Introduction
Esophageal cancer (EC) has the sixth highest
mortality and the eighth highest incidence rate worldwide (1). Its incidence rate in China is the
highest in the world (2). The
primary histological type is esophageal squamous-cell carcinoma
(ESCC), which accounts for ~90% of all EC cases in China. Patients
with ESCC are usually diagnosed at an advanced stage, and their
5-year survival rate is therefore low (~10-20%) (3,4). It has
been reported that smoking and alcohol consumption are major
causative factors of ESCC, as they promote gene mutations
associated with processes including tumor initiation, progression
and even metastasis. Thus, it is important to understand the
molecular mechanisms of the tumorigenesis process to identify
targets and develop novel treatments for ESCC.
Various genes, mRNAs and micro (mi)RNAs have been
reported to form a network regulating the tumorigenesis and
development of EC. Numerous studies have indicated that certain
genes act as tumor suppressors, and that several genes inhibit
cancer cell migration, invasion and tumor progression in ESCC
(5–8). High-throughput sequencing technologies,
including microarrays, which are able to detect changes in the
expression of a vast amount of genes, have been widely used in
cancer diagnosis and cancer research. In a previous study, numerous
differentially expressed genes (DEGs) were detected in the tumor
tissues of patients with ESCC relative to those in normal tissue or
normal epithelial cells by microarrays (9). These hundreds of DEGs are involved in
signalling pathways in ESCC, which encompass biological processes
(BP), molecular functions (MF) and cellular components (CC). Hu
et al (10) examined DEGs in
tumor and matched normal adjacent tissue samples from patients with
ESCC using microarrays. However, the regulatory roles of these
DEGs, including the pathways in their interaction network, have
remained to be elucidated (10).
Therefore, in the present study, bioinformatics
methods were used to analyze the DEGs and their interaction
networks. Original data were downloaded from the Gene Ontology
(GEO) database (http://www.ncbi.nlm.nih.gov/geo/). The DEGs were
identified from tumor tissues of patients with ESCC compared with
those in matched normal adjacent tissues. The 200 top DEGs were
then selected for further bioinformatics analysis, including
analysis of Gene Ontology (GO) terms, Kyoto Encyclopedia of Genes
and Genomes (KEGG) pathways, protein-protein interaction (PPI)
networks and Spearman's correlation tests (11). In general, the present study may help
identify potential therapeutic targets and provide valuable
information to further illuminate the molecular mechanisms of
ESCC.
Materials and methods
Microarray data
Gene expression profiles of GSE20347 were downloaded
from the GEO repository collated by Hu et al (10). These data were based on the
AgilentGPL571 platform (Affymetrix Human Genome U133A 2.0 Array,
HG-U133A_2; Affymetrix; Thermo Fisher Scientific, Inc., Waltham,
MA, USA), which included 13 samples of normal adjacent esophageal
tissues (with the ID nos. GSM509787-GSM509803) and 17 samples of
tumor tissues (with the ID nos. GSM509804-GSM509820) from patients
with ESCC. Total RNA had been extracted using the PureLink
Micro-to-Midi RNA Purification System (Invitrogen; Thermo Fisher
Scientific, Inc.) and was detected by Affymetrix HG-U133A 2.0 gene
expression arrays (Affymetrix; Thermo Fisher Scientific, Inc.). R
(Bioconductor; http://www.bioconductor.org/) was used for background
correction and normalization of the data.
Identification of DEGs
The raw data files used for analysis included TXT
files (Agilent platform). The files were used to create heat maps
with the Morpheus online software (https://software.broadinstitute.org/morpheus/). The
data were classified into two groups, namely the normal and tumor
groups. The top 200 DEGs (100 upregulated and 100 downregulated
genes) were screened by their signal-to-noise ratio (SNR).
GO and pathway enrichment analysis of
DEGs
The top 200 DEGs were analyzed using with the
database for annotation, visualization, integrated discovery
(DAVID) online tool (https://david.ncifcrf.gov/; DAVID bioinformatics
resources 6.8 of the National Institute of Allergy and Infectious
Diseases/National Institutes of Health), which may be used for the
high-throughput functional analysis of genes (12). The database includes GO and KEGG
pathway analyses. GO is a useful tool for identifying
characteristic biological information by using high-throughput
genome transcriptome data (13) and
was used in the present study for GO enrichment analysis in the
categories BP, cellular component (CC) and MF. KEGG pathway
analysis was also performed to gain insight into the signaling
pathways regulated by the DEGs (14–16).
PPI network and module analysis
The Search Tool for the Retrieval of Interacting
Genes/Proteins (STRING; http://version10.string-db.org/; version 10.0)
database is a powerful online tool that overlays 9.6 million
proteins from 2,034 organisms and 184 million interactions. The top
200 DEGs were analyzed by STRING for the PPI. Subsequently,
interactions with a combined score of >0.4 (the value that was
considered statistically significant) were selected to construct
the PPI network using Cytoscape software 3.4.0 (http://www.cytoscape.org). Finally, the plug-in
Molecular Complex Detection (MCODE) of Cytoscape was used to
construct the modules of the PPI network. The top modules with an
MCODE score of >3 and a node number of >4 were selected for
further pathway analysis with KEGG.
Spearman correlation test of the top
200DEGs in patients with ESCC
To further determine DEGs that has the most
connections to the other top DEG, a Spearman's correlation test was
performed in Excel 2007 (Microsoft Corp., Redmond, WA, USA).
Subsequently, the genes were screened with the following settings:
0.95<Pearson correlation coefficient (PCC) <1 and
−1<PCC<-0.95. The gene with the highest correlation was
selected as the hub gene to construct the co-expression network in
Cytoscape. Finally, GO enrichment analyses in the categories BP, CC
and MF for the hub gene and their associated genes were further
performed by (Biological Networks Gene Ontology) BiNGO tool
(17) and the KEGG analysis was
performed with the ClueGO plug-in of Cytoscape.
