Introduction
Cutaneous malignant melanoma (CMM) is a type of skin
tumor. This malignant type of neoplasm evolves from melanoma cells
at the base of the epidermis. Its incidence rate accounts for 1–3%
of all malignant tumor types and 5–10% of malignant skin tumors,
and has demonstrated a clear upward trend in recent years (1). It is currently thought that the
pathogenesis of MM is primarily associated with solar irradiation
of the skin. Ultraviolet rays in the sunlight burn the skin to
induce DNA mutations. Mutations in P16 or cyclin-dependent kinase
inhibitor 2A located on the short arm of chromosome 9 have been
identified as the major cause of genetic pre-disposition to MM
(2). Surgery, radiotherapy and
chemotherapy are the major treatments of MM; however, the efficacy
is low. The discovery of melanoma-associated mutations in genes in
recent years and in-depth study may provide approaches for targeted
therapies for a new era of MM treatment.
Normal cells gather together and adhere to the
extracellular matrix (ECM). They survive through mutual substances
and signal conduction. Once they lose contact with the ECM, the
cells undergo programmed death, also known as apoptosis. This form
of cell death was first named anoikis in 1994 (3). Anoikis is indispensable to maintain the
stability of the body tissue. Its main role is to prevent abnormal
cell growth or adhesion of cells to the abnormal ECM. Resistance to
anoikis is a hallmark of tumor metastasis, allowing tumor cells to
spread through the circulatory system to other distant organs.
Tumor cells are resistant to apoptosis through autocrine and
paracrine mechanisms after losing cell-to-ECM and cell-to-cell
contact, and regain the ability to diffuse, metastasize and invade
(4–6). Studies have demonstrated that integrins
as inhibitors of anoikis have a profound effect on cell survival
(7,8). The phosphoinositide 3-kinase (PI3K)/Akt
and mitogen-associated protein kinase kinase/extracellular
signal-regulated kinase pathways are involved in the critical step
of resistance to anoikis (4).
Muscle intestine and stomach expression 1 (MIST1),
also referred to as basic helix-loop-helix (bHLH) family, member
a15, is a transcriptional factor. The bHLH motif, which is
consistent throughout this family, contains ~60 amino acids and
consists of a basic region and a HLH motif that binds to DNA
(9). It has been reported that MIST1
has an important role in maintaining normal cell numbers and
promoting terminal differentiation of acinar cells in pancreatic
exocrine glands. These effects indicate that MIST1 is a key
anti-cancer factor in pancreatic tumorigenesis (10).
The transcription factor SNAI1 is involved not only
in the regulation of primordial germ cell migration, gastrulation
and neural crest migration during normal embryonic development, but
also has an important role in the occurrence, recurrence, invasion
and metastasis of organ tumors and organ fibrosis (11–13).
SNAI1 inhibits the expression of E-cadherin and has a crucial role
in epithelial-to-mesenchymal transition (EMT) (14,15).
Interfering with SNAI1 expression may increase the death rate of
cancer cells (16). In addition,
SNAI1 inhibits the expression of phosphatase and tensin homologue
(PTEN) through binding to the promoter of PTEN on chromosome 10.
This leads to resistance to gamma-ray-induced apoptosis (17).
PI3K/AKT is an important signaling pathway that
facilitates the immortalization of cells and promotes the
development of normal vascular and tumor angiogenesis (18–21).
PTEN is a lipid phosphatase that dephosphorylates
phosphatidylinositol (3,4,5)-trisphosphate and blocks the PI3K/AKT
pathway. The effect of PTEN on p27 appears to be mediated via
inhibition of the PI3K/Akt pathway. After the expression of PTEN is
inhibited, cell cycle progression is accelerated through
phosphorylation of Akt and glycogen synthase kinase-3 (22). It is reported that SNAI1 directly
represses PTEN and leads to activation of the PI3K/Akt pathway, and
then leads to inactivation and phosphorylation of the pro-apoptotic
protein Bcl-2-associated death promoter. This contributes to
anoikis resistance (23).
The present study initially explored the association
between MIST1 and SNAI1 in human melanoma cells. It was found that
MIST1 and SNAI1 contributed to the detachment of cells from the
cell matrix by altering the expression of certain proteins. Their
upregulation was beneficial for survival after suspension culture.
The expression of SNAI1 changed with the alteration of MIST1. It
was demonstrated that the PTEN/AKT signaling pathway also
participated in their function; SNAI1 was found to inhibit the
expression of PTEN and activate Akt. This helped cells to bypass
anoikis. MIST1 was identified bind to the promoter of SNAI1 and
activate the expression of SNAIL1 as a transcription factor, as
indicated using chromatin immunoprecipitation (ChIP) and luciferase
reporter gene technology.
Materials and methods
Cell culture
HUVEC, NHEM 2493, A375 and MV3 cell lines were
obtained from the American Type Culture Collection (ATCC; Manassas,
VA, USA). HUVEC was maintained in endothelial cell growth-2 medium
supplemented with 2% fetal bovine serum, 15 ng/ml insulin-like
growth factor-1, 5 ng/ml epidermal growth factor, 0.75 Units/ml
heparin sulfate, 5 ng/ml vascular endothelial growth factor, 5
ng/ml basic fibroblast growth factor, 10 mM L-glutamine, 1 ng/ml
hydrocortisone hemisuccinate and 50 µg/ml ascorbic acid (all from
Lonza Group, Ltd., Basel, Switzerland). NHEM was maintained in
serum- and phorbol myristate acetate-free melanocyte growth medium
M2 (Promocell GmbH, Heidelberg, Germany). MV3 and A375 were
maintained in Dulbecco's modified Eagle's medium supplemented with
15% fetal bovine serum (both from Gibco; Thermo Fisher Scientific,
Inc., Waltham, MA, USA). Cells were maintained in an incubator at
37°C in a humidified atmosphere containing 5% CO2.
Adhesion assay
Six-well plates were coated with 10 g/ml fibronectin
(cat. no. F2006; Sigma Aldrich; Merck KGaA, Darmstadt, Germany).
Then they were blocked at room temperature with 0.2% BSA for 2 h. A
total of 1×105 cells were placed in each well and
incubated in the 37°C cell incubator for 30 min. Medium was
discarded and excess cells were rinsed using PBS. Adhered cells
were stained with crystal violet at room temperature for 10 min and
photographed under a light microscope.
Cell death detection ELISA
A total of 5×104 cells were placed in
normal and low-attachment surface 24-well plates. A Cell Death
Detection ELISAPLUS (cat. no. 11774425001; Roche
Diagnostics, Basel, Switzerland) was used to detect apoptosis level
according to the manufacturer's instructions.
RNA isolation and polymerase chain
reaction (PCR) analysis
TRIzol (Invitrogen; Thermo Fisher Scientific, Inc.)
was used for extracting total RNA from cells. ReverAid First Strand
cDNA Synthesis kit (cat. no. K1622; Fermentas; Thermo Fisher
Scientific, Inc.) was applied to reverse-transcribe mRNA with a
polyA tail into complementary (c)DNA. PCR Master Mix (cat. no.
