inhibitor, or scr-miR,
conditioned culture medium was collected and centrifuged to remove
cell debris. Samples were incubated with Sirius Red color dye for
10 min at room temperature. After precipitation in a 96-well plate,
data analysis was performed on a microplate reader (Multiskan MK3;
Thermo Labsystems, Franklin, MA, USA) based on the absorbance at
540 nm. The experiment was performed in triplicate.
Statistical analysis
Data are presented as means ± SD. All in
vitro experiments included at least 3 replicates per group.
Groups were compared using the two-tailed Student's t-test for
parametric data. When comparing multiple groups, data were analyzed
by ANOVA with Bonferroni's post-hoc test. P<0.05 was considered
to indicate a statistically significant difference.
Results
miR-29b expression was significantly
increased on PGE2 treatment
In our earlier study, we found that upon
PGE2 incitement, miR-29b was significantly upregulated
in ASMCs, unusual undifferentiated muscle cell possessing the
capacity to produce ECM. In order to further confirm this, ASMCs
were treated with 500 ng/ml PGE2 or 10 mmol/l
indomethacin, a non-steroidal anti-inflammatory drug (NSAID), in
prior to the evaluation of miR-29b expression in vitro
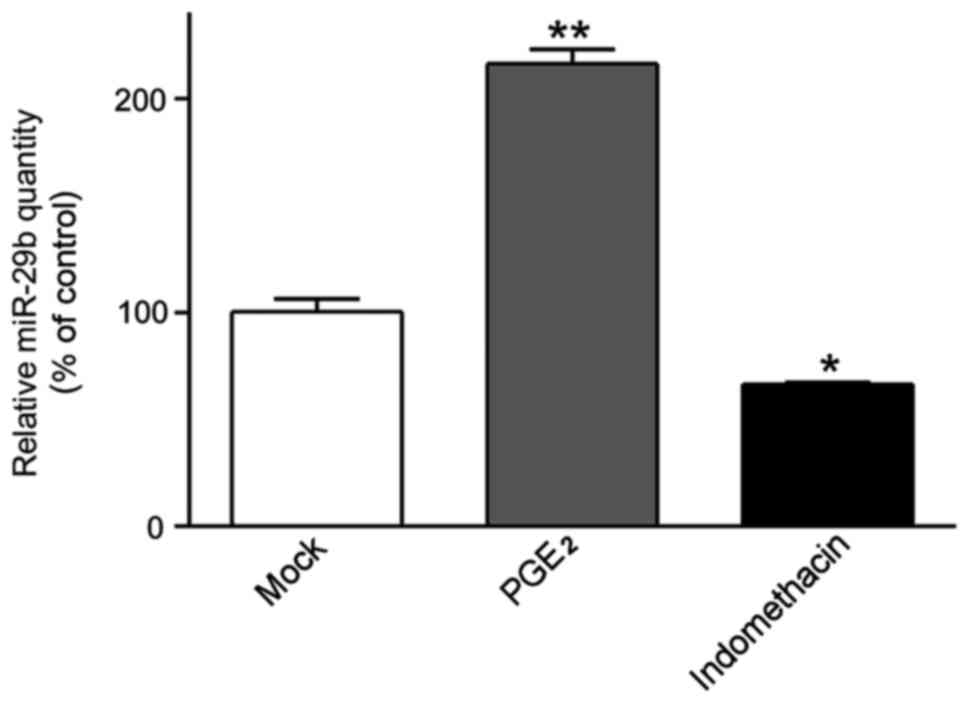
(Fig. 1). Strictly consistent with
previous results, miR-29b was dramatically increased in
PGE2-treated ASMCs compared to untreated cells
(2.162±0.117 vs. 1.004±0.010, P<0.001), whereas miR-29b was
significantly downregulated in the presence of indomethacin
(0.665±0.015 vs. 1.004±0.010, when compared with untreated cells;
P<0.01).