Sample collection
ESCC and normal tissue samples were collected from
the Panyu Central Hospital and the Third Affiliated Hospital of
Southern Medical University (Guangzhou, China) from June 30, 2015
to May 2, 2017. All patients were male, with a mean age of
62.20±5.98 years, and diagnosed by clinical pathology (14 cases
were squamous carcinoma and 1 was adenocarcinoma). All samples were
stored at −80°C after collection. A total of 30 samples, which
included 15 tumor samples and 15 normal adjacent tissue samples
(>3 cm from the tumor tissue) as controls, were used to detect
the gene expression of SLURP by reverse transcription-quantitative
polymerase chain reaction (RT-qPCR).
RT-qPCR assay
Total RNA was extracted from all samples using
TRIzol (Invitrogen; Thermo Fisher Scientific, Inc.) according to
the manufacturer's instructions. First-strand complementary (c)DNA
was synthesized with 1 µg total RNA per sample using the
All-in-One™ First-Strand cDNA Synthesis kit (Gene Copoeia, Inc.,
Rockville, MD, USA). Subsequently, the cDNA sample was amplified
using All-in-One qPCR mix (Gene Copoeia, Inc.) in a final volume of
20 µl in an ABI Vii7 dx reactor (Applied Biosystems; Thermo Fisher
Scientific, Inc.). The amplifications were performed as follows:
Initial incubation for 2 min at 50°C, denaturation for 30 sec at
95°C, and 45 cycles of 95°C for 5 sec and 65.6°C for 34 sec. The
experiments were performed in triplicate and quantified using
melting curve analysis. β-actin was used as an endogenous reference
control. The relative gene expression levels were calculated using
the 2−ΔΔCq method (18).
The primer pairs for SLURP-1 and β-actin were as follows: SLURP-1
forward, 5′-GCTCCTGTGTGGCCACCGAC-3′ and reverse,
5′-GAGCCAGGCCCCGTCAGAGA-3′; β-actin forward,
5′-ACTCTTCCAGCCTTCCTTCC-3′ and reverse,
5′-GCGGCGCAATACGAATGCCCC-3′.
Statistical analysis
P<0.05 was considered to indicate a statistically
significant difference. The P-value was adjusted by using the false
discovery rate (FDR) method for multiple hypothesis testing. FDR
<0.05 was established as the threshold (15–17). The
data are expressed as mean ± standard deviation. Independent
t-tests were used to analyze the PCR results and performed using
SPSS software (version 16.0; SPSS, Inc., Chicago, IL, USA).
Results
Identification of DEGs
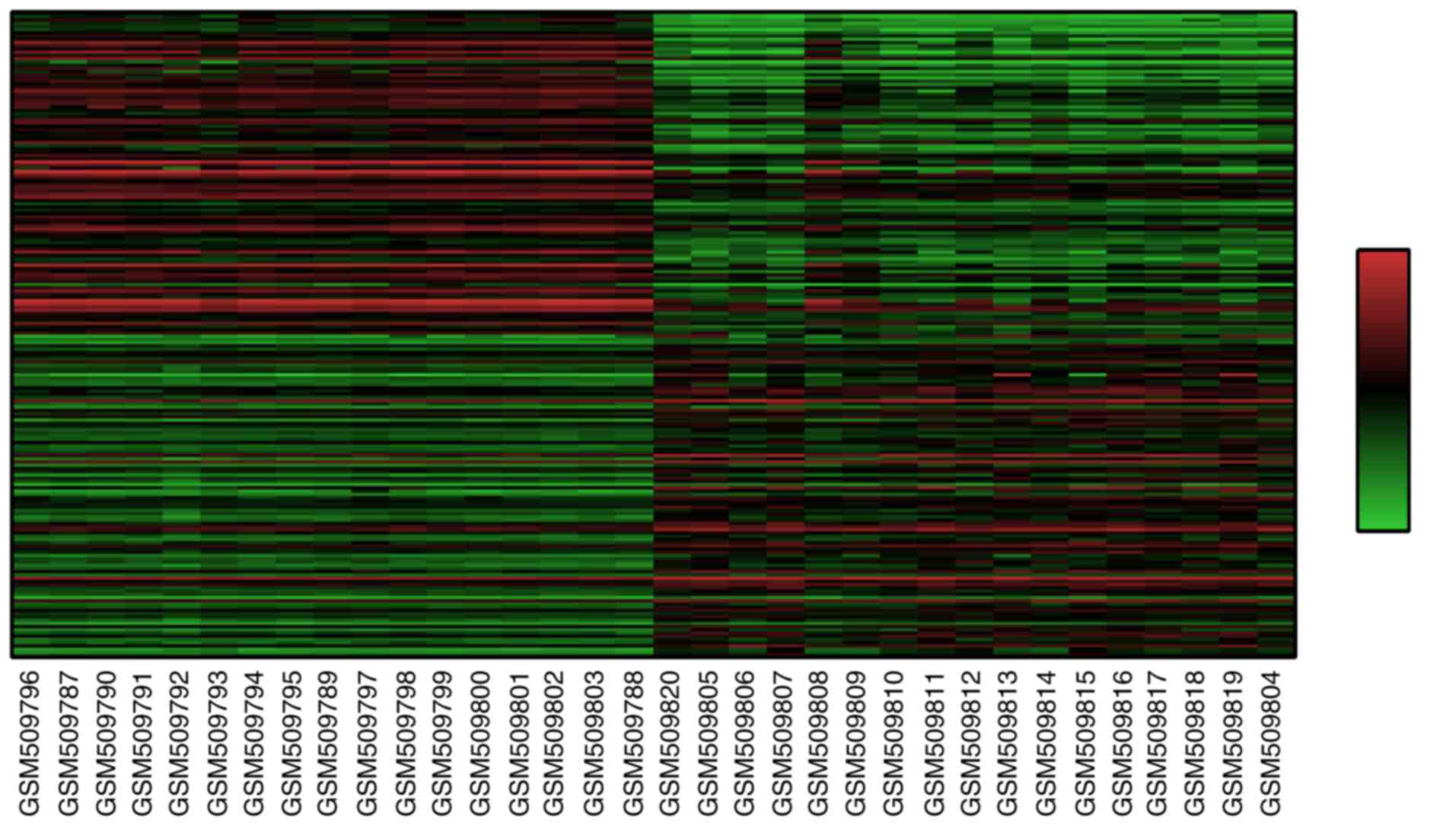
A total of 22,277 DEGs were identified. The
expression of these genes was analyzed with the Morpheus online
tool to form a heat map (top 100 upregulated and 100 downregulated
genes), which were selected according to their SNR value. The heat
map is presented in Fig. 1.