K0172; Invitrogen; Thermo Fisher Scientific, Inc.) was used for
PCR. Cycling conditions for the PCR reaction were as follows:
Initial denaturation step of 3 min at 94°C, followed by 31 cycles
consisting of 30 sec at 94°C, 30 sec at 55°C and 30 sec at 72°C,
with a final extension period of 7 min at 72°C.
Sequences of the primers were as follows: MIST1
forward, 5′-GCGGATGCACAAGCTAAATAAC-3′ and reverse,
5′-CTCGATCTTGGAGAGCTTCTTG-3′; SNAI1 forward,
5′-CCTTCGTCCTTCTCCTCTACTT-3′ and reverse,
5′-GGCACTGGTACTTCTTGACATC-3′; GAPDH forward,
5′-GCGGATGCACAAGCTAAATAAC-3′ and reverse,
5′-CTCGATCTTGGAGAGCTTCTTG-3′.
Protein extraction and western blot
analysis
Laemmli sample buffer containing 5%
2-mercaptoethanol (both from Bio-Rad Laboratories, Inc., Hercules,
CA, USA) was used to extract total protein from cells.
Pierce™ BCA Protein assay kit (cat. no. 23227;
Invitrogen; Thermo Fisher Scientific, Inc.) was used to detect
protein concentration and 20 µg protein was loaded in each well.
Protein samples were separated by 8–15% SDS-PAGE, followed by
transfer onto a polyvinylidene fluoride membrane (cat. no.
PVM020C-160; Pall Life Sciences, Port Washington, NY, USA). The
following primary antibodies were used: Rabbit MIST1 monoclonal
antibody (mAb) (cat. no. ab180889; 1:2,000 dilution in 5% w/v milk;
Abcam, Cambridge, MA, USA), goat SNAI1 mAb (cat. no. ab53519;
1:2,000 dilution in 5% w/v milk; Abcam), rabbit Akt mAb [cat. no.
9272L; 1:2,000 dilution in 3% w/v bovine serum albumin (BSA); Cell
Signaling Technology, Inc., Danvers, MA, USA], rabbit
phospho-(p)Akt (Ser473) mAb (cat. no. 4060; 1:1,000 dilution in 3%
w/v BSA; Cell Signaling Technology, Inc.), rabbit PTEN mAb (cat.
no. 9188; 1:2,000 dilution in 3% w/v BSA; Cell Signaling
Technology, Inc.) and mouse GAPDH mAb (cat. no. ab8245; 1:5,000
dilution in 5% w/v milk; Abcam). The primary antibodies were
incubated at 4°C overnight. Goat Anti-Mouse IgG H&L (cat. no.
ab6789; 1:2,000), Goat Anti-Rabbit IgG H&L (cat. no. ab6721;
1:2,000 dilution in 3% w/v BSA) and Rabbit Anti-Goat IgG H&L
(cat. no. ab6741; 1:2,000 dilution in 5% w/v milk; all from Abcam)
were used as secondary antibodies. The secondary antibodies were
incubated at room temperature for 4 h. Bands were visualized using
a Chemiluminescent horseradish peroxidase substrate (cat. no.
P90720; EMD Millipore; Billerica, MA, USA) and images were captured
using X-OMAT BT Film (cat. no. 6535876; Carestream Health, Inc.,
Rochester, NY, USA). The concentration of LY294002 (cat. no. 9901;
Cell Signaling Technology, Inc., Boston, MA, USA) was 10 µM to
suppress p-AKT (Ser473).
Construction of overexpression and
knockdown vectors
cDNA of HUVEC was used as a template to amplify the
full-length open reading frames of MIST1 and SNAI1 by PCR, which
were cloned in the expression vector pcDNA3.1 (cat. no. V79020;
Invitrogen; Thermo Fisher Scientific, Inc.). pLKO.1 containing
small hairpin (sh)RNAs specifically targeting MIST1, SNAI1 and PTEN
(Shanghai GeneChem Co., Ltd., Shanghai, China) were used to
knockdown these genes in the cell lines. The shRNAs had the
following sequences: shRNA-1 targeting MIST1,
5′-CCGGCCAAGGGTCTGCGGAGC-3′; shRNA-2 targeting MIST1,
5′-CCATGTCCAGCAGCCGCCTCC-3′; shRNA-1 targeting SNAI1,
5′-TTTACCTTCCAGCAGCCCTAC-3′; shRNA-2 targeting SNAI1,
5′-ACCTCAGCCTGGGTGCCCTCA-3′; shRNA-1 targeting PTEN,
5′-AGAGTTGCACAATATCCTTTT-3′; shRNA-2 targeting PTEN,
5′-GAGGAAACCTCAGAAAAAGTA-3′. A 293T cell lentiviral packaging
system was used. A total of 2.5 µg Rev, 3 µg Rev, 3,5 µg Rev and 12
µg target plasmid were transfected into 293T cells in a 10 cm cell
culture dish with polyethyleneimine (cat. no. B600070; ProteinTech
Group, Inc., Chicago, IL, USA) at a concentration of 1.7 µg/µl.
After 4 h of transfection, the medium was changed to complete
medium to collect the virus. The collected virus infected the
target cells 3 times for 8 h each.
ChIP assay
Direct combination and transcriptional activation of
SNAI1 by MIST1 was predicted by the Encyclopedia of DNA Elements in
the UCSC database (http://genome.ucsc.edu/ENCODE/). The JASPAR database
(http://jaspar.genereg.net/) was used to
predict the MIST1 binding motif. Cells were fixed with 1%
formaldehyde and terminated with 2.5 mM glycine. The scraped cells
were sonicated for lysis in PBS with sodium thiosulfate. The
lysates were divided into three aliquots, one of which was a
positive control and received no treatment and one of which was a
negative control, which was incubated with immunoglobulin G (cat.
no. I5006) and protein G PLUS-Agarose (cat no. P7700) 9both from
Sigma-Aldrich; Merck KGaA). One third of the cell lysate was used
as the test group, and was incubated with antibody against MIST1
(1:100) and Protein G PLUS-Agarose. After removal of RNA and
protein, DNA was extracted with phenol-chloroform, respectively.
Next, the degree of enrichment was detected using real-time
quantitative PCR.
Luciferase reporter gene
technology
At 24 h prior to transfection, 1×105 A375
and MV3 cells were seeded into 24-well plates. pGL3 luciferase
reporters vector containing target fragments (~800 ng) were
complexed with 1 µl Lipofectamine 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.), and then added to the cells. These target
fragments included −1,199 to −753, −2,135 to −753, −3,235 to −753,
−4,024 to −753, −2,135 to −1,200, −3,235 to −1,200 and −4,024 to
−1,200 upstream of TSS site of SNAI1. Renilla reniformis
luciferase (pRL-TK) was used as the transfection control. Cells
were harvested and lysed 24 h later. The luciferase activity was
measured using a Dual-Luciferase Reporter Assay System in a Promega
GloMax 20/20 Luminometer (Promega Corp., Madison, WI, USA).