miR-29b directly targets the 3′-UTR of
COL1A1 gene in cultured SVMCs
Although various ECM components have been identified
as targets of miR-29b in other tissues, the downstream target genes
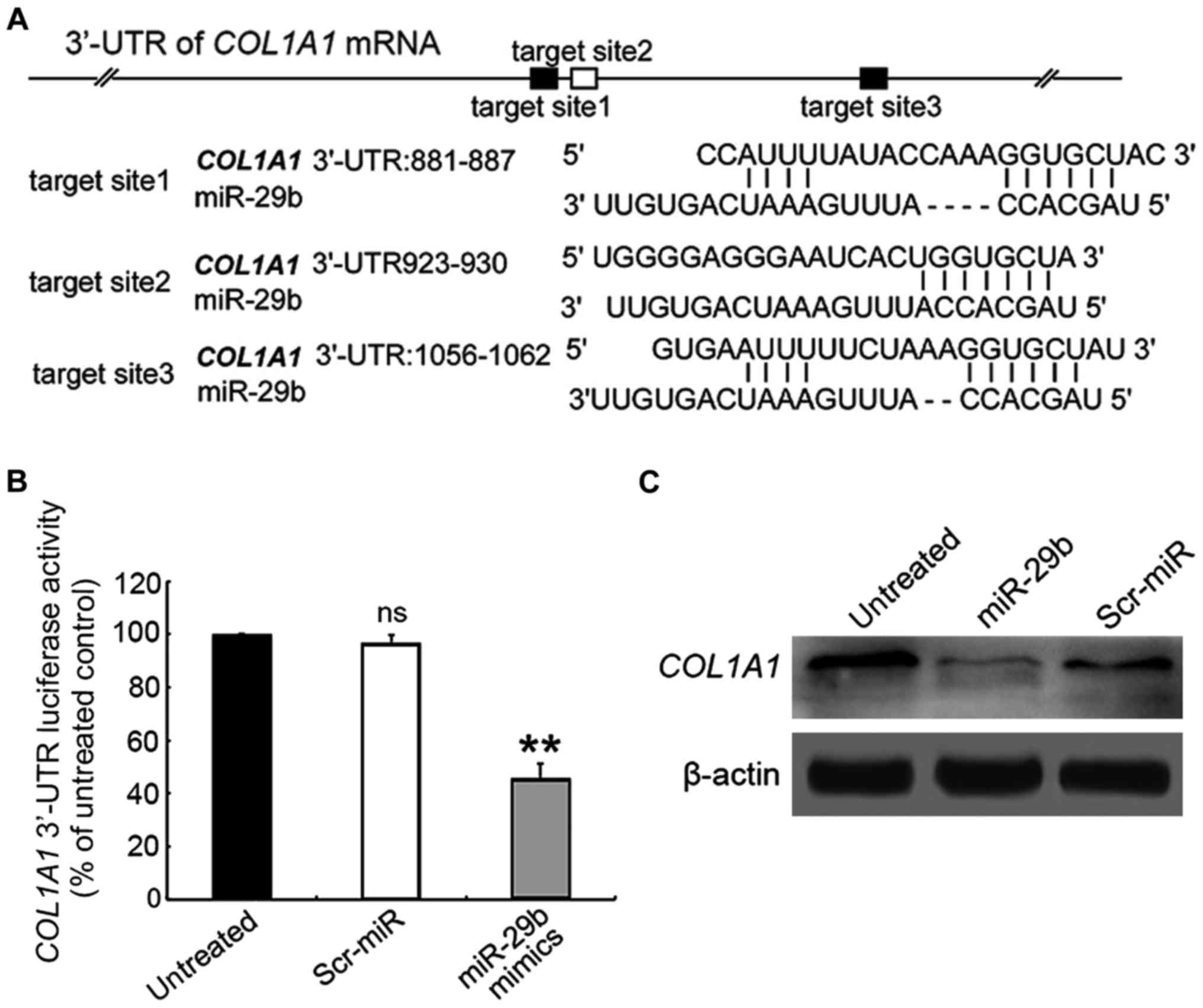
in ASMCs remain unknown. As shown in Fig. 2A, the putative miR-29 binding sites
are enriched in the 3′-UTR region of COL1A1 gene. To confirm
whether miR-29b could directly target COL1A1, the 3′-UTR region of
COL1A1 gene was cloned into a luciferase reporter vector, which was
co-transfected into ASMCs with the miR-29b mimics or Scr-miR, prior
to the performance of the luciferase reporter assay. In the
presence of miR-29b mimics, the relative luciferase activity of
COL1A1-3′-UTR-transfected ASMCs was significantly decreased
compared with those untransfected cell control (Fig. 2B), whereas Scr exposure had no
significant effect on the fluorescence intensity of the
COL1A1-3′-UTR-transfected ASMCs.