GO term enrichment and KEGG pathway
analysis of DEGs
GO term enrichment analysis indicated that the
upregulated DEGs were most significantly enriched in membrane-bound
vesicles in the category CC, but no significant enrichment was
identified in the categories BP and MF. The downregulated DEGs were
most significantly enriched in the regulation of DNA metabolic
processes, nucleotide binding and chromosomes in the categories BP,
MF and CC, respectively (Table I).
The KEGG analysis indicated that the downregulated DEGs were
enriched in the regulation of cell cycle pathways (Table II).
 | Table I.GO analysis of differentially
expressed genes in esophageal squamous cell carcinoma. |
Table I.
GO analysis of differentially
expressed genes in esophageal squamous cell carcinoma.
| A, Upregulated
genes in the category cellular component |
|---|
| GO term | Function | N (%) | P-value |
|---|
| GO:0031988 | Membrane-bound
vesicle | 33 (0.25) |
4.02×10−8 |
| GO:0070062 | Extracellular
exosome | 29 (0.22) |
1.27×10−7 |
| GO:1903561 | Extracellular
vesicle | 29 (0.22) |
1.45×10−7 |
| GO:0043230 | Extracellular
organelle | 29 (0.22) |
1.46×10−7 |
| GO:0044421 | Extracellular
region part | 30 (0.23) |
3.16×10−5 |
|
| B, Downregulated
genes |
|
| GO term |
Function | N (%) | P-value |
|
| Biological
process |
|
|
|
|
GO:0006259 | DNA metabolic
process | 17 (0.11) |
1.81×10−7 |
|
GO:0051276 | Chromosome
organization | 17 (0.11) |
1.33×10−6 |
|
GO:0010564 | Regulation of cell
cycle process | 12 (0.08) |
7.36×10−6 |
|
GO:1903047 | Mitotic cell cycle
process | 12 (0.08) |
2.68×10−5 |
| Cellular
component |
|
|
|
|
GO:0005694 | Chromosome | 20 (0.13) |
2.80×10−8 |
|
GO:0044427 | Chromosomal
part | 19 (0.12) |
4.98×10−8 |
|
GO:0098687 | Chromosomal
region | 11 (0.07) |
1.35×10−6 |
|
GO:0000228 | Nuclear
chromosome | 14 (0.09) |
1.78×10−6 |
|
GO:0000793 | Condensed
chromosome | 8 (0.05) |
1.00×10−5 |
|
GO:0044454 | Nuclear chromosome
part | 12 (0.08) |
3.14×10−5 |
| Molecular
function |
|
|
|
|
GO:0000166 | Nucleotide
binding | 27 (0.17) |
6.38×10−6 |
|
GO:1901265 | Nucleoside
phosphate binding | 27 (0.17) |
6.38×10−6 |
|
GO:0035639 | Purine
ribonucleoside triphosphate binding | 23 (0.15) |
1.03×10−5 |
|
GO:0032550 | Purine
ribonucleoside binding | 23 (0.15) |
1.08×10−5 |
|
GO:0001883 | Purine nucleoside
binding | 23 (0.15) |
1.09×10−5 |
|
GO:0032549 | Ribonucleoside
binding | 23 (0.15) |
1.12×10−5 |
|
GO:0001882 | Nucleoside
binding | 23 (0.15) |
1.18×10−5 |
|
GO:0032555 | Purine
ribonucleotide binding | 23 (0.15) |
1.41×10−5 |
|
GO:1901363 | Heterocyclic
compound binding | 44 (0.28) |
1.48×10−5 |
|
GO:0017076 | Purine nucleotide
binding | 23 (0.15) |
1.50×10−5 |
|
GO:0032553 | Ribonucleotide
binding | 23 (0.15) |
1.62×10−5 |
|
GO:0005524 | ATP binding | 20 (0.13) |
1.70×10−5 |
|
GO:0036094 | Small molecule
binding | 27 (0.17) |
1.97×10−5 |
|
GO:0097159 | Organic cyclic
compound binding | 44 (0.28) |
2.02×10−5 |
|
GO:0032559 | Adenyl
ribonucleotide binding | 20 (0.13) |
2.15×10−5 |
|
GO:0030554 | Adenyl nucleotide
binding | 20 (0.13) |
2.28×10−5 |
| Table II.Kyoto Encyclopedia of Genes and
genomes pathway analysis of differentially expressed genes in
esophageal squamous cell carcinoma. |
Table II.