Statistical analysis
Statistical analyses were performed using SPSS 19.0
software (SPSS Inc., Chicago, IL, USA). Data are presented as the
mean ± standard deviation for continuous data. One-way analysis of
variance was performed for comparison analysis. Dunnett's test was
used for pairwise comparisons of multiple treatment groups. All
experimental groups were compared with the control groups.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Expression of MIST1 positively
correlates with SNAI1 in normal and melanoma cells
MIST1 is the first transcription factor identified
as a protein specifically expressed in serous exocrine cells
(24). MIST1 was identified to have
an important role in pancreatic cancer. SNAI1 is involved in EMT
and is closely associated with tumor metastasis, recurrence and
prognosis as a classic zinc finger protein. However, the
interaction between these two proteins and their function in human
melanoma cells has remained to be fully elucidated. The present
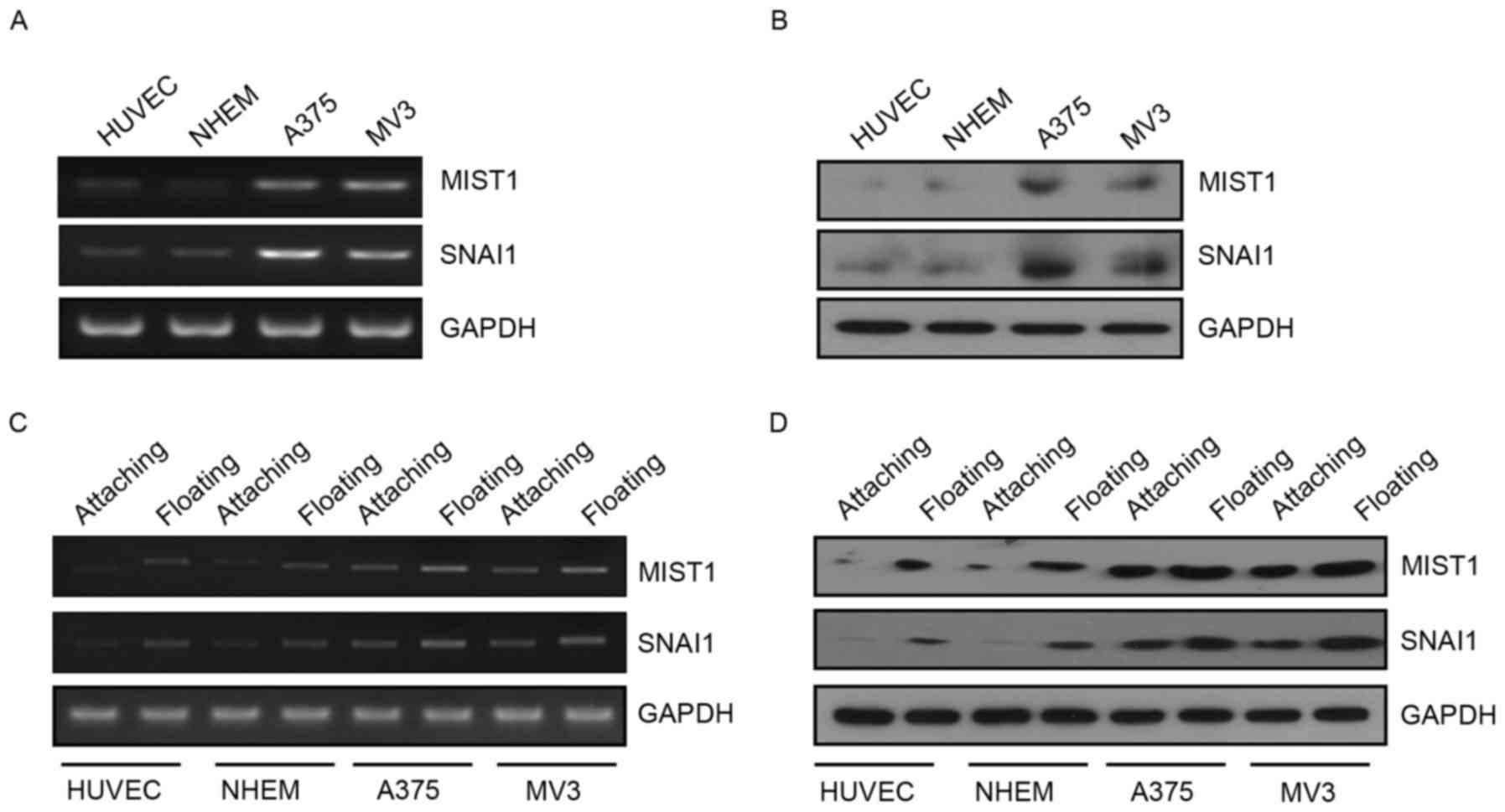
study found that the expression of MIST1 and SNAI1 was upregulated
in the human melanoma cell lines A375 and MV3 at the RNA and
protein level (Fig. 1A and B). This
indicated that MIST1 and SNAI1 are likely to have a role in the
development and progression of melanoma. Furthermore, the
expression of MIST1 and SNAI1 in normal or melanoma cells was
substantially increased after culture in a 24-well plate with a low
attachment surface for 24 h (Fig. 1C and
D). Leaving the matrix stimulated the cells to express MIST1
and SNAI1. These results indicated that MIST1 and SNAI1 may help
cells to bypass anoikis after loss of anchorage to their physical
environment.
MIST1 and SNAI1 disrupt cell-matrix adhesion and
promote anchorage independence. Previous studies have reported that
SNAI1 has a pro-apoptotic function and helps cells to bypass
anoikis (23). However, the
association of MIST1 with anoikis has remained elusive.
