Introduction
Optic neuritis (ON) refers to a spectrum of
inflammatory lesions involving the optic nerve; it is a blinding
optic nerve disease that occurs most frequently in young and
middle-aged individuals. The most common type is idiopathic ON,
which is further subdivided into idiopathic demyelinating optic
neuritis (IDON), also known as multiple sclerosis-associated optic
neuritis (MS-ON), neuromyelitis optica-associated optic neuritis
(NMO-ON) and ON associated with other central nervous system
demyelinating diseases (1). NMO-ON
has a higher disability rate compared with IDON, and an aquaporin-4
(AQP4) immunoglobulin G (IgG) (+) status has a decisive role in the
diagnosis of NMO-ON. The probability of progressing to NMO-ON in
AQP4 IgG (+) patients is significantly higher than that of AQP4 IgG
(−) patients, and the rate of progression to NMO-ON is faster in
AQP4 IgG (+) patients. Detection of AQP4 IgG, as a clinical
biomarker for identifying NMO-ON and other demyelinating diseases,
is helpful for the early diagnosis and prognostication of NMO-ON
patients. However, a clinical study indicated that 10–20% of NMO-ON
patients have an AQP4 IgG (−) status (2).
New diagnostic criteria for NMO-ON were developed by
the International NMO Diagnostic Group in 2015 (3); diagnostic criteria for serum-negative
NMO-ON were proposed, and it was suggested to actively search for
potential alternative biomarkers for AQP4 IgG (−) NMO-ON patients.
It has been reported that AQP4 IgG (−) NMO-ON patients are myelin
oligodendrocyte glycoprotein (MOG) IgG (+) (4,5), and it
is therefore speculated that MOG IgG is involved in the
pathogenesis in these serum AQP4 IgG (−) NMO-ON patients. Saadoun
et al (6) microinjected MOG
IgG, sourced from patients with NMO, into mouse brains and compared
the results with those obtained by AQP4 IgG injection. The results
indicated that MOG IgG caused myelin changes and altered the
expression of axonal proteins within two weeks, but did not produce
any inflammation, axonal loss or neuronal or astrocyte death, while
AQP4 IgG produced complement-mediated myelin loss, and neuronal and
astrocyte death with limited recovery at two weeks. In addition,
MOG IgG is also detected in the serum of certain adolescent
patients with acute disseminated encephalomyelitis and multiple
sclerosis; besides intracranial lesions, these patients also suffer
from ON. Therefore, MOG IgG should be actively screened for ON
patients with an unknown cause.
Patients and methods
Patients
A total of 43 RON patients (11 males and 32 females;
age, 30–51 years) admitted to the Neurology Department of Beijing
Tongren Hospital Affiliated to Capital Medical University (Beijing,
China) from December 2014 to May 2015 were enrolled in the present
study. At the same time, 8 healthy individuals (4 males and 4
females; age, 32–49 years) admitted to the same hospital from
December 2014 to May 2015 were included randomly. The present study
was approved by the ethics committee of Beijing Tongren Hospital
(Beijing, China). Written informed consent was obtained from the
patients and/or their guardians, as well as the healthy
individuals.
Inclusion/exclusion criteria
Patients with an acute ON attack meeting the
diagnostic criteria of the American Optic Neuritis Study Group
(1) were included. They were
required to have had two or more ON attacks previously according to
their complete medical data. Patients with contraindications for
intravenous application of methylprednisolone and/or
immunosuppressors, and patients with other ophthalmic diseases
(including anterior segment lesions, vitreous lesions, retinopathy,
refractive errors and glaucoma), other types of ON (including
ischemic, oppressive, invasive, traumatic, toxic, nutritional
metabolic and hereditary disease), diminution of vision due to
intracranial diseases (e.g., other cerebrovascular, infectious,
traumatic, degenerative, genetic metabolic or nutritional metabolic
disease, or poisoning), alcohol encephalopathy or Alzheimer's
disease, as well as pregnant or lactating patients were excluded
from the study.
Diagnostic criteria for ON
The diagnosis of ON was based on the diagnostic
criteria of the American Optic Neuritis Study Group (1): Acute vision loss accompanied with or
without eye pain, nerve fibre bundle damage-associated visual field
anomaly and at least one of the following two criteria: Relative
afferent pupillary defect and visual evoked potential abnormality;
no clinical and laboratory evidence of compressive, ischemic,
toxic, hereditary, metabolic and invasive optic neuropathy;
clinical and laboratory evidence of retinal disease and other
ocular and neurological disorders without leading to acute vision
loss; patients with typical onset of acute ON for >24 h via
objective examination with an interval of at least one month since
the last episode.
Data collection
The demographic data (onset age and sex) and
clinical features, including the course of the disease,
monocular/binocular involvement, eye pain, optic disc edema, worst
visual acuity at onset, worst videofluoroscopic swallowing study
(VFSS) at onset, visual recovery after treatment with prednisone
and/or immunosuppressive agents, VFSS score after treatment, onset
frequency of ON, involvement of intraorbital segment, canal segment
and intracranial segment of optic nerve and optic chiasma, the
presence of other lesions involving the central nervous system,
other autoimmune abnormalities (e.g., anti-nuclear antibody,
anti-extractable nuclear antigens antibody spectrum,
anti-double-stranded DNA antibody, human leukocyte antigen-B27,
anti-neutrophil cytoplasmic antibodies and cardiolipin antibody) of
patients were collected. AQP4 and MOG IgG in the serum of patients
were detected via the cell-based assay (CBA). All patients were
treated with methylprednisolone in the acute phase, and certain
patients received the immunosuppressive therapy during the process
of hormone reduction. One patient was diagnosed with multiple
sclerosis and treated with interferon. All patients were followed
up, and the visional recovery of patients at 6 months after the
last onset was recorded via outpatient review upon visitation to
the clinc.