Kyoto Encyclopedia of Genes and
genomes pathway analysis of differentially expressed genes in
esophageal squamous cell carcinoma.
| Expression | Term | Function | N (%) | P-value |
|---|
| Downregulated | cfa04110 | Cell cycle
pathway | 8 (0.05) |
1.62×10−5 |
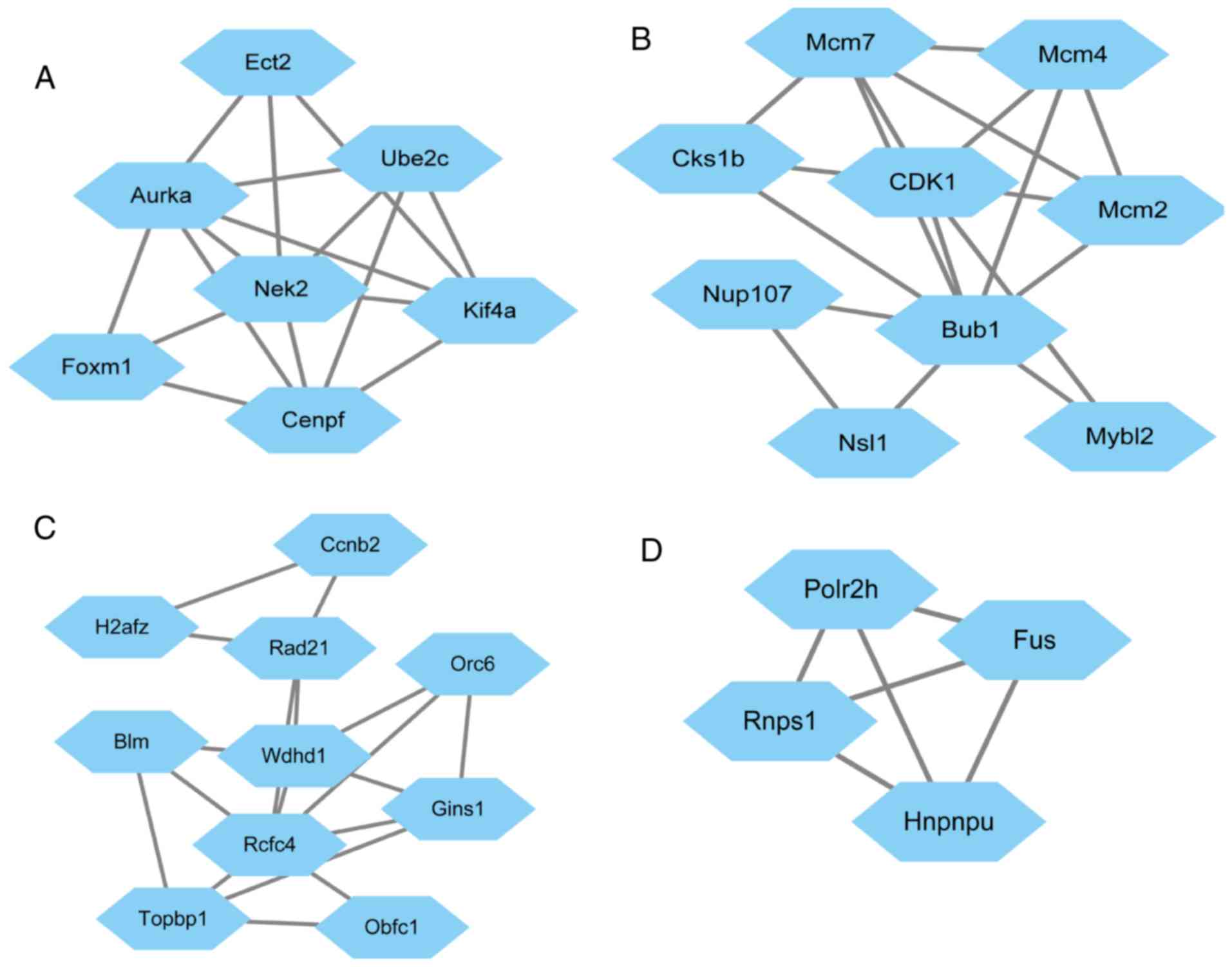
PPI network and top module
construction
Based on the STRING database, the PPI network of the
top 200 DEGs was constructed with Cytoscape software (Fig. 2). In the PPI networks, nodes with a
high degree of connectivity were defined as hub proteins. The top
10 hub proteins included cyclin-dependent kinase 4 (CDK4), budding
uninhibited by benzimidazoles 1 (BUB1) and cyclin B2 (CCNB2). The
degree of connectivity of CDK4 was 30, and it was therefore the
most highly connected node. The network consisted of 110 nodes and
262 edges. The top four significant modules were selected for
further pathway enrichment analysis (MCODE score, >3; number of
nodes, >4) and the results indicated that the genes were
significantly enriched in cell cycle pathways (Table III).
| Table III.The enriched pathway of module 2. |
Table III.
The enriched pathway of module 2.