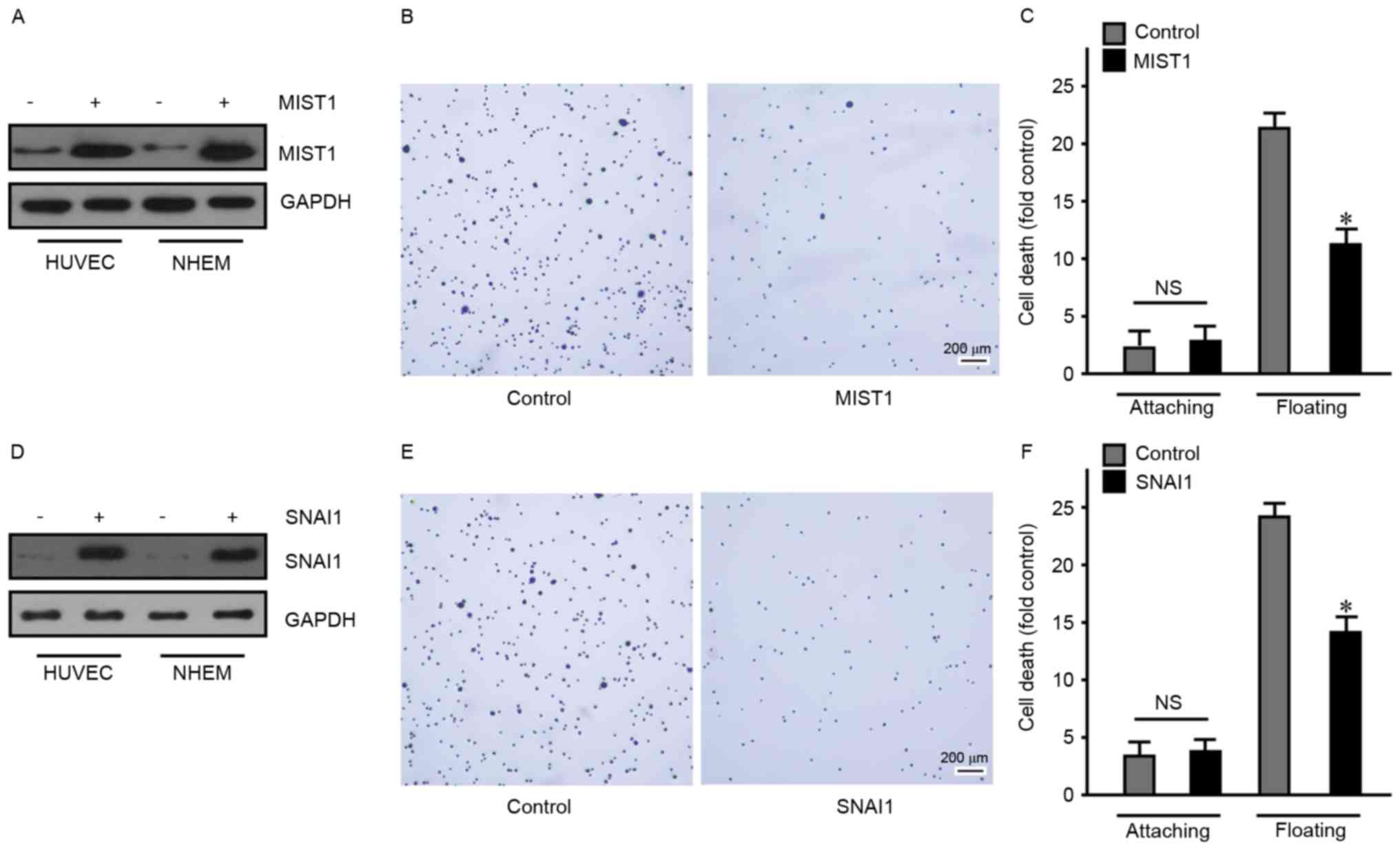
To overexpress MIST1 and SNAI1, a 293T cell
lentiviral packaging system was used, with which HUVEC and NHEM
were transfected to endogenously express MIST1 and SNAI1 (Fig. 2). First, overexpression of MIST1 was
confirmed by western blot analysis (Fig.
2A). Subsequent cell behavioral experiments revealed that MIST1
expression resulted in a decreased adherence of normal human HUVEC
and NHEM to fibronectin (Fig. 2B).
As anoikis-sensitive cells, HUVEC and NHEM were used to assess the
influence of MIST1 on anoikis. The ratio of dead cells in HUVEC and
NHEM overexpressing MIST1 was detected after floating for 24 h by
using a BD Cell Death Detection ELISA kit. After overexpression of
MIST1, resistance to anoikis was obviously improved (Fig. 2C). Furthermore, HUVEC and NHEM were
transfected with SNAI1 and subjected to the above experiments,
revealing a similar effect of SNAI1 to that of MIST1 (Fig. 2D-F). These results indicated that
MIST1 and SNAI1 promoted anchorage independence in normal human
cells. Following loss of anchorage, MIST1 and SNAI1 protected cells
from anoikis.
Knockdown of MIST1 and SNAI1 increases
adhesion and induces anoikis in melanoma cells
Conversely, to determine the role of MIST1 and SNAI1
in human melanoma cells, their expression was altered in tumor cell
lines. shRNAs were utilized to reduce the expression of MIST1 and
SNAI1 in the A375 and MV3 human melanoma cell lines (Fig. 3). In order to avoid off-target
effects, two target sequences were used to reduce expression of
MIST1 and SNAI1, respectively, namely vector-shMIST1-1,
vector-shMIST1-2, vector-shSNAI1-1 and vector-shSNAI1-2. The
knockdown of MIST1 was detected by immunoblot (Fig. 3A). A fibronectin adhesion assay was
also performed to confirm the influence of MIST1 on the attachment
to the ECM. Knockdown of MIST1 increased the binding of A375 and
MV3 human melanoma cell lines to fibronectin (Fig. 3B). After suspension culture for 24 h,
the apoptotic rate of A375 and MV3 cells was significantly
increased following MIST1 knockdown (Fig. 3C). Furthermore, knockdown of SNAI1
was revealed to have a similar effect to that of MIST1 (Fig. 3D-F). It was apparent that knockdown
of endogenous MIST1 and SNAI1 inhibited anchorage independence of
human melanoma cells and increased their sensitivity to anoikis.
The results of the gain- and loss-of-function studies suggested
that MIST1 and SNAI1 help human melanoma cells to part from the ECM
and bypass anoikis, which may contribute to the formation of
distant metastases.