CBA for detection of AQP4 and MOG IgG
status
The 293 cell line (widely known as Human Embryonic
Kidney 293 cells) was obtained from the Cell Bank of the Chinese
Academy of Sciences (Shanghai, China). The 293 cells with stable
expression of MOG were established as the substrate for the CBA.
AQP4 IgG and MOG IgG in the serum specimens of recurrent ON (RON)
patients was detected via indirect immunofluorescence assay
(4). The expression vectors of the
full-length sequences of AQP4 and MOG were constructed, and
plasmids were transferred into 293 cells with the Neofect
transfection reagent (Nofectbiotech, Beijing, China). At 24 h after
transfection, the cells were fixed with immune fixative (4%
Polyoxymethylene; Sigma-Aldrich; Merck KGaA, Darmstadt, Germany)
and blocked with 10% goat serum (Gibco; Thermo Fisher Scientific,
Inc., Waltham, MA, USA) for 30 min. Patient serum was diluted at
1:100 and incubated with 293 cells at room temperature overnight.
The cells were incubated with Alexa Fluor 488-conjugated sheep
anti-human antibody (1:250; cat. no. ab201540; Abcam, Cambridge,
UK) for 30 min at 20°C, and then washed three times with PBS
containing Tween-20, followed by staining with DAPI (0.1 ug/ml) for
10 min at 20°C. The cells were then observed under a fluorescence
microscope after mounting. The AQP4 and MOG IgG status of RON
patients was then determined; AQP4 and MOG are transmembrane
glycoproteins, and NMO-associated IgGs are marked by green
fluorescence in the cell membrane. A positive and negative control
were set up in the experiment as follows: Rabbit anti-human MOG (1
µg/ml; cat. no. M0695) and AQP4 (1 µg/ml) antibody (cat. no. G9626;
both Sigma-Aldrich; Merck KGaA) was added to the serum obtained
from a healthy individual as the positive control at room
temperature overnight, while serum obtained from a healthy
individual was used as a negative control.
RON grouping
AQP4 antibody and MOG IgG in patients meeting the
diagnostic criteria for RON were detected via the CBA. According to
the results, they were divided into MOG IgG (+) AQP4 IgG (−) group
[MOG (+) group], MOG IgG (−) AQP4 IgG (+) group [AQP4 IgG (+)
group], MOG IgG (−) and AQP4 IgG (−) group [MOG/AQP4 IgG (−) group]
and MOG IgG (+) and AQP4 IgG (+) group [MOG/AQP4 IgG (+) group].
The demographic data, degree of visual impairment, brain and optic
nerve magnetic resonance imaging (MRI) and other laboratory test
data were collected. The clinical characteristics of each subgroup
were analyzed.
Observational indexes
The visual function data of the 43 patients enrolled
were collected and the visual impairment of 83 affected eyes was
observed. A corrected visual acuity of the affected eye of >0.1
indicated relatively good visual acuity, while that of ≤0.1