| Pathway | P-value | False discovery
rate | Nodes |
|---|
| Cell cycle |
1.71×10−6 |
9.35×10−4 | Cyclin-dependent
kinases regulatory subunit 1, Cyclin-dependent kinase 1, MCM7,
Nuclear pore complex protein Nup107, MCM4, MCM2, Mitotic checkpoint
serine/threonine-protein kinase BUB1, KAT8 regulatory NSL complex
subunit 1, Myb-related protein B |
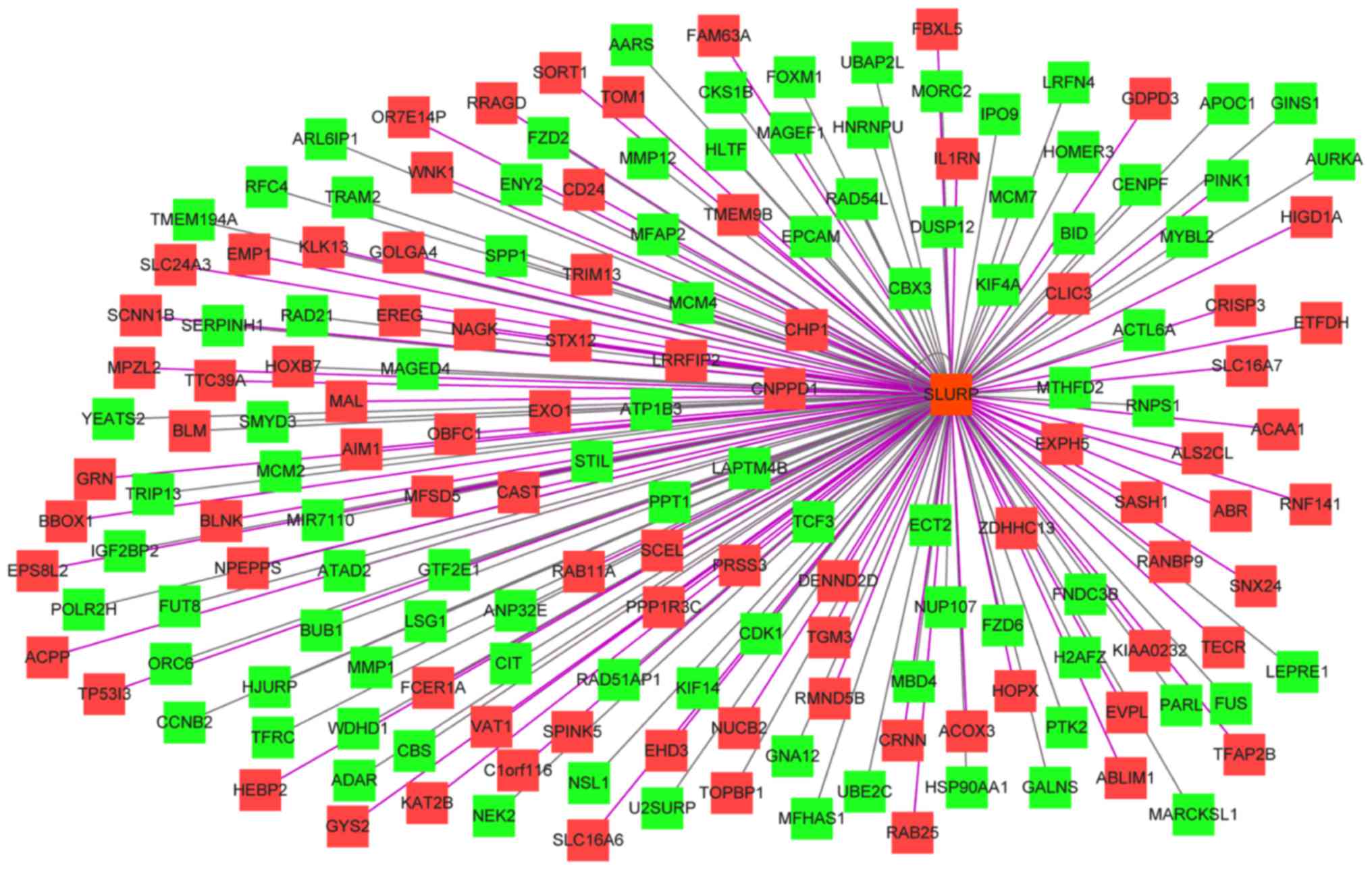
Spearman correlation analysis of the
top 200 DEGs
The results of the Spearman correlation analysis
indicated that the most connected gene was secreted LY6/PLAUR