We next assessed the effect of miR-29b on the COL1A1
expression level in ASMCs by using RT-qPCR assay. The results
demonstrated that the expression of COL1A1 in miR-29b
mimics-transfected ASMCs was reduced at mRNA level compared to
untransfected cells (Fig. 3A;
0.587±0.178-fold, P<0.05), which was corroborated by
immunoblotting assay (Fig. 2C).
These results suggest that miR-29b exerts an inhibitory effect on
COL1A1 expression through directly targeting the 3′-UTR.
PGE2 promotes miR-29b-mediated ECM
degradation in ASMCs
It has been reported that PGE2
participates in vascular wall remodeling via modulating MMP
activities (21), thus we speculated
whether PGE2 is able to regulate other
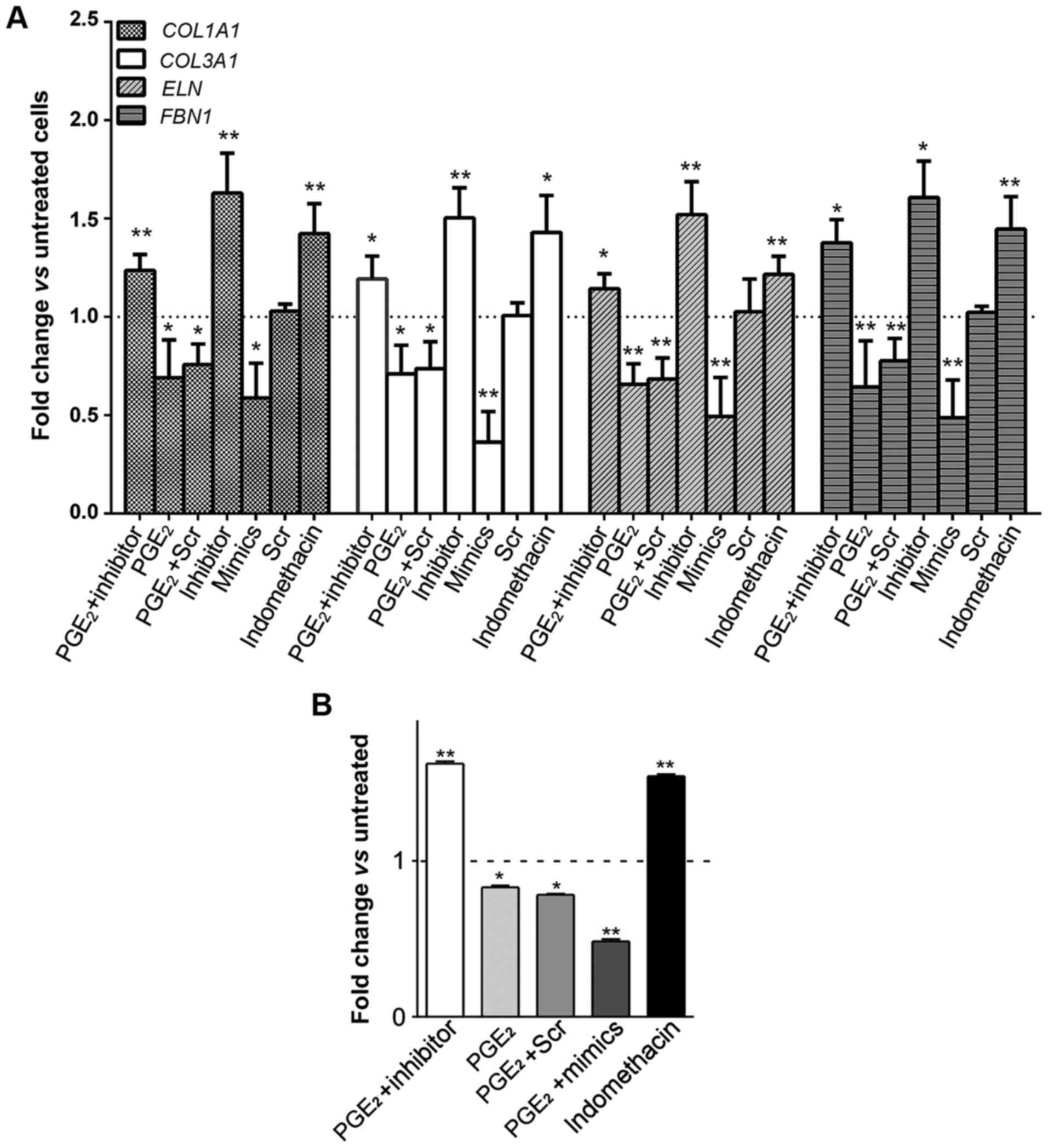
profibrosis-associated ECM components. Collagen gene expression
alterations (represented by COL1A1 and COL3A1) in AMSCs were
assessed in the presence of PGE2. As shown in Fig. 3A, upon PGE2 treatment, the
mRNA expression of COL1A1 and COL3A1 was significantly repressed
(0.690±0.193- and 0.710±0.145-fold, P<0.05, respectively).