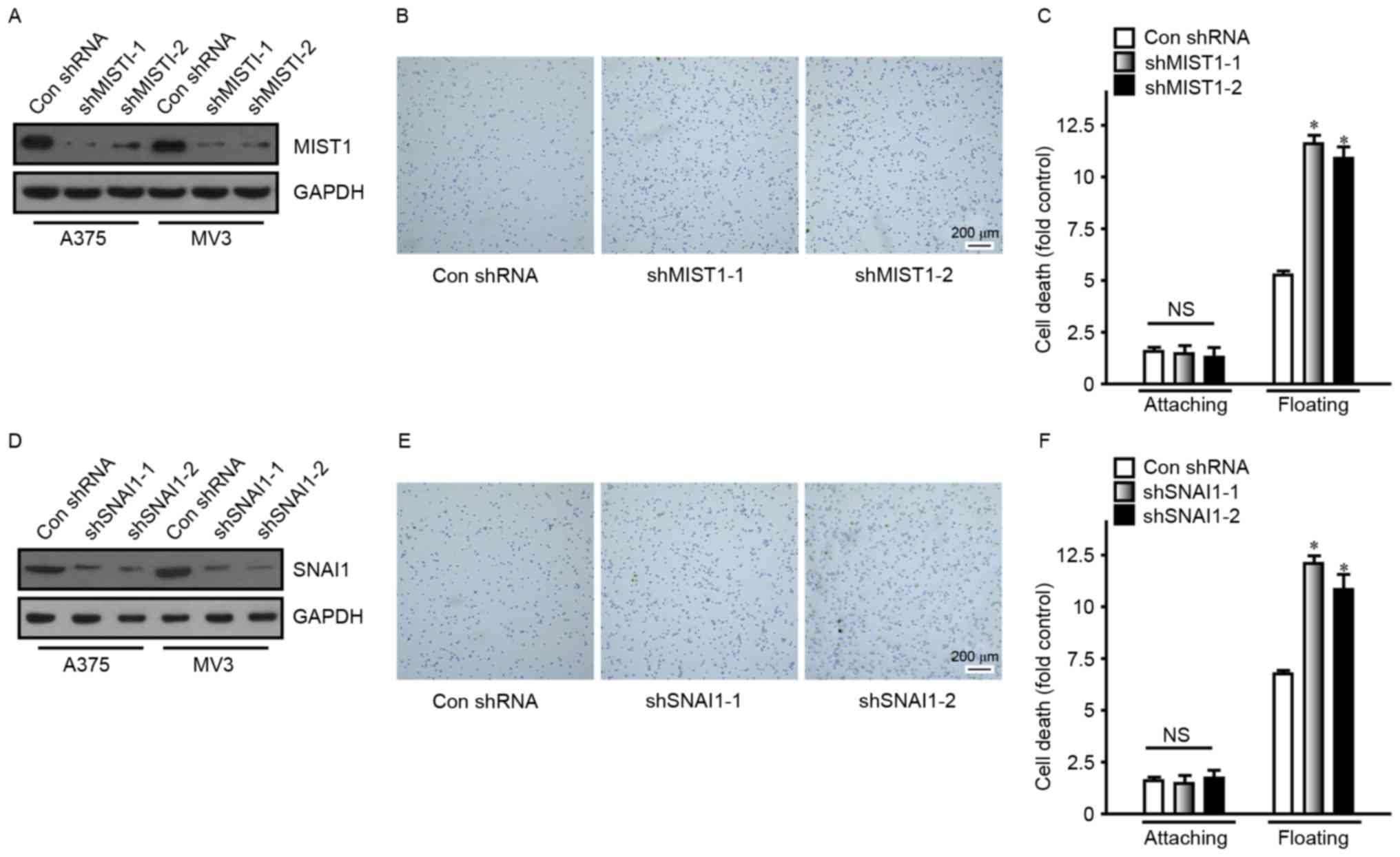 | Figure 3.Knockdown of MIST1 and SNAI1 increases
adhesion and induces cells to undergo anoikis. (A) MIST1 was
knocked down in the A375 and MV3 cell lines, as confirmed by
western blot analysis. (B) Knockdown of MIST1 improved the adhesion
ability of A375 cells (scale bar, 200 µm). Findings for MV3 were
similar, data not shown. (C) Inhibition of MIST1 decreased the
viability of A375 cells in suspension culture, indicating increased
anoikis. Findings for MV3 were similar, data not shown. (D) SNAI1
was knocked down in the A375 and MV3 cell lines, as confirmed by
western blot analysis. (E) Knockdown of SNAI1 improved the adhesion
ability of A375 cells (scale bar, 200 µm). (F) Inhibition of SNAI1
decreased the viability of A375 cells in suspension culture,
indicating increased anoikis. NS, no significance; *P<0.05 vs.
control. shMIST1, small hairpin RNA targeting muscle intestine and
stomach expression 1; Con, control. |
MIST1 confers anoikis resistance
through regulating SNAI1
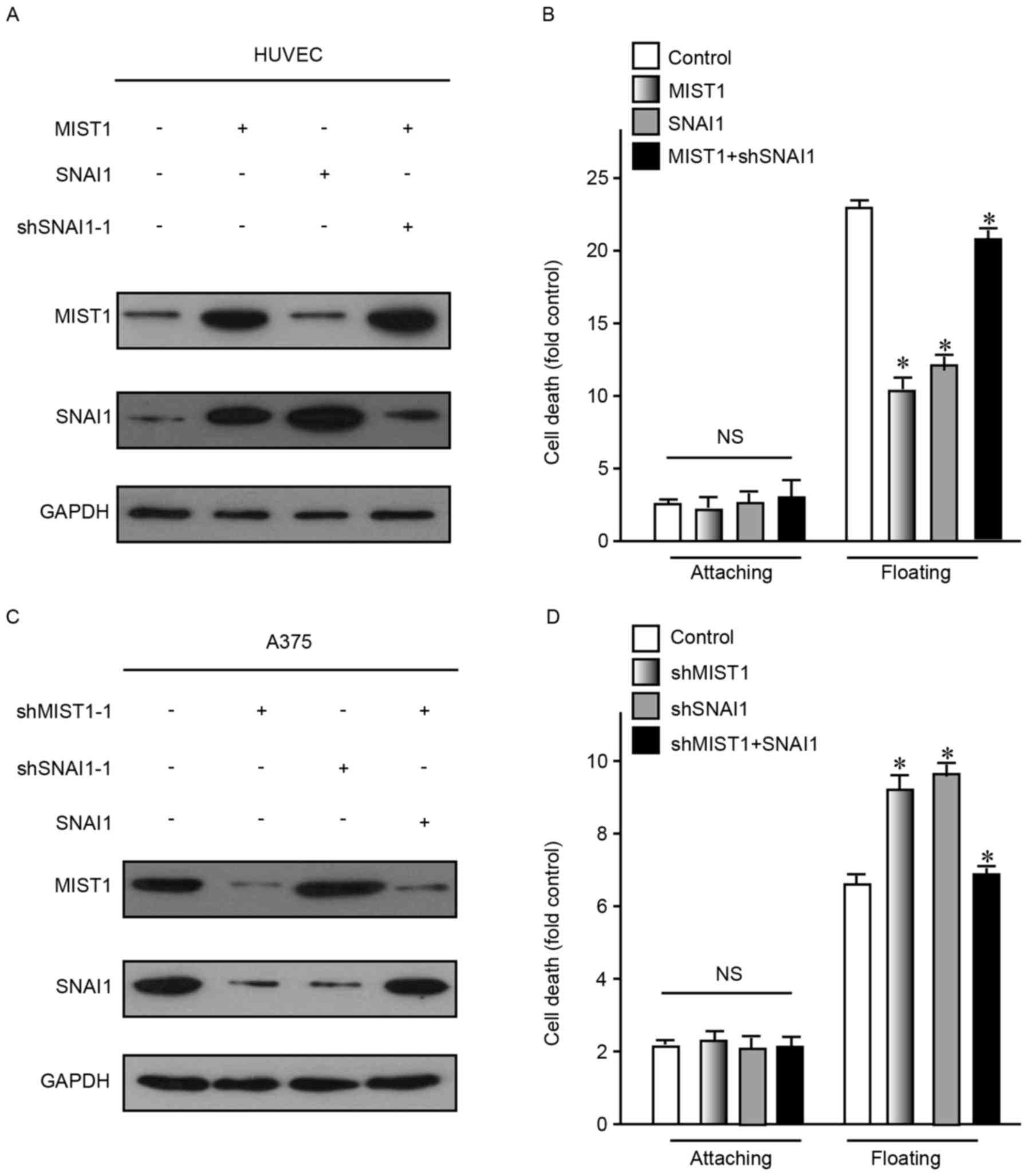
As the abovementioned results indicated that MIST1
and SNAI1 had a role in the development and progression of human
melanoma cells, the association between them was then assessed. To
define the correlation between MIST1 and SNAI1, these two genes
were ectopically overexpressed in HUVEC and NHEM cells, which do
not highly express them. The results demonstrated that only MIST1
overexpression caused an increase in the expression of SNAI1, but
not vice versa (Fig. 4A). After
enhancement of the expression of MIST1 or SNAI1, the resistance to
anoikis was obviously increased (Fig.