indicated poor visual acuity. Visual function VFSS score were
determined prior to and at 6 months after treatment with
methylprednisolone and/or immunosuppressive agents.
Statistical analysis
Statistical analysis was performed using SPSS 18.0
software (SPSS, Inc., Chicago, IL, USA). A normality test was
performed for continuous variables; data with a normal distribution
were expressed as the mean ± standard deviation, while those with a
skewed distribution were expressed as the median [25% quartile-75%
quartile (Q25-Q75)]. One-way analysis of variance was used for
intergroup comparisons, followed by the Least-Significant
Difference test as a post-hoc test, while the Kruskal-Wallis test
followed by a Mann-Whitney U post-hoc test with Bonferroni's
correction was applied in the case of an abnormal distribution. The
chi-square test was used for categorical variables. The
demographics, clinical features, characteristics of laboratory and
imaging examinations, vision at onset and visual function recovery
at 6 months after treatment were compared among the MOG IgG (+)
group, AQP4 IgG (+) group, MOG/AQP4 IgG (+) and MOG/AQP4 IgG (−)
groups. P<0.05 was considered to indicate a statistically
significant difference.
Results
Case presentation of a NMO-ON patient
with MOG IgG (+) status
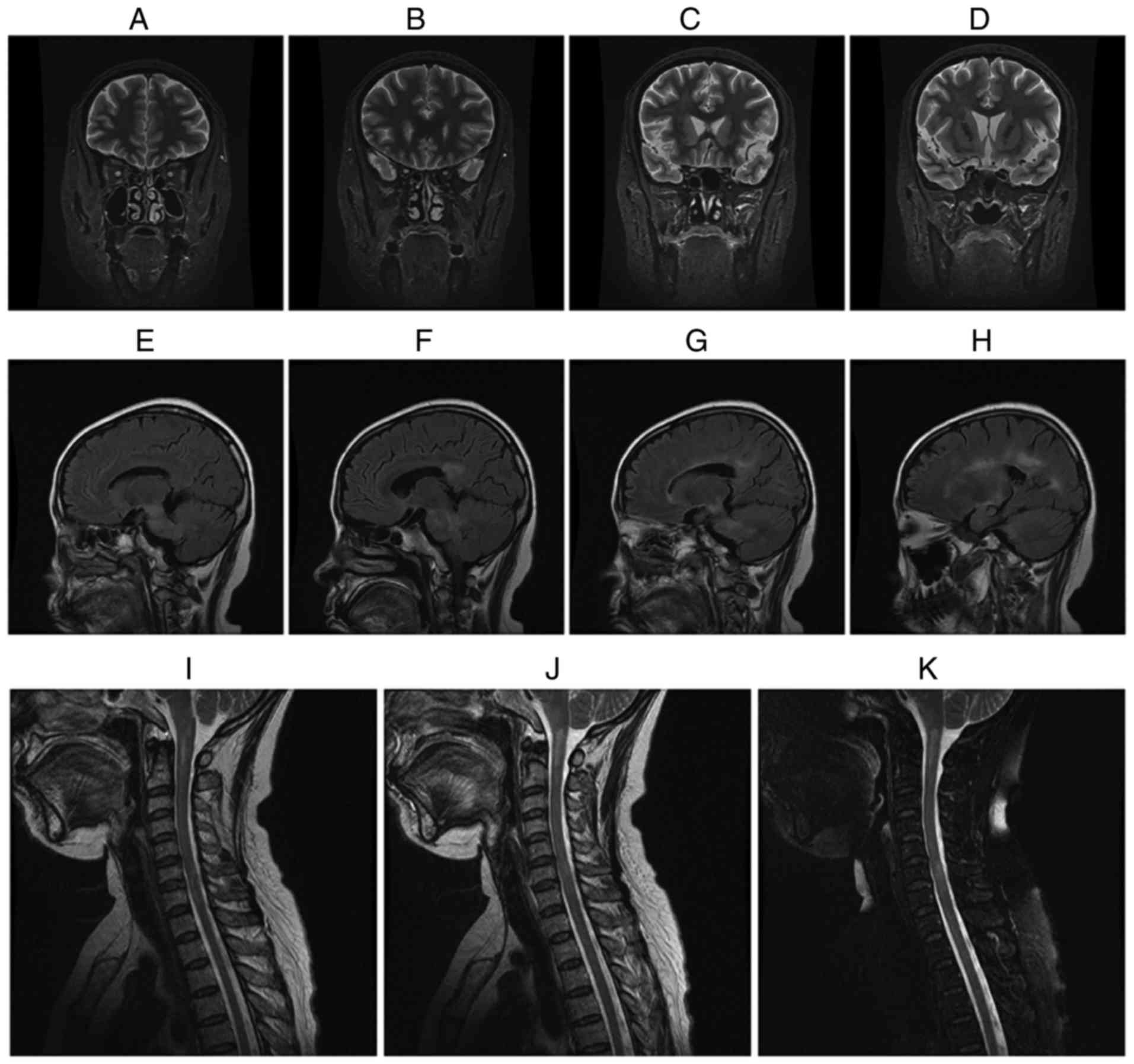
Fig. 1 displays
representative MRI images of a female MOG IgG (+) NMO-ON patient
with the onset age of 38 years, course of disease of 16 years and a
history of 15 attacks who had acute ON in both eyes. The patient
presented with acute myelitis involving >3 segments, postrema
syndrome and acute diencephalic syndrome. As presented in Fig. 1A-D, the brain MRI coronal short-time
inversion recovery revealed that the intraorbital segment, canal
segment and intracranial segment of the optic nerve and optic
chiasma had become thinner and the signal was increased when
compared with earlier images obtained from patients prior to the
study. As presented in Fig. 1E-H,
the brain MRI sagittal T2 fluid-attenuated inversion recovery
revealed an abnormal signal in the bilateral basal ganglia,
thalamus, lateral ventricle, brain stem and corpus callosum. As
presented in Fig. 1I-K, cervical MRI
sagittal T2-weighted images revealed an abnormal signal in
medullary-C2 and C5-T1.
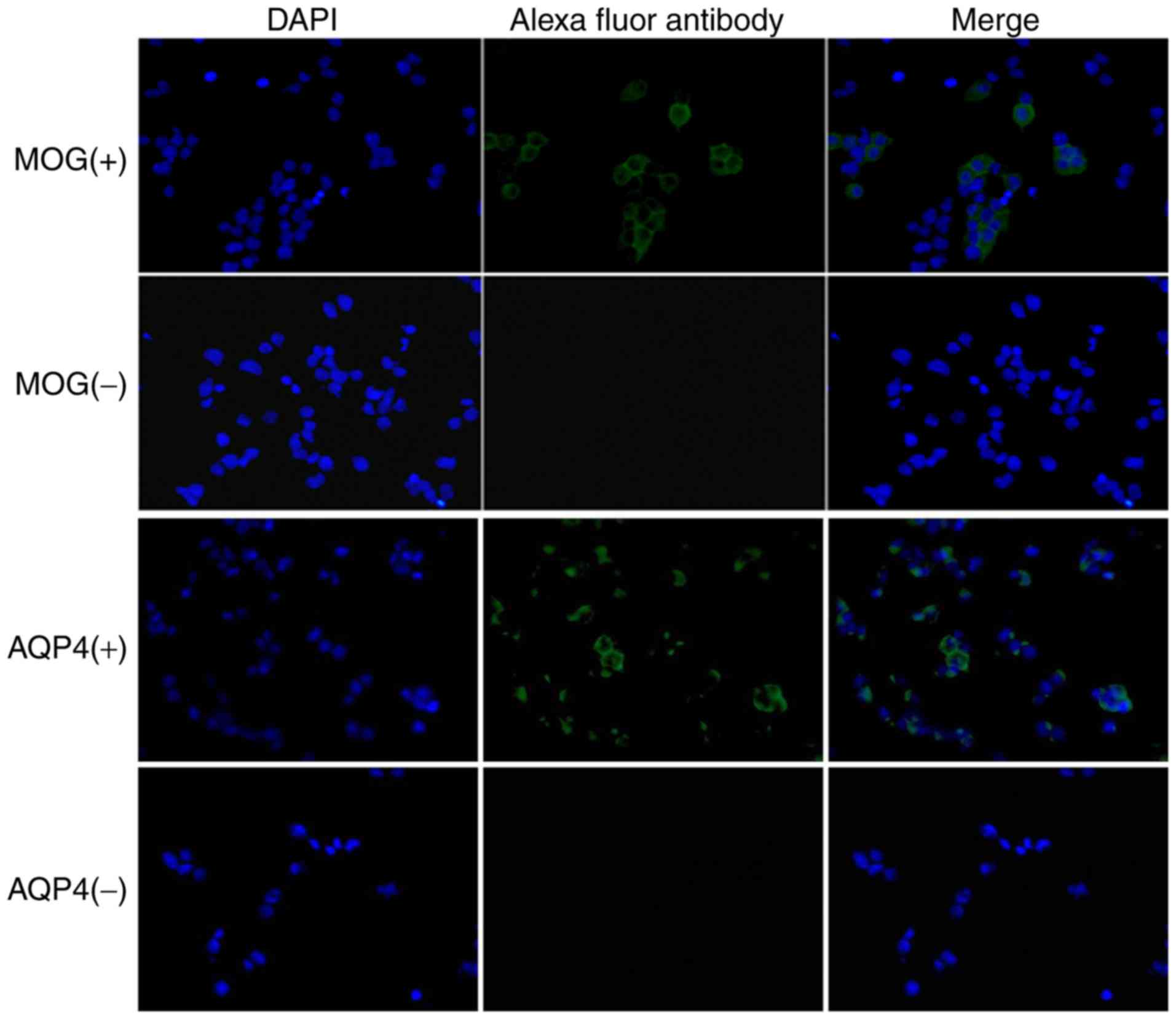
Detection of MOG IgG via CBA
The 293 cells with stable expression of the complete
genomic sequence of MOG were successfully constructed. MOG IgG was
detected via a CBA and observed under a fluorescence microscope. As
presented in Fig. 2, the
representative images demonstrated that the cells incubated with
serum from the MOG IgG (+) and AQP4 (+) patients had green
fluorescence while DAPI-stained nuclei exhibited blue
fluorescence.