Likewise, PGE2 also inhibited the expression of ELN and
FBN1 in ASMCs (0.657±0.105-fold and 0.643±0.235-fold, P<0.01,
respectively). Of note, ECM levels were augmented in response to
indomethacin.
To further investigate whether PGE2
exerts inhibitory effects on ECM expression via upregulating
miR-29b, miR-29b in ASMCs was then suppressed by transfecting with
its inhibitor, followed by assessing the ECM expression profiles.
Successful inhibition of miR-29b (<30% of expression),
regardless of PGE2 treatment, was confirmed by RT-qPCR
analysis (data not shown). Interestingly, expression levels of ECM
genes in PGE2-stimulated and miR-29b
inhibitor-transfected ASMCs were not inhibited but even increased
compared to the untreated control (Fig.
3A). These collectively indicated that PGE2
regulates ECM production by altering miR-29 expression.
The promoted effect of PGE2 on
miR-29b-mediated ECM downregulation in ASMCs was re-confirmed by
monitoring soluble collagen synthesis after incitement. As shown in
Fig. 3B, soluble collagen production
was decreased in PGE2-treated ASMCs in comparison to
that in untreated control. Notably, transfection of miR-29b
inhibitor significantly compromised the inhibitory effect of
PGE2 upon soluble collagen synthesis, whereas miR-29b
mimics exacerbated ECM degradation caused by PGE2
treatment.
Discussion
The pathogenesis of an aneurysm as well as its
progression and ultimate rupture involve series of complicated
pathological processes. The determination of underlying mechanisms
and identification of effective medical therapy remains a major
challenge in recent aneurysmal medicine. In particular, novel
molecular therapies in conquering AAA seem to be of vital
importance. As far, patients with larger aneurysms depend on
elective surgery. Understanding the miR-mediated regulation of ECM
perturbations in pathological conditions will potentially provide
insightful prospective in seeking innovative medical strategies to
fight AAA.
In a separate case-control study, the expansion of
AAAs in patients taking NSAIDs was significantly repressed compared
with the control subjects (18),
suggesting that inhibition of PGs synthesis in AAA development. PGs
are ubiquitously generated in all cell types and function as
autocrine and paracrine regulators to maintain local homeostasis or
inflammatory response. Among them, PGE2 is the most
prevalent and bioactive of the mammalian PG, which was found
previously to be abundantly generated in the aneurysm wall and
involved in the regulation of collagen synthesis (19). It has been demonstrated that during
viral infection, there was a dramatic increase in the expression
level of miR-29, which inhibited DNA methyl transferase (DNMT)
activity and thereby contribute to the activation of COX2 and
consequent enhancement of PGE2 production (23). Collectively, we conclude that miR-29
promotes PGE2 accumulation in response to inflammation
through epigenetic modification-induced COX2 activation. However
the effects of PGE2 on miR-29 and the sequential
ECM-mediated inflammatory signaling pathways have rarely been
studied.