4B). Conversely, knockdown of MIST1 decreased the expression of
SNAI1 in the human melanoma cell lines A375 and MV3, which express
the two genes at relatively high levels, while knockdown of SNAI1
did not affect the levels of MIST1 (Fig.
4C). After knockdown of MIST1 or SNAI1, the resistance to
anoikis was obviously decreased (Fig.
4D). As alteration of MIST1 caused a change of SNAIL1 but not
vice versa, it was indicated that MIST1 is an upstream gene of
SNAIL1. Furthermore, the results indicated that MIST1 promotes
anchorage independence of cells through regulating SNAI1.
MIST1 hijacks the PTEN/AKT signaling
pathway via SNAI1
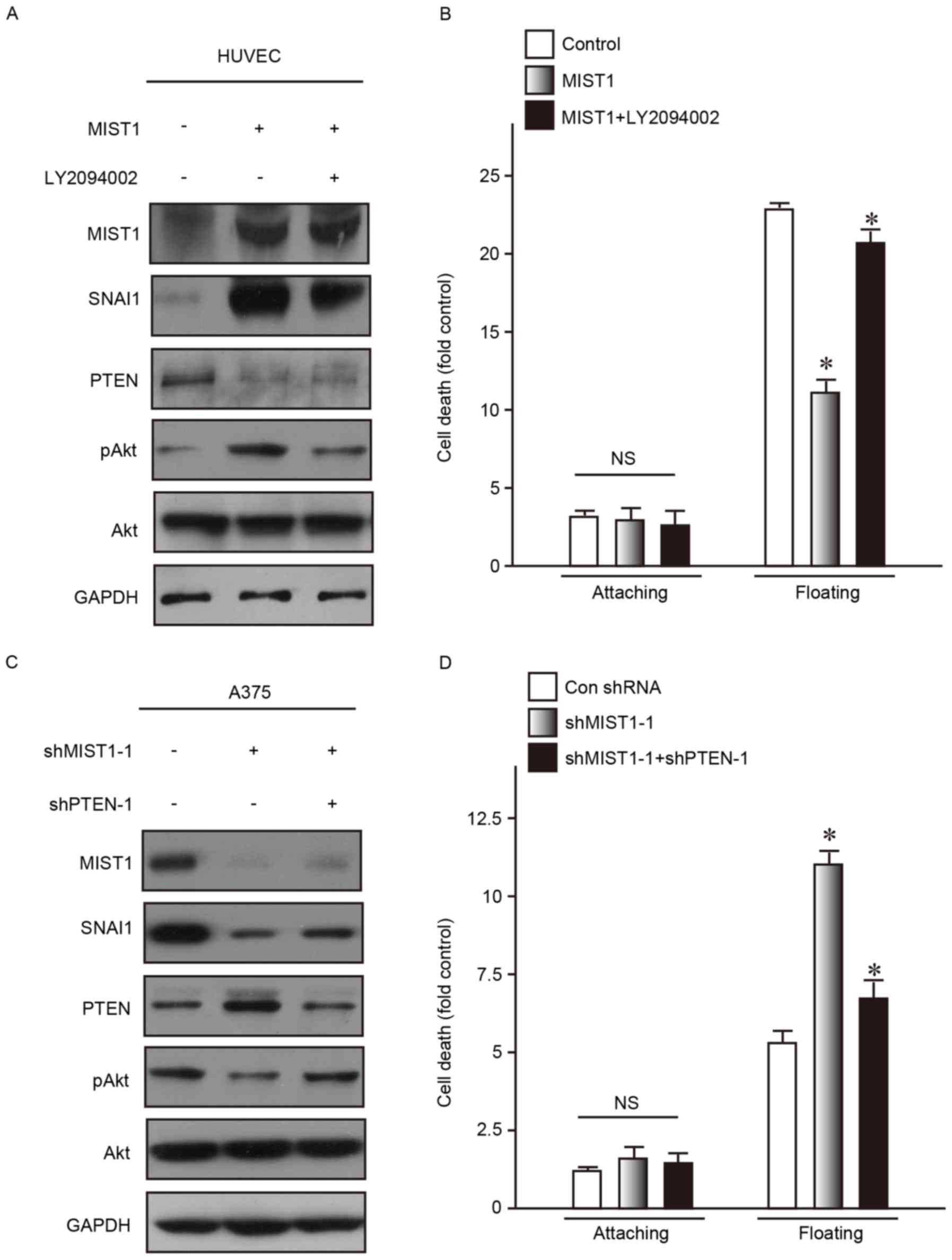
To investigate the molecular mechanisms via which
MIST1 and SNAI1, which are highly expressed in the A375 and MV3
human melanoma cell lines, reduce their sensitivity to anoikis, the
activation of Akt kinase, the downstream substrate of PI3K, was
examined. Dysregulation of Akt is commonly found in tumor cells and
has an important role in resistance to anoikis and promoting cell
survival (25). AKT phosphorylation
at the Ser473 residue was obviously increased following MIST1
overexpression in the normal human HUVEC cell line. In addition, it
was previously reported that SNAI1 directly repressed PTEN, which
then led to PI3K/Akt pathway activation, thus contributing to
anoikis resistance (23). Consistent
with these previous findings, the present study discovered
downregulation of PTEN by MIST1 and SNAI1 (Fig. 5A). As a competitive inhibitor of
PI3K, LY294002 is generally used to suppress p-AKT (Ser473), and
was applied in the present study at 10 µM to reduce p-AKT (Ser473).
After inhibition of AKT phosphorylation (Ser473), the MIST1-induced
reduction in sensitivity to anoikis was restored (Fig. 5A and B).
The expression of PTEN was increased in the A375 and
MV3 human melanoma cell lines after knockdown of MIST-1.
Correspondingly, the levels of p-AKT (Ser473) were obviously
decreased (Fig. 5C). shRNA targeting
PTEN was used to reduce its expression, which abrogated the
decrease in the levels of p-AKT (Ser473) caused by MIST1 knockdown
(Fig. 5C). In addition, the MIST1
knockdown-associated increases in anoikis were inhibited by
simultaneous PTEN knockdown.
The results of the gain- and loss-of-function study
suggested that the PTEN/Akt signaling pathway is involved in the
mechanism by which MIST1 and SNAI1 mediat anoikis resistance in
human melanoma cells.