domain (SLURP). SLURP was selected as a hub gene and further
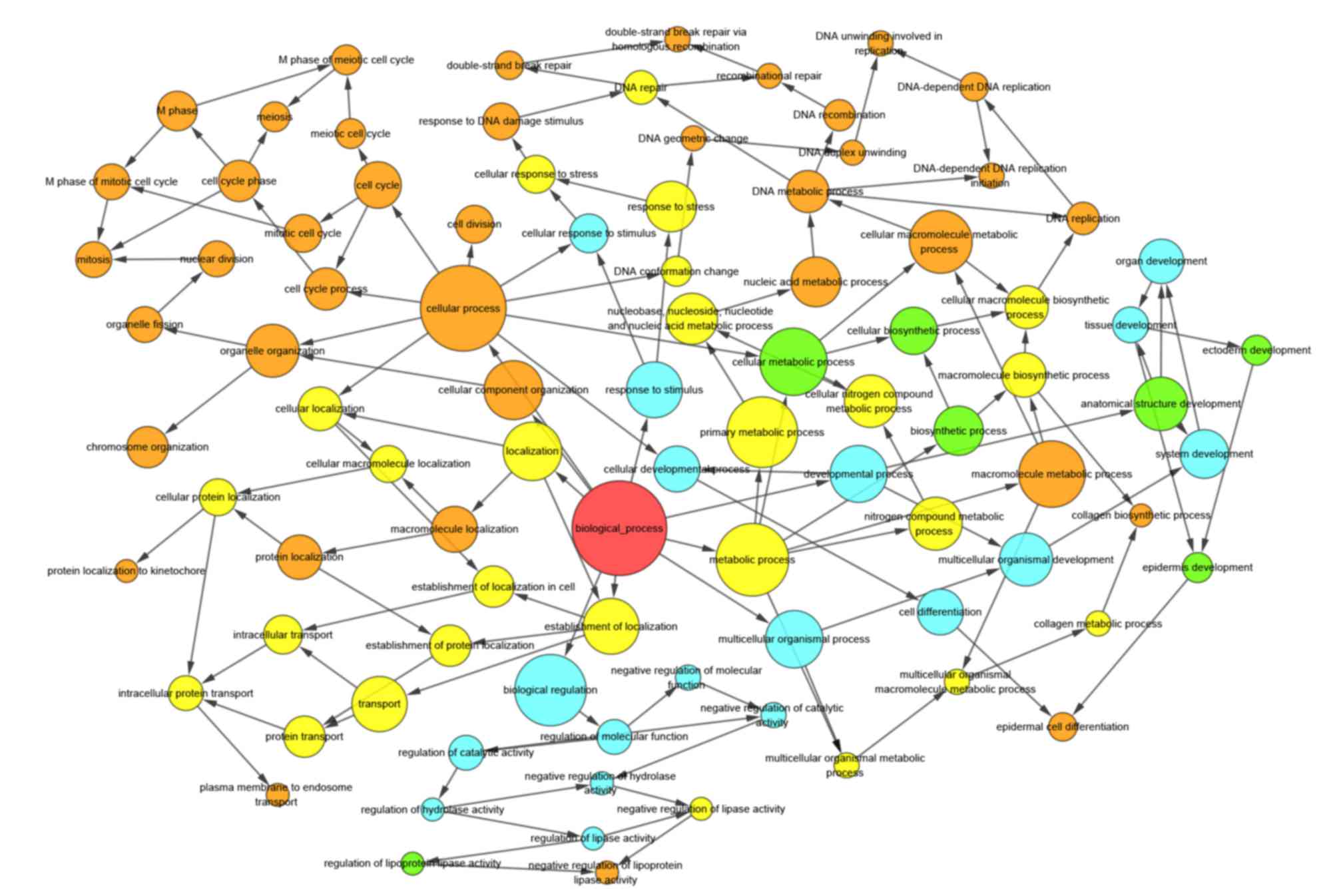
analysis was performed on SLURP and its associated genes (Fig. 3). The results of the bioinformatics
analysis suggested that these genes were most significantly
enriched in the chromosomal part, organelle organization and
protein binding in the categories CC, BP and MF, respectively
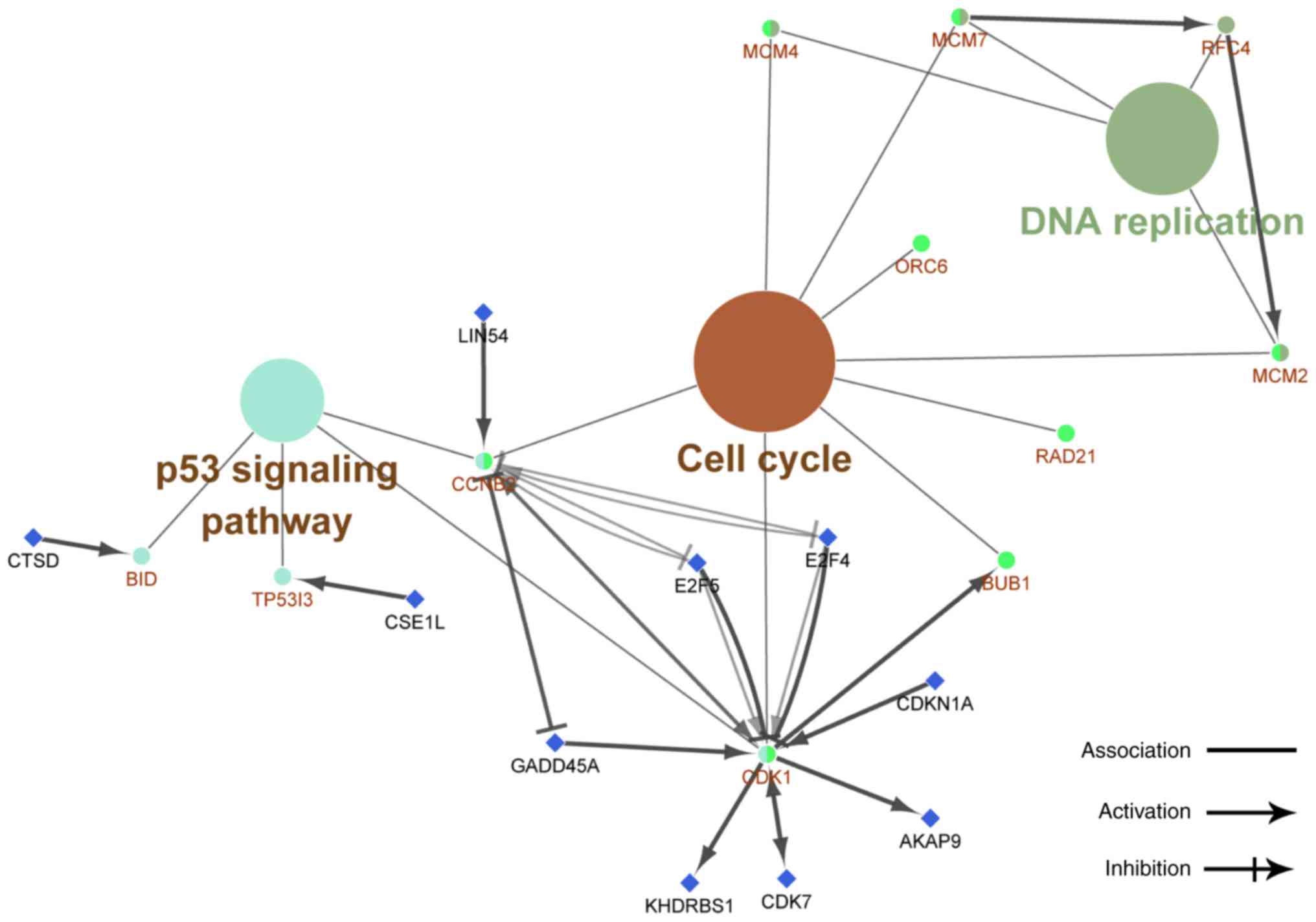
(Figs. 4–6). KEGG pathway analysis revealed that the
genes were involved in DNA replication, cell cycle and P53
signaling pathways (Fig. 7).