In this study, we investigated the underlying
mechanism of PGE2 and the pharmacology of its blockade,
by introduction of an NSAID indomethacin, in orchestrating the
inflammatory response, with particular regard to the AAA. Data from
our experiments indicated that treatment of ASMCs with
PGE2 induced an increase in miR-29b level, whereas
indomethacin resulted in a decrease in miR-29b expression, hence a
profibrotic response in AAA.
The miR-29 family is well characterized by its
capacity to inhibit the expression of ECM components and thereby
block the related fibrosis in a variety of organs. Previously
published studies have already manifested the therapeutic
implication of miR-29 as a target for fibrosis in heart (11), lung (24), liver (25), and kidney (26), and systemic sclerosis (27). In fibrotic conditions, decreased
miR-29b directly results in an aggrandized collagen gene
expression. Unfortunately, a profibrotic response in these organs
is often pathologic and related to several serious diseases.
Whether miR-29b downregulation plays a beneficial role in
conquering certain fibrosis-related diseases is not known Although
the precise mechanisms remain unclear, repression of miR-29b
expression in experimental AAA development has been widely reported
in previously published studies (9–11).
Fig. 3 shows that modulation of
miR-29b levels in ASMCs, especially overexpression with miR-29b
mimics or suppression by miR-29b inhibitor, resulted in significant
alteration in expression profiles of target collagen genes.
Treatment of smooth muscle cells with PGE2 elevated
miR-29b expression and thereby suppressed the ECM genes expression
compared to untreated cells, while an inhibitor of PG synthesis
termed as indomethacin counteracts the promotion by PGE2
of ECM genes expression. Of note, introduction of miR-29b inhibitor
compromised the effects of PGE2 on collagen gene
expression profiles, suggesting that PGE2 plays an
inhibitory role in ECM expression by targeting miR-29b.
Taken together, we proposed that there is a novel
bidirectional positive-feedback loop between PGE2
synthesis and miR-29b, functioning as a node in the regulation of
ECM homeostasis. When AAA disease occurs, miR-29b expression level
is obviously elevated, which inhibits methylation degree of COX2
and enhances its activity, and this will ultimately promote
PGE2 accumulation. On the contrary, increased
PGE2 can further accelerate miR-29b augmentation and
therefore induce the pathological degradation of ECM proteins.
Data from a previous study suggested that a decline
in the miR-29b expression triggers a profibrotic process in AAAs
(28), which is usually deemed as a
pathologic response to aneurismal dilatation. As the aneurysms
dilate is companied by an increase in the risk of rupture, the
aortic wall may tend, like a balloon, to become thinner and weaker
compared to smaller aneurysms. In this case, deposition of new more
soluble collagen fibers in aortic wall, leading to the thickening
of aneurysmal walls, will occur to compensate the attenuated media
and to reduce arterial wall tension (29). Although the new generated soluble
collagens are more susceptible to hydrolysis by MMPs, the fibrotic
thickening of the adventitia will hinder aneurysm expansion. Our
observations highlighted the extent of soluble collagens
degradation that took place in ASMCs as exposed to PGE2
treatment, which was rescued by introduction of indomethacin.
Furthermore, upon PGE2 treatment, there was a decline in
ELN content, which is synthesized constitutively by smooth muscle
cells in the media. Since the destruction of ELN fibers has been
suggested as a significant pathological process in aortic
dilatation and being related to the loss of elastic capacities of
the aortic media (30,31), the above suggest that PGE2
as well as miR-29b are crucially involved in the aneurysmal
expansion. However, the application of agents that modulate miR-29b
expression is relatively less efficient in the aorta compared to
that in other organs such as kidney, liver and heart (28), likely due to preferential uptake in
these organs. In this study, drugs targeting PGE2 are
potentially more effective in control of aneurysmal dilatation.
In conclusion, in this study we propose that more
expandable anti-inflammatory drugs that inhibit PGE2
synthesis may emerge as a promising avenue to trigger fibrosis in
the aortic wall and thereafter protect the aorta from expansion in
human patients with AAA. The in vivo experiments underlying
their therapeutic potential to inhibit aneurysm expansion and
ultimate rupture are being carried out in our laboratory and
further evidence will be published in the future.
Acknowledgements
Not applicable.