MIST1 promotes SNAI1 transcription by
directly binding to its promoter region
The aforementioned results confirmed that MIST1
stimulates the expression of SNAI1, but the specific mechanism
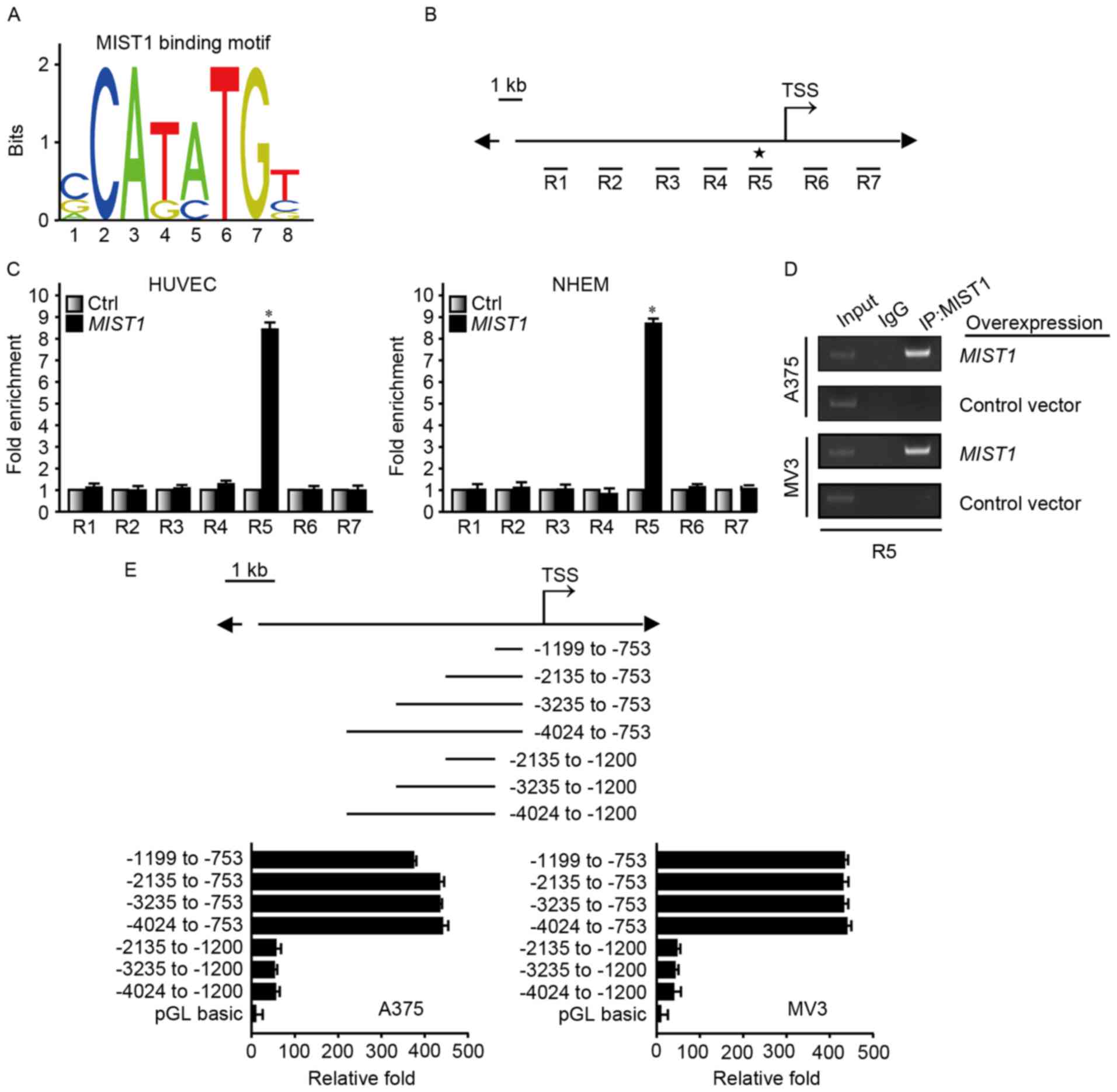
required further exploration. The MIST1 binding motif predicted by
the JASPAR database (http://jaspar.genereg.net/) exists in a binding
sequence predicted by the ENCODE database (https://genome.ucsc.edu/ENCODE/) (Fig. 6A). ChIP was used to confirm that
MIST1 directly regulated SNAI1. For this experiment, seven regions
close to the transcription start site were selected (Fig. 6B). Protein G PLUS-Agarose binding
with MIST1 antibody was used to pull down DNA-binding fragments. A
ChIP assay using HUVEC and NHEM overexpressing MIST1, the
amplification intensity of the R5 fragment from the SNAI1 promoter
was significantly higher than that of the other DNA segments, as
confirmed by PCR (Fig. 6C and D).
A375 and MV3, which highly express MIST1, were transfected with
luciferase reporter vectors. Vectors containing the −1,199 to −753
bp fragment demonstrated enhanced luciferase reporter signaling
(Fig. 6E). Taken together, these
results strongly indicated that MIST1 regulates the expression of
SNAI1 through binding to its promoter to ultimately confer
resistance to anoikis in human melanoma cells.
Discussion
Melanoma is a melanocytic malignancy derived from
the pigmented areas of the skin, mucous membrane, eye and central
nervous system, with a high degree of malignancy and a poor
prognosis. According to statistics, ~160,000 patients are newly
diagnosed with melanoma worldwide and ~48,000 succumb to melanoma
each year (26). The 5-year survival
rate of patients with early, advanced and late-stage melanoma,
respectively, was 91.2, 61.7 and 15.2%. In addition, the recurrence
rate in melanoma patients in remission is 9 times that of other
cancer types (26). Current
treatments used for melanoma are surgery, radiation therapy,
chemotherapy and immunotherapy. The clinical application of the
cytotoxic T-lymphocyte-associated protein 4 antibody ipilimumab and
the BRAF inhibitor vemurafenib, which were approved for the
treatment of advanced melanoma and BRAF mutant melanoma in 2011,
improved the treatment of melanoma to a certain degree, a
requirement for more in-depth study of the mechanisms underlying
the development and metastasis of melanoma remains, as more
effective targets to combat melanoma require to be developed.
The present study focused on the key step in the
metastasis of melanoma, namely resistance to anoikis. Anoikis helps
tumor cells to survive during migration from the original matrix
into the blood and lymph nodes. Studies have indicated that ECM as
an inhibitor of anoikis and integrins has a profound effect on cell
survival. The most important factors in integrin-regulated signal
transduction pathways include focal adhesion kinase,
integrin-linked protein kinase, tyrosine kinase, PI3K,
extracellular signal-regulated kinase and connexin Shc (27). The present study elucidated the
upstream signaling of the PTEN/Akt signaling pathway with regard to
anoikis.
MIST1 exists in numerous tissue types, but regarding
the cell type, mainly in serous exocrine cells. MIST1 is the first
transcription factor identified to be a protein specifically
expressed by serous exocrine cells and has a key role in the
formation and maintenance of serous secretory exocytosis. As a
well-known transcription factor, SNAI1 has a critical role in EMT
of tumor cells. However, the association between MIST1 and SNAI1
and their role in tumor progression has remained to be
elucidated.
In the present study, MIST1 was found to be
upregulated in the A375 and MV3 human melanoma cell lines. Ectopic
overexpression of MIST1 increased the resistance of normal cells to
anoikis. Conversely, knockdown of MIST1 expression reduced the
resistance of melanoma cells to anoikis. These suggested that MIST1
has a critical role in the resistance of melanoma cells to anoikis.