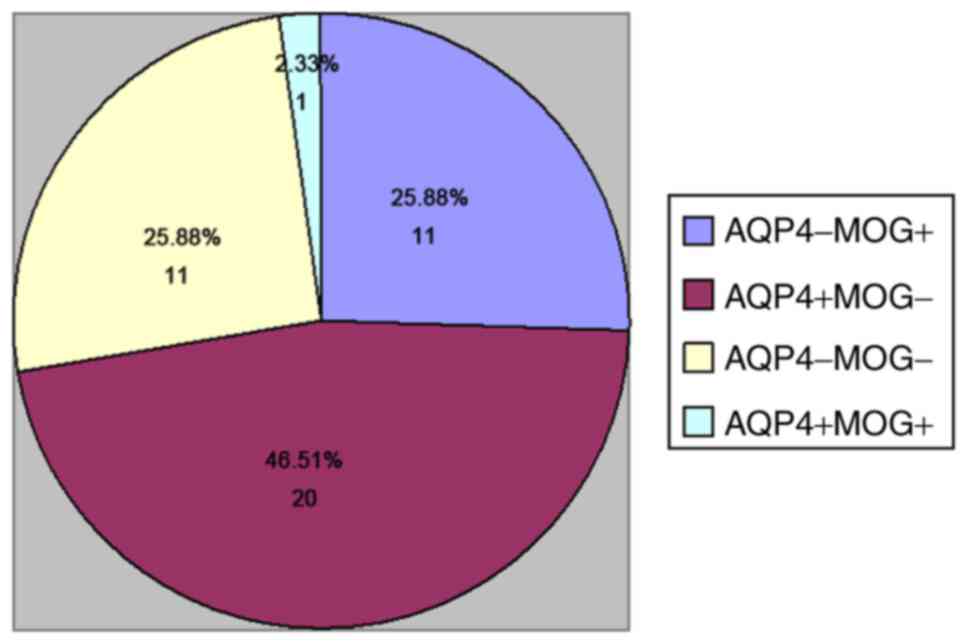
Basic data and clinical features of
NMO-ON patients in each group
A total of 43 patients hospitalized from December
2014 to May 2015 met the inclusion criteria. The cohort comprised
11 cases of MOG IgG (+) (25.58%), 20 cases of AQP4 IgG (+)
(46.51%), 11 cases of MOG/AQP4 IgG (−) (25.58%) and 1 case of
MOG/AQP4 IgG (+) (2.33%; Fig. 3). A
total of 32 females (74.42%) and 11 males (25.58%) with an average
age at onset of 33±12 years and the total course of disease of 24
months (Q25-Q75, 12–111) were included. A total of 27 cases
(62.79%) had eye pain and 17 cases (39.53%) presented with optic
disk edema after onset. In the 43 patients, 83 eyes were affected,
including 20 eyes in the MOG IgG (+) group, 39 eyes in the AQP4 IgG
(+) group, 22 eyes in the MOG/AQP4 IgG (−) group and 2 eyes in the
MOG/AQP4 IgG (+) group. The brain MRI indicated that there were 15
cases (34.88%) of demyelinating lesions in the brain parenchyma,
and optic nerve MRI revealed that there were 43 cases (100.00%)
with intraorbital segment involvement of the optic nerve, 32 cases
(74.42%) with canal segment involvement, 18 cases (18.60%) with
intracranial segment involvement and 8 cases (18.60%) with optic
chiasma involvement. Furthermore, the spinal MRI confirmed that
there were 5 cases (11.62%) of spinal cord involvement, including 4
cases (9.30%) of spinal cord involvement of ≥3 segments.
Serological examination indicated that 18 cases (41.86%) had
abnormalities in other autoimmune indexes.
As presented in Tables
I and II, the AQP4 IgG (+)
group was mostly comprised of females compared with the MOG IgG (+)
group and the MOG/AQP4 IgG (−) group (P<0.05). Compared with the
MOG/AQP4 IgG (−) group, optic disk edema was rarer with fewer
attacks in the AQP4 IgG (+) group (P<0.05); however, no
statistically significant difference was identified between the MOG
IgG (+) and the AQP4 IgG (+) and MOG/AQP4 IgG (−) groups. Optic
nerve MRI indicated that the intraorbital segments of the optic
nerve were involved in the three groups, but the canal segment and
intracranial segment of the optic nerve in the AQP4 IgG (+) group
were more significantly involved than those in the other two groups
(P<0.05). The optic chiasma was more significantly involved in
the AQP4 IgG (+) group than that in the MOG/AQP4 IgG (−) group
(P<0.05), and there was no statistically significant difference
in the involvement of optic chiasma between the MOG IgG (+) group
and the other two groups. There were no statistically significant
differences in the age at onset, occurrence of eye pain, course of
disease, intracranial lesions, spinal cord involvement and other
immune indexes among the three groups.
 | Table I.Comparisons of general features and
clinical manifestation between different groups. |
Table I.