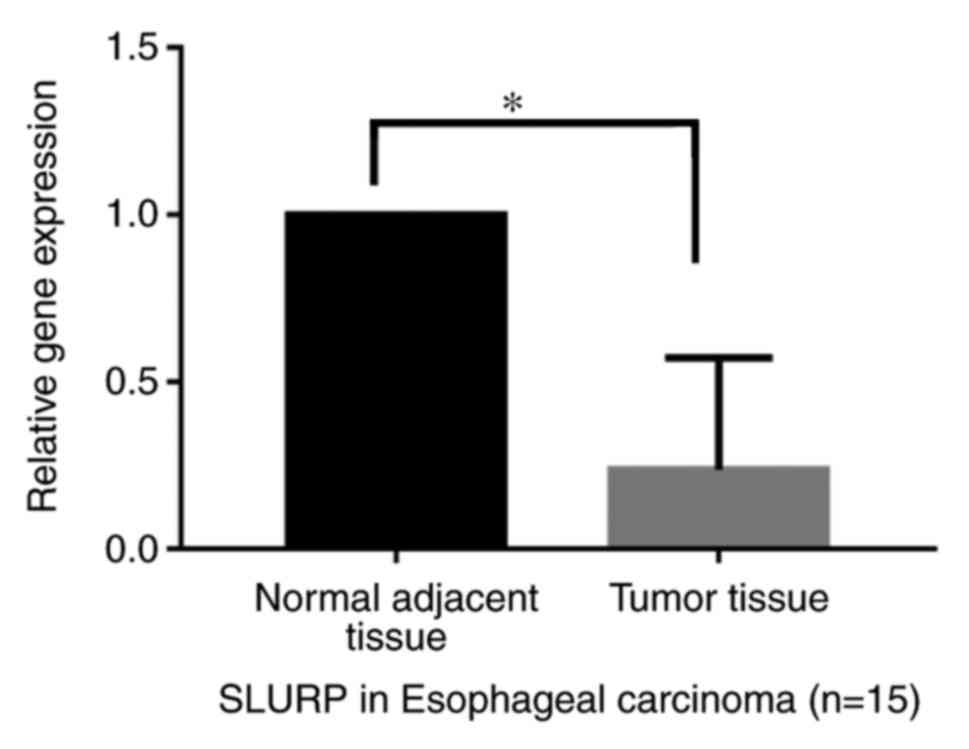
PCR analysis of SLURP
The results indicated that the expression of the hub
gene SLURP-1 was significantly decreased in the tumor samples
relative to that in the normal adjacent tissues in patients with
esophageal carcinoma (P<0.05; Fig.
8).
Discussion
ESCC is caused by external factors leading to gene
mutations. Thus, understanding the underlying molecular mechanisms
is of pivotal importance for ESCC diagnosis and treatment. In the
present study, a dataset downloaded from the GEO database was
analyzed and the bioinformatics tools Morpheus, DAVID and STRING
were used to obtain DEGs, hub proteins, hub genes and major
deregulated pathways in ESCC.
A total of 22,277 DEGs were identified from the
dataset GSE20347. To better understand the interactions of DEGs,
the top 200 DEGs were selected for further analysis. The results of
the GO term enrichment revealed that these DEGs were most highly
involved in membrane-bound vesicles, DNA metabolic processes,
nucleotide binding and chromosomes. KEGG analysis indicated that
the DEGs were enriched in cell cycle pathways. These results
suggest that the DEGs may be mainly involved in regulating the cell
cycle (19,20) and organogenesis, which are closely
associated with tumorigenesis (21)
and tumor progression (22–29). The results from a study by Lin et
al (30) confirmed that the
dysregulation of genes that regulate the G1/S transition is common
in ESCC. Reduced expression of the protein p21WAF1/Cip1
was reported to predict a shorter overall survival time of patients
with ESCC (27,28). The present results indicated that the
deregulation of certain genes is involved in ESCC, and that various
DEGs may be associated with the genesis of ESCC.
The analysis of PPI networks indicated that the top
10 hub proteins included CDK4, BUB1, CCNB2, heat shock protein
(HSP)90AA1, aurora kinase (AURK)A, H2A histone family member Z
(H2AFZ), replication factor C subunit 4 (RFC4), as well as
minichromosome maintenance complex component 7 (MCM7), MCM4 and
MCM2. These proteins are closely associated with the cell cycle,
tumorigenesis (31), transferase
signaling pathway (32,33), transforming growth factor β-mediated
cell cycle control (34,35), embryonic development (36), DNA-dependent ATPase activity
(37) and DNA unwinding enzymes
(38–40). CDK4 was the node with the highest
degree of interaction and was the most connected hub protein,
interacting with 30 genes in the regulatory network. This result
was consistent with that by Su et al (41), which also indicated that CDK4 was the
most significantly upregulated gene by analyzing 5 mRNA expression
datasets of EC tissues/cell lines from GEO. A recent study revealed
that CDK4 had a negative association with EC-related gene 4, which
has a tumor suppressor function in ESCC (42). CDK4/6 inhibitor-SHR6390 was reported
to exert an antitumor effect against ESCC (43). AURKA, MCM7 and MCM4 were closely
associated with cell proliferation and migration in ESCC (44–46).