Funding
This study did not receive any specific grant from
funding agencies in the public, commercial, or not-for-profit
sectors.
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
ZH and XJ contributed to the conception of the
study. TZ contributed significantly to the data analysis and study
preparation. YH and GL performed the data analyses and wrote the
study. GL helped perform the analysis with constructive
discussions. All authors have read and approved the final
study.
Ethics approval and consent to
participate
The Ethics Committee of the Provincial Hospital
Affiliated to Shandong University approved the study (Jinan,
China).
Consent for publication
Not applicable.
Competing interests
All authors declare that they have no financial or
other conflicts of interest in relation to this study and its
publication.
References
|
1
|
Weintraub NL: Understanding abdominal
aortic aneurysm. N Engl J Med. 361:1114–1116. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kent KC: Clinical practice. Abdominal
aortic aneurysms. N Engl J Med. 371:2101–2108. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Thompson RW, Liao S and Curci JA: Vascular
smooth muscle cell apoptosis in abdominal aortic aneurysms. Coron
Artery Dis. 8:623–631. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhang C: MicroRNomics: A newly emerging
approach for disease biology. Physiol Genomics. 33:139–147. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Seeger T and Boon RA: MicroRNAs in
cardiovascular ageing. J Physiol. 594:2085–2094. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Small EM and Olson EN: Pervasive roles of
microRNAs in cardiovascular biology. Nature. 469:336–342. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Urbich C, Kuehbacher A and Dimmeler S:
Role of microRNAs in vascular diseases, inflammation, and
angiogenesis. Cardiovasc Res. 79:581–588. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kin K, Miyagawa S, Fukushima S, Shirakawa
Y, Torikai K, Shimamura K, Daimon T, Kawahara Y, Kuratani T and
Sawa Y: Tissue- and plasma-specific microRNA signatures for
atherosclerotic abdominal aortic aneurysm. J Am Heart Assoc.
1:e0007452012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Roderburg C, Urban GW, Bettermann K, Vucur
M, Zimmermann H, Schmidt S, Janssen J, Koppe C, Knolle P, Castoldi
M, et al: Micro-RNA profiling reveals a role for miR-29 in human
and murine liver fibrosis. Hepatology. 53:209–218. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Qin W, Chung AC, Huang XR, Meng XM, Hui
DS, Yu CM, Sung JJ and Lan HY: TGF-β/Smad3 signaling promotes renal
fibrosis by inhibiting miR-29. J Am Soc Nephrol. 22:1462–1474.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Van Rooij E, Sutherland LB, Thatcher JE,
DiMaio JM, Naseem RH, Marshall WS, Hill JA and Olson EN:
Dysregulation of microRNAs after myocardial infarction reveals a
role of miR-29 in cardiac fibrosis. Proc Natl Acad Sci USA.
105:13027–13032. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
De Paepe A, Nuytinck L, Hausser I,
Anton-Lamprecht I and Naeyaert JM: Mutations in the COL5A1 gene are
causal in the Ehlers-Danlos syndromes I and II. Am J Hum Genet.
60:547–554. 1997.PubMed/NCBI
|
|
13
|
Rahkonen O, Su M, Hakovirta H, Koskivirta
I, Hormuzdi SG, Vuorio E, Bornstein P and Penttinen R: Mice with a
deletion in the first intron of the Col1a1 gene develop
age-dependent aortic dissection and rupture. Circ Res. 94:83–90.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Menashi S, Campa JS, Greenhalgh RM and
Powell JT: Collagen in abdominal aortic aneurysm: Typing, content,
and degradation. J Vasc Surg. 6:578–582. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Rizzo RJ, McCarthy WJ, Dixit SN, Lilly MP,
Shively VP, Flinn WR and Yao JS: Collagen types and matrix protein
content in human abdominal aortic aneurysms. J Vasc Surg.
10:365–373. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chen KC, Wang YS, Hu CY, Chang WC, Liao
YC, Dai CY and Juo SH: OxLDL up-regulates microRNA-29b, leading to
epigenetic modifications of MMP-2/MMP-9 genes: A novel mechanism
for cardiovascular diseases. FASEB J. 25:1718–1728. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Funk CD: Prostaglandins and leukotrienes:
Advances in eicosanoid biology. Science. 294:1871–1875. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Walton LJ, Franklin IJ, Bayston T, Brown
LC, Greenhalgh RM, Taylor GW and Powell JT: Inhibition of
prostaglandin E2 synthesis in abdominal aortic aneurysms:
Implications for smooth muscle cell viability, inflammatory
processes, and the expansion of abdominal aortic aneurysms.