Comparisons of general features and
clinical manifestation between different groups.
|
|
|
|
|
| P-value |
|---|
|
|
|
|
|
|
|
|---|
| Item | MOG antibody (+) | AQP4 antibody
(+) | MOG/AQP4 antibody
(−) | MOG/AQP4 antibody
(+) | MOG antibody (+) vs.
AQP4 antibody (+) | MOG antibody (+) vs.
MOG/AQP4 antibody (−) | AQP4 antibody (+) vs.
MOG/AQP4 antibody (−) |
|---|
| Cases (n) | 11 | 20 | 11 | 1 |
|
|
|
| Sex
(male/female) | 4/7 | 1/19 | 6/5 | 0/1 | 0.041 | 0.691 | 0.005 |
| Age at onset
(years) | 31.55±13.03 | 33.65±11.29 | 32.55±12.17 | 28 | 0.386 | 0.963 | 0.442 |
| Median course of
disease (months) | 24 | 30 | 12 | 156 | 0.531 | 0.158 | 0.334 |
| Eye pain n (%) | 7 (63.6%) | 13 (65%) | 7 (63.6%) | 0 (0%) | 1.000 | 1.000 | 1.000 |
| Optic disk edema
(n) | 5 (45.5%) | 4 (20%) | 8 (72.3%) | 0 (0%) | 0.228 | 0.369 | 0.006 |
| Median onset
frequency (time) | 2 | 3 | 2 | 5 | 0.224 | 0.273 | 0.006 |
| Maximum VFSS score
prior to treatment | 5 | 6 | 5 | 5 | 0.009 | 0.164 | 0.055 |
| Maximum VFSS score at
6 months after treatment | 1 | 3 | 1 | 3 | 0.001 | 0.053 | 0.014 |
| Affected eyes n
(%) | 20 (90.1%) | 39 (97.5%) | 22 (100%) | 2 (100%) |
|
|
|
| Vision ≤0.1 at onset
n (%) | 10 (45.5%) | 33 (82.5%) | 13 (59.1) | 1 (50%) | 0.013 | 0.733 | 0.034 |
| Vision ≤0.1 after
treatment n (%) | 2 (9.1%) | 20 (50%) | 2 (9.1%) | 0 (0%) | 0.002 | 1.000 | 0.001 |
| Table II.Comparison of imaging characteristics
and treatments between different groups. |
Table II.
Comparison of imaging characteristics
and treatments between different groups.
|
|
|
|
|
| P-value |
|---|
|
|
|
|
|
|
|
|---|
| Item | MOG antibody (+) | AQP4 antibody
(+) | MOG/AQP4 antibody
(−) | MOG/AQP4 antibody
(+) | MOG antibody (+) vs.
AQP4 antibody (+) | MOG antibody (+) vs.
MOG/AQP4 antibody (−) | AQP4 antibody (+) vs.
MOG/AQP4 antibody (−) |
|---|
| Optic nerve MRI |
|
|
|
|
|
|
|
|
Intraorbital segment of optic
nerve n (%) | 11 (25.6) | 20 (46.5) | 11 (25.6) | 1 (2.3) | – | – | – |
| Canal
segment of optic nerve n(%) | 6 (54.5) | 19 (95) | 6 (54.5) | 0 (0) | 0.011 | 1.000 | 0.012 |
|
Intracranial segment of optic
nerve n (%) | 2 (18.2) | 14 (70) | 1 (9.1) | 0 (0) | 0.008 | 1.000 | 0.002 |
| Optic
chiasma n (%) | 1 (9.1) | 7 (35) | 0 (0) | 0 (0) | 0.213 | 1.000 | 0.036 |
| Brain MRI |
|
|
|
|
|
|
|
|
Intracranial lesions n
(%) | 3 (27.3) | 9 (45) | 2 (18.2) | 1 (100) | 0.472 | 1.000 | 0.211 |
| Spinal MRI |
|
|
|
|
|
|
|
| Spinal
involvement n (%) | 1 (9.1) | 4 (20) | 0 (0) | 0(0) | 0.655 | 1.000 | 0.287 |
|
Abnormality in other immune
indexes n (%) | 3 (27.3) | 10 (50) | 4 (36.4) | 1 (100) | 0.285 | 1.000 | 0.735 |
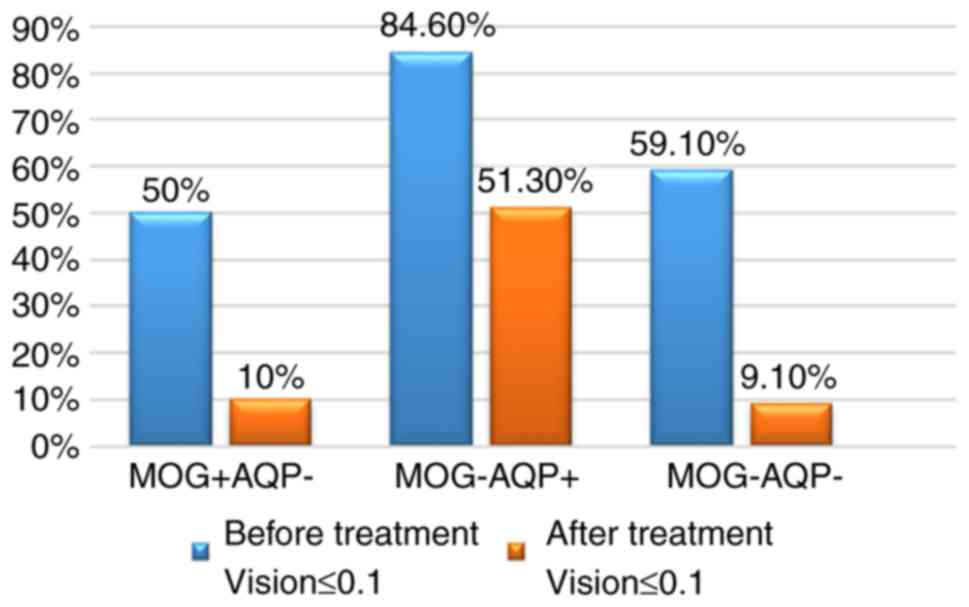
Comparison of visual function
prognosis of patients
All patients enrolled were followed up at the clinic
6 months after onset, with items assessed including visual acuity,
visual field and ocular fundus. All of the patients received the
same treatment. Prior to treatment, poor visual acuity was
determined in 10 eyes (50.0%) of the MOG IgG (+) group, 33 eyes
(84.6%) of the AQP4 IgG (+) group and 13 eyes (59.1%) of the
MOG/AQP4 IgG (−) group. At 6 months after treatment, poor visual
acuity was still present in 2 eyes (10.0%) of the MOG IgG (+), 20
eyes (51.3%) of the AQP4 IgG (+) and 2 eyes (9.1%) of the MOG/AQP4
IgG (−) group (Fig. 4). The vision
at onset was worse and the recovery was poorer in the AQP4 IgG (+)
group compared with those in the MOG IgG (+) group and MOG/AQP4 IgG
(−) groups (P<0.05; Table I).