Gain- and loss-of-function experiments and ChIP confirmed that
SNAI1 was the downstream gene of MIST1. Furthermore, a luciferase
reporter assay located the MIST1 recognition sequence at −1,199 to
−753 bp of the SNAI1 gene promoter. It was further demonstrated
that MIST1 promotes the evasion of anoikis by melanoma cells
through the PTEN/Akt pathway.
To the best of our knowledge, the present study was
the first to report that MIST1 binds directly to the promoter
region of SNAI1 and regulates its expression. It was previously
demonstrated that SNAI1 inhibits the phosphorylation of Akt by
inhibiting the expression of PTEN (23). The present study provided an update
regarding the upstream signaling of the PTEN/Akt pathways. This
provided a novel target for the clinical treatment of melanoma.
However, the specific mechanism underlying the upregulation of
MIST1 in melanoma has remained elusive, and its study may provide
an approach for the clinical treatment of melanoma. In addition,
the role of MIST1 and SNAI1 in anoikis resistance requires further
verification in a wide range of tumor types.
References
|
1
|
Jemal A, Siegel R, Ward E, Hao Y, Xu J and
Thun MJ: Cancer statistics, 2009. CA Cancer J Clin. 59:225–249.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
High WA and Robinson WA: Genetic mutations
involved in melanoma: A summary of our current understanding. Adv
Dermatol. 23:61–79. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Giancotti FG and Tarone G: Positional
control of cell fate through joint integrin/receptor protein kinase
signaling. Annu Rev Cell Dev Biol. 19:173–206. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Paoli P, Giannoni E and Chiarugi P:
Anoikis molecular pathways and its role in cancer progression.
Biochim Biophys Acta. 1833:3481–3498. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tan K, Goldstein D, Crowe P and Yang JL:
Uncovering a key to the process of metastasis in human cancers: A
review of critical regulators of anoikis. J Cancer Res Clin Oncol.
139:1795–1805. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yang J, Zheng Z, Yan X, Li X, Liu Z and Ma
Z: Integration of autophagy and anoikis resistance in solid tumors.
Anat Rec (Hoboken). 296:1501–1508. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Thompson EW, Newgreen DF and Tarin D:
Carcinoma invasion and metastasis: A role for
epithelial-mesenchymal transition? Cancer Res. 65:5991–5995. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zanotti S, Gibertini S, Bragato C,
Mantegazza R, Morandi L and Mora M: Fibroblasts from the muscles of
Duchenne muscular dystrophy patients are resistant to cell
detachment apoptosis. Exp Cell Res. 317:2536–2547. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kadesch T: Consequences of heteromeric
interactions among helix-loop-helix proteins. Cell Growth Differ.
4:49–55. 1993.PubMed/NCBI
|
|
10
|
Jia D, Sun Y and Konieczny SF: Mist1
regulates pancreatic acinar cell proliferation through p21
CIP1/WAF1. Gastroenterology. 135:1687–1697. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wakamatsu Y, Sun Y and Konieczny SF:
Comparative gene expression analyses reveal heterochrony for Sox9
expression in the cranial neural crest during marsupial
development. Evol Dev. 16:197–206. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wu ZQ, Rowe RG, Lim KC, Lin Y, Willis A,
Tang Y, Li XY, Nor JE, Maillard I and Weiss SJ: A Snail1/Notch1
signalling axis controls embryonic vascular development. Nat
Commun. 5:39982014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Usova EV, Kopantseva MR, Egorov VI,
Kopantzev EP and Sverdlov ED: SNAI1 and SNAI2-transcriptional
master-regulators of epithelial-mesenchimal transition). Patol
Fiziol Eksp Ter. 59:76–87. 2015.(In Russian). PubMed/NCBI
|
|
14
|
Herranz N, Pasini D, Díaz VM, Francí C,
Gutierrez A, Dave N, Escrivà M, Hernandez-Muñoz I, Di Croce L,
Helin K, et al: Polycomb complex 2 is required for E-cadherin
repression by the Snail1 transcription factor. Mol Cell Biol.
28:4772–4781. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Moreno-Bueno G, Peinado H, Molina P,
Olmeda D, Cubillo E, Santos V, Palacios J, Portillo F and Cano A:
The morphological and molecular features of the
epithelial-to-mesenchymal transition. Nat Protoc. 4:1591–1613.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wu Y and Zhou BP: Snail: More than EMT.
Cell Adh Migr. 4:199–203. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Escrivà M, Peiró S, Herranz N, Villagrasa
P, Dave N, Montserrat-Sentís B, Murray SA, Francí C, Gridley T,
Virtanen I and García de Herreros A: Repression of PTEN phosphatase
by Snail1 transcriptional factor during gamma radiation-induced
apoptosis. Mol Cell Biol. 28:1528–1540. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang L, Wu J, Ling MT, Zhao L and Zhao
KN: The role of the PI3K/Akt/mTOR signalling pathway in human
cancers induced by infection with human papillomaviruses. Mol
Cancer. 14:872015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Guidetti GF, Canobbio I and Torti M:
PI3K/Akt in platelet integrin signaling and implications in
thrombosis. Adv Biol Regul. 59:36–52. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Fokas E, McKenna WG and Muschel RJ: The
impact of tumor microenvironment on cancer treatment and its
modulation by direct and indirect antivascular strategies. Cancer
Metastasis Rev. 31:823–842. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Safdari Y, Khalili M, Ebrahimzadeh MA,
Yazdani Y and Farajnia S: Natural inhibitors of PI3K/AKT signaling
in breast cancer: Emphasis on newly-discovered molecular mechanisms
of action. Pharmacol Res. 93:1–10. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chung JH and Eng C: Nuclear-cytoplasmic
partitioning of phosphatase and tensin homologue deleted on
chromosome 10 (PTEN) differentially regulates the cell cycle and
apoptosis. Cancer Res. 65:8096–8100. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Barrallo-Gimeno A and Nieto MA: The Snail
genes as inducers of cell movement and survival: Implications in
development and cancer. Development. 132:3151–3161. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Pin CL, Bonvissuto AC and Konieczny SF:
Mist1 expression is a common link among serous exocrine cells
exhibiting regulated exocytosis. Anat Rec. 259:157–167. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sheng S, Qiao M and Pardee AB: Metastasis
and AKT activation. J Cell Physiol. 218:451–454. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lozano R, Naghavi M, Foreman K, Lim S,
Shibuya K, Aboyans V, Abraham J, Adair T, Aggarwal R, Ahn SY, et
al: Global and regional mortality from 235 causes of death for 20
age groups in 1990 and 2010: A systematic analysis for the Global
Burden of Disease Study 2010. Lancet. 380:2095–2128. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hanks SK, Ryzhova L, Shin NY and Brábek J:
Focal adhesion kinase signaling activities and their implications
in the control of cell survival and motility. Front Biosci.
8:d982–d996. 2003. View
Article : Google Scholar : PubMed/NCBI
|