Circulation. 100:48–54. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Diaz A, Munoz E, Johnston R, Korn JH and
Jimenez SA: Regulation of human lung fibroblast alpha 1(I)
procollagen gene expression by tumor necrosis factor alpha,
interleukin-1 beta, and prostaglandin E2. J Biol Chem.
268:10364–10371. 1993.PubMed/NCBI
|
|
20
|
Holmes DR, Wester W, Thompson RW and
Reilly JM: Prostaglandin E2 synthesis and cyclooxygenase expression
in abdominal aortic aneurysms. J Vasc Surg. 25:810–815. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yokoyama U, Ishiwata R, Jin MH, Kato Y,
Suzuki O, Jin H, Ichikawa Y, Kumagaya S, Katayama Y, Fujita T, et
al: Inhibition of EP4 signaling attenuates aortic aneurysm
formation. PLoS One. 7:e367242012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Gonzalez P, Luna C, Li G, Qiu J and
Epstein DL: miR-29 is induced by prostaglandin E and forms negative
feedback loops with the Wnt and TGFbeta pathways in human
trabecular meshwork cells. Invest Ophthalmol Vis Sci.
51:32102010.PubMed/NCBI
|
|
23
|
Fang J, Hao Q, Liu L, Li Y, Wu J, Huo X
and Zhu Y: Epigenetic changes mediated by microRNA miR29 activate
cyclooxygenase 2 and lambda-1 interferon production during viral
infection. J Virol. 86:1010–1020. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Xiao J, Meng XM, Huang XR, Chung AC, Feng
YL, Hui DS, Yu CM, Sung JJ and Lan HY: miR-29 inhibits
bleomycin-induced pulmonary fibrosis in mice. Mol Ther.
20:1251–1260. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhang Y, Wu L, Wang Y, Zhang M, Li L, Zhu
D, Li X, Gu H, Zhang CY and Zen K: Protective role of
estrogen-induced miRNA-29 expression in carbon
tetrachloride-induced mouse liver injury. J Biol Chem.
287:14851–14862. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang B, Komers R, Carew R, Winbanks CE, Xu
B, Herman-Edelstein M, Koh P, Thomas M, Jandeleit-Dahm K,
Gregorevic P, et al: Suppression of microRNA-29 expression by
TGF-β1 promotes collagen expression and renal fibrosis. J Am Soc
Nephrol. 23:252–265. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Maurer B, Stanczyk J, Jüngel A,
Akhmetshina A, Trenkmann M, Brock M, Kowal-Bielecka O, Gay RE,
Michel BA, Distler JH, et al: MicroRNA-29, a key regulator of
collagen expression in systemic sclerosis. Arthritis Rheum.
62:1733–1743. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Maegdefessel L, Azuma J, Toh R, Merk DR,
Deng A, Chin JT, Raaz U, Schoelmerich AM, Raiesdana A, Leeper NJ,
et al: Inhibition of microRNA-29b reduces murine abdominal aortic
aneurysm development. J Clin Invest. 122:497–506. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sakalihasan N, Heyeres A, Nusgens BV,
Limet R and Lapière CM: Modifications of the extracellular matrix
of aneurysmal abdominal aortas as a function of their size. Eur J
Vasc Surg. 7:633–637. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Martufi G, Satriano A, Moore RD, Vorp DA
and Di Martino ES: Local quantification of wall thickness and
intraluminal thrombus offer insight into the mechanical properties
of the aneurysmal aorta. Ann Biomed Eng. 43:1759–1771. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hance KA, Tataria M, Ziporin SJ, Lee JK
and Thompson RW: Monocyte chemotactic activity in human abdominal
aortic aneurysms: Role of elastin degradation peptides and the
67-kD cell surface elastin receptor. J Vasc Surg. 35:254–261. 2002.
View Article : Google Scholar : PubMed/NCBI
|