As presented in Table
I, the VFSS score was relatively lower with the worst visual
function after onset and the visual function recovery after
treatment was better in the MOG IgG (+) group compared with that in
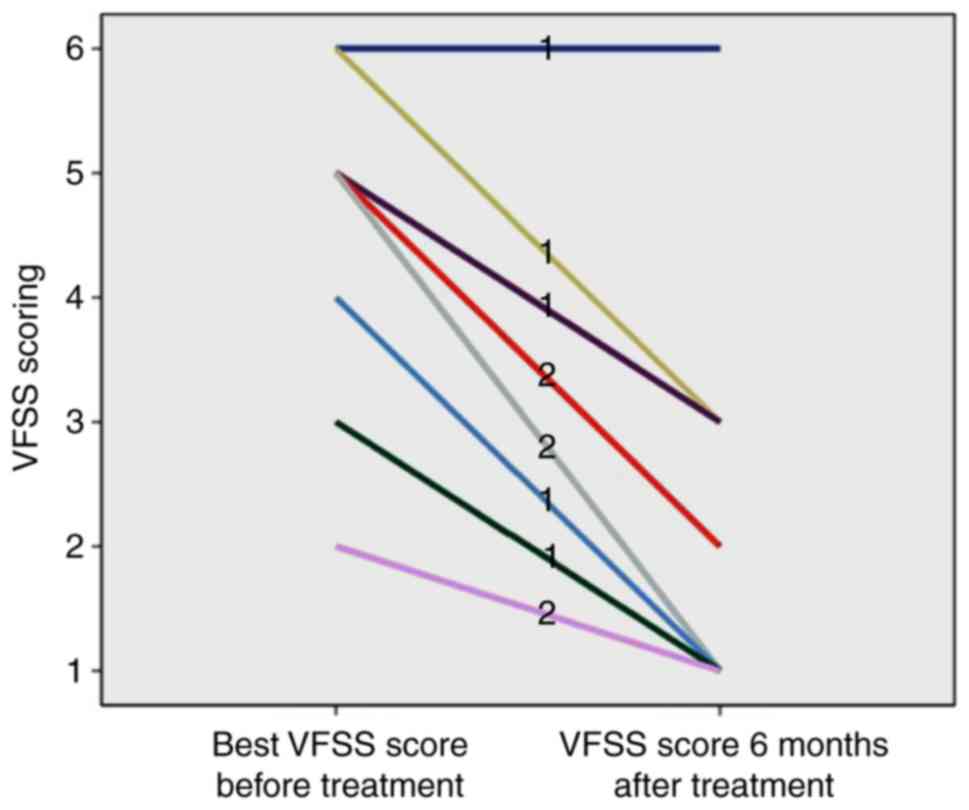
the AQP4 IgG (+) group (P<0.05). As presented in Figs. 5 and 6, although 1 case in the MOG IgG (+) group
(9.09%) had a poor recovery of visual function, the remaining 10
cases (90.91%) had a better recovery of visual function, and the
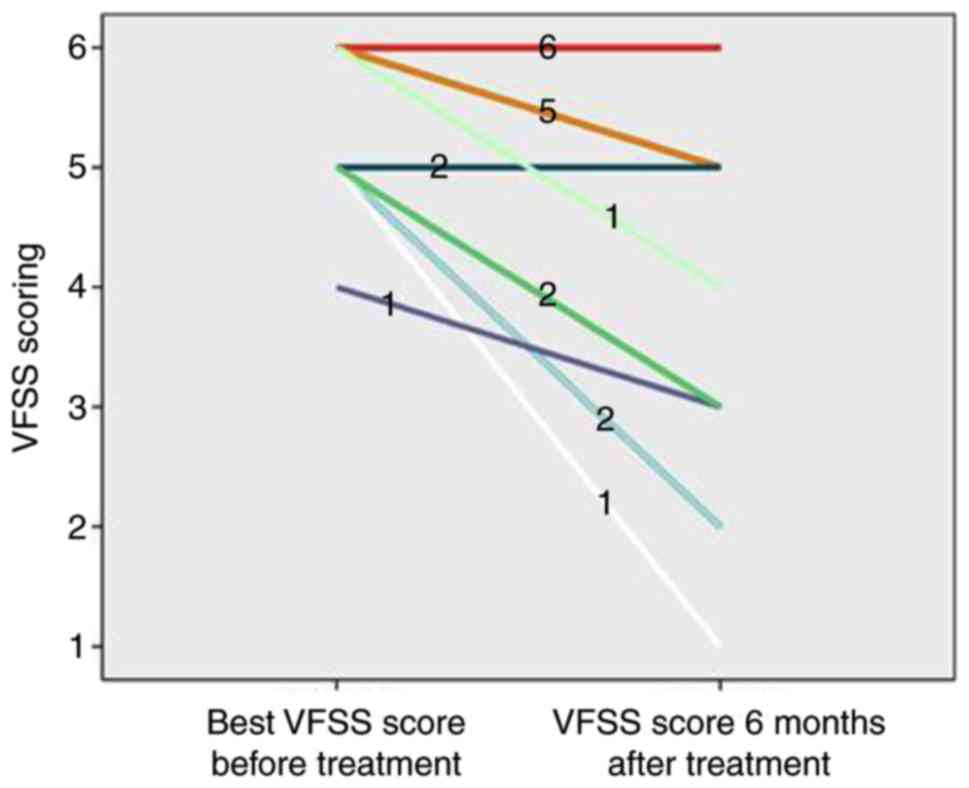
VFSS score after treatment ranged between 1 and 3. The majority of
patients in the AQP4 IgG (+) group (14 cases, 70.00%) had a poor
visual function recovery. After treatment, the VFSS score ranged
between 4 and 6; a small number of AQP4 IgG (+) patients (6 cases,
30.00%) had a better recovery, and the score ranged between 1 and
3.
Association between MOG IgG and
NMO-ON
Out of the 12 MOG IgG (+) patients, 2 met the
diagnostic criteria for NMO-ON (Table
I) (2), including 1 case of AQP4
IgG (+) NMO-ON; this patient had a course of disease of 13 years, a
right vision at onset of ≤0.1 and a left vision at onset of
>0.1, and presented with a recurrent attack of ON in both eyes.
After hormone and immunosuppressive therapy, the vision was
recovered to be >0.1 in both eyes, and the VFSS score was
recovered from 5 to 3 points; the intraorbital segment of the optic
nerve was involved, but the intracranial segment, canal segment and
optic chiasma were not involved; there were demyelinating lesions
in the intracranial semi-oval center, but the spinal cord was not
involved. The other case as aforementioned was an AQP4 IgG (−)
NMO-ON patient with a course of disease of 16 years and a history
of 15 attacks, who presented with a recurrent attack of ON in both
eyes, acute myelitis involving >3 segments, postrema syndrome
and acute diencephalic syndrome, which used to be diagnosed as
‘multiple sclerosis’. After interferon treatment, the patient was
diagnosed with NMO-ON and treated with hormone and
immunosuppressive therapy. The recovery of vision on both eyes was
poor (≤0.1); The VFSS score prior to and after treatment was 6
points. Imaging indicated involvement of the intraorbital segment,
canal segment, intracranial segment and optic chiasma of bilateral
optic nerves, demyelinating lesions in the intracranial medulla
oblongata, thalamus, midbrain, corpus callosum, basal ganglia,
frontal white matter, lateral ventricle, and medullary C2 and
C5-T1.
Discussion
In the present study, 27.91% of RON patients were
MOG IgG (+) [including 1 case that was also AQP4 IgG (+)] with a
male/female ratio of 1:1.75. In 2015, Matsuda et al
(7) detected the MOG antibody in 70
patients with ON, and the results indicated that 18 cases were MOG
IgG (+), including 7 males and 11 females (1:1.57), and the MOG IgG
(+) rate was 25.7%. The above results were similar to those of the
present study.
In the present study, the age at onset, course of
disease, onset frequency and other immune indexes of MON IgG (+)
RON patients were not significantly different compared with those
of AQP4 IgG (+) RON and MON/AQP4 IgG (−) RON patients. However,
another study indicated that compared with AQP4 IgG (+) patients,
MOG IgG (+) patients were younger at onset, and the prevalence was
not higher in females as in the present study (8). Other studies have reported that the age
at onset was not significantly different between MOG IgG (+) and
AQP4 IgG (+) patients (9). In 2015,
Nakajima et al (10) detected
the MOG antibody in patients with idiopathic ON and determined that
MOG IgG (+) patients with idiopathic ON present with eye pain. In
this study, 63.6% patients in the MOG IgG (+) group had eye pain,
but there was no significant difference compared with the AQP4 IgG
(+) and MON/AQP4 IgG (−) groups. In 2014, Sato et al
(11) detected that 16 out of 215
NMO-ON patients had a MOG IgG (+) status. It was revealed that
those MOG IgG (+) patients had a monophase course compared with
AQP4 IgG (+) patients (11).
Furthermore, in this study, the onset frequency was not
significantly different between the MOG IgG (+) group and the AQP4
IgG (+) group, which may be due to the fact that all patients in
the experiment had a relapsing course, and there was a lack of
research on the monophase course.
Through imaging analysis, intraorbital segment
involvement of RON was identified in each group of the present
study. Compared with those in the MOG IgG (+) group and MOG/AQP4
IgG (−) group, more canal segment and intracranial segment
involvement was observed in the AQP4 IgG (+) group. Compared with
the MOG/AQP4 IgG (−) group, the optic chiasma was more frequently
involved in the AQP4 IgG (+) group. Thus, the involved part of the
optic nerve in MOG IgG (+) RON was more forward, but that in the
AQP4 IgG (+) RON was more backward, which was similar to the
results of Ramanathan et al (12).
Two protocols were adopted in the present study for
the assessment of the worst vision at onset and visual function
after recovery. First, the visual changes were directly observed,
and the worst vision after onset and better vision after recovery
were evaluated with 0.1 as the threshold. Second, the VFSS score
was given for the degree of visual impairment prior to and after
treatment, and the changes in score were observed. Regardless of
the evaluation protocol, the vision at onset of MOG IgG (+) RON
patients was not as poor as that of AQP4 IgG (+) RON patients, and
the visual recovery was better in MOG IgG (+) RON patients after
hormone with/without immunosuppressive therapy. A number of studies
have reported similar results (13–15).
In the present study, two cases met the diagnostic
criteria for NMO-ON, including 1 case of AQP4 antibody-negative
NMO-ON with RON, acute myelitis, postrema syndrome and diencephalic
syndrome, which also suggested that MOG may be a potential
biomarker for AQP4 IgG (−) NMO-ON. Therefore, for clinical patients
with suspected NMO-ON with AQP4 IgG (−) status, the detection of
serum MOG antibody may be performed, so as to facilitate early
diagnosis and treatment, and thereby improve their prognosis.
Compared with the AQP IgG (+) group, the higher
prevalence in females in the MOG IgG (+) RON group was less
pronounced, the vision at onset was better, the recovery of visual
function after treatment was better, and the involved part of the
optic nerve was more forward. MOG IgG may be present in the serum
of NMO-ON patients, which may be utilized as a potential biomarker
for NMO-ON with AQP4 IgG (−) status.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JW and YP designed the present study; LL and YZ
collected the data, YZ and JW analysed the data; YP, ZQ and KF
performed the experiments, and prepared the manuscript. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the ethics
committee of Beijing Tongren Hospital (Beijing, China). Written
informed consent was obtained from the patients and/or their
guardians, as well as the healthy individuals.
Consent for publication
Written informed consent was obtained from the
patients and/or their guardians, as well as the healthy
individuals.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Schneider E, Zimmermann H, Oberwahrenbrock
T, Kaufhold F, Kadas EM, Petzold A, Bilger F, Borisow N, Jarius S,
Wildemann B, et al: Optical coherence tomography reveals distinct
patterns of retinal damage in neuromyelitis optica and multiple
sclerosis. Plos One. 8:e661512013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wan H, He H, Zhang F, Sha Y and Tian G:
Diffusion-weighted imaging helps differentiate multiple sclerosis
and neuromyelitis optica-related acute optic neuritis. J Magn Reson
Imaging. 45:1780–1785. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tan CT, Mao Z, Qiu W, Hu X, Wingerchuk DM
and Weinshenker BG: International consensus diagnostic criteria for
neuromyelitis optica spectrum disorders. Neurology. 86:491–492.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kitley J, Waters P, Woodhall M, Leite MI,
Murchison A, George J, Kuker W, Chandratre S, Vincent A and Palace
J: Neuromyelitis optica spectrum disorders with aquaporin-4 and
myelin-oligodendrocyte glycoprotein antibodies: A comparative
study. Jama Neurol. 71:276–283. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Giacoppo S, Soundara RT, Galuppo M,
Pollastro F, Grassi G, Bramanti P and Mazzon E: Purified
Cannabidiol, the main non-psychotropic component of Cannabis
sativa, alone, counteracts neuronal apoptosis in experimental
multiple sclerosis. Eur Rev Med Pharmacol Sci. 19:4906–4919.
2015.PubMed/NCBI
|
|
6
|
Saadoun S, Waters P, Owens GP, Bennett JL,
Vincent A and Papadopoulos MC: Neuromyelitis optica MOG-IgG causes
reversible lesions in mouse brain. Acta Neuropathol Commun.
2:352014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Matsuda R, Kezuka T, Umazume A, Okunuki Y,
Goto H and Tanaka K: Clinical profile of Anti-Myelin
oligodendrocyte glycoprotein antibody seropositive cases of optic
neuritis. Neuroophthalmology. 39:213–219. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hoftberger R, Sepulveda M, Armangue T,
Blanco Y, Rostasy K, Calvo AC, Olascoaga J, Ramio-Torrenta L,
Reindl M, Benito-Leon J, et al: Antibodies to MOG and AQP4 in
adults with neuromyelitis optica and suspected limited forms of the
disease. Mult Scler. 21:866–874. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kim SM, Woodhall MR, Kim JS, Kim SJ, Park
KS, Vincent A, Lee KW and Waters P: Antibodies to MOG in adults
with inflammatory demyelinating disease of the CNS. Neurol
Neuroimmunol Neuroinflamm. 2:e1632015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Nakajima H, Motomura M, Tanaka K, Fujikawa
A, Nakata R, Maeda Y, Shima T, Mukaino A, Yoshimura S, Miyazaki T,
et al: Antibodies to myelin oligodendrocyte glycoprotein in
idiopathic optic neuritis. BMJ Open. 5:e77662015. View Article : Google Scholar
|
|
11
|
Sato DK, Callegaro D, Lana-Peixoto MA,
Waters PJ, de Haidar JF, Takahashi T, Nakashima I,
Apostolos-Pereira SL, Talim N, Simm RF, et al: Distinction between
MOG antibody-positive and AQP4 antibody-positive NMO spectrum
disorders. Neurology. 82:474–481. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ramanathan S, Prelog K, Barnes EH, Tantsis
EM, Reddel SW, Henderson AP, Vucic S, Gorman MP, Benson LA, Alper
G, et al: Radiological differentiation of optic neuritis with
myelin oligodendrocyte glycoprotein antibodies, aquaporin-4
antibodies, and multiple sclerosis. Mult Scler. 22:470–482. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Akaishi T, Nakashima I, Takeshita T,
Kaneko K, Mugikura S, Sato DK, Takahashi T, Nakazawa T, Aoki M and
Fujihara K: Different etiologies and prognoses of optic neuritis in
demyelinating diseases. J Neuroimmunol. 299:152–157. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Siritho S, Sato DK, Kaneko K, Fujihara K
and Prayoonwiwat N: The clinical spectrum associated with myelin
oligodendrocyte glycoprotein antibodies (anti-MOG-Ab) in thai
patients. Mult Scler. 22:964–968. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Martinez-Hernandez E, Sepulveda M, Rostasy
K, Hoftberger R, Graus F, Harvey RJ, Saiz A and Dalmau J:
Antibodies to aquaporin 4, myelin-oligodendrocyte glycoprotein, and
the glycine receptor α1 subunit in patients with isolated optic
neuritis. JAMA Neurol. 72:187–193. 2015. View Article : Google Scholar : PubMed/NCBI
|