BUB1-related protein kinase was significantly higher in cancerous
tissue than in adjacent normal tissue, and after radiochemotherapy,
it was significantly decreased in the tissue of patients with ESCC
(47). HSP90A and CCNB1 protein were
reported to be associated with tumor malignancy and prognosis in
patients with ESCC (48). It was
demonstrated that abnormal levels of H2AF may be associated with
poor survival of ESCC patients (49). However, as the involvement of RFC4
and MCM2 in ESCC has been rarely investigated, further study is
necessary. Analysis of Hub protein functions indicated that these
proteins have a key role in the regulation of ESCC, including the
genesis development and progression of tumors, and that these hub
proteins may serve as therapeutic targets in ESCC.
The present study used the plug-in MCODE to
construct the modules. The results indicated that the functions of
genes in the top 4 modules were mainly associated with cell cycle
pathways. Li et al (50)
indicated that overall, the DNA repair pathways were significantly
associated with a risk of ESCC. Roncalli et al (51) studied cell cycle-associated genes in
patients with EC, and their results demonstrated that these were
significantly associated lymph node metastasis and unfavorable
survival rates. These results indicate that the pathways associated
with the top modules mainly regulate tumor progression in ESCC and
that certain genes in theses pathways may serve as potential
prognostic biomarkers.
In the present study, Spearman's correlation test
was used to analyze the correlation between the top 200 DEGs. The
results revealed that SLURP was the hub gene that was most highly
connected to the other genes. The results of the bioinformatics
analysis indicated that this hub gene and its associated genes are
significantly enriched in the chromosomal part, organelle
organization and protein binding in the GO categories CC, BP and
MF, respectively. These genes are also involved in DNA replication,
cell cycle and P53 signaling pathways. The expression of SLURP-1
was the assessed in 15 patients with EC, and the results revealed
that SLURP-1was significantly decreased in the tumor samples
relative to that in the normal adjacent tissues. SLURP-1 has been
reported to participate in signal transduction, immune activation
and cell adhesion to exert its antitumor activity (52). This was consistent with the results
of recent studies, which indicated that DEGs screened by
RNA-sequencing data or The Cancer Genome Atlas analysis have
important roles in regulating growth, invasion and metastasis of
tumors as well as immune responses, and the DEGs included collagen
type I α 1, matrix metallopeptidases, keratin 4, cysteine-rich
secretory protein2 and 3, mucin 21 and cyclin D1 (53,54). The
present results indicated that the hub gene SLURP-1 may have a key
role in regulating the tumorigenesis of ESCC and that it may serve
as a potential biomarker in tumor diagnosis.
In conclusion, the present study identified DEGs and
the hub proteins (CDK4, BUB1, CCNB2, HSP90AA1, AURKA, H2AFZ, RFC4,
MCM7, MCM4 and MCM2) and a hub gene (SLURP) in ESCC. These genes
are primarily involved in regulating the tumorigenesis and
progression of ESCC. The hub proteins and gene may be considered as
candidate therapeutic targets and may provide information for
further studies on the molecular biological functions and
mechanisms of ESCC. However, the present study had certain
limitations, including the fact that only GEO 1 dataset of
microarray data was used and that the sample size was relatively
small.
Acknowledgements
The authors would like to thank Professor Edward I.
Wong (Milton International Education Group, Hong Kong) for revising
the manuscript.
Funding
This work was funded by grants of the Technical New
Star of Zhujiang, Pan Yu district, Guangzhou (grant no.
2013-special-15-6.10), the Science and Technology program of Pan Yu
(grant no. 2015-Z03-09), the Traditional Chinese Medicine Bureau of
Guangdong Province (grant no. 20161186) and the Science and
Technology Program of Guangzhou (grant no. 201804010012).
Availability of data and materials
The analyzed data sets generated during the study
are available from the corresponding author on reasonable
request.
Authors' contributions
XC and SC performed the analysis of the data and
writing of the paper. BL, XZ, WL and HL collected the samples and
performed the PCR. XC performed the data analysis of GEO.
Ethical approval and consent to
participate
This study was approved by the Ethics Committee of
Panyu Central Hospital (ethical approval no. 20180001). All
patients provided informed consent to participate in this
study.
Patient consent for publication
All patients gave consent for the publication on
their data in the current study.
Competing interests
The authors declared they have no competing
conflicts.
References
|
1
|
Chen W, Zheng R, Zhang S, Zeng H, Xia C,
Zuo T, Yang Z, Zou X and He J: Cancer incidence and mortality in
China, 2013. Cancer Lett. 401:63–71. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Tran GD, Sun XD, Abnet CC, Fan JH, Dawsey
SM, Dong ZW, Mark SD, Qiao YL and Taylor PR: Prospective study of
risk factors for esophageal and gastric cancers in the Linxian
general population trial cohort in China. Int J Cancer.
113:456–463. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yang Q, Wang YX, He M, Li J, Qi Z, Zhu SC
and Qiao XY: Factors affecting on long-time survival in patients
with stageIII thoracic esophageal carcinoma after esophagectomy.
Zhonghua Zhong Liu Za Zhi. 38:530–537. 2016.(In Chinese).
PubMed/NCBI
|
|
4
|
Yu S, Zhang W, Ni W, Xiao Z, Wang X, Zhou
Z, Feng Q, Chen D, Liang J, Fang D, et al: Nomogram and recursive
partitioning analysis to predict overall survival in patients with
stage IIB-III thoracic esophageal squamous cell carcinoma after
esophagectomy. Oncotarget. 7:55211–55221. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yi Y, Lu X, Chen J, Jiao C, Zhong J, Song
Z, Yu X and Lin B: Downregulated miR-486-5p acts as a tumor
suppressor in esophageal squamous cell carcinoma. Exp Ther Med.
12:3411–3416. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Osako Y, Seki N, Kita Y, Yonemori K,
Koshizuka K, Kurozumi A, Omoto I, Sasaki K, Uchikado Y, Kurahara H,
et al: Regulation of MMP13 by antitumor microRNA-375 markedly
inhibits cancer cell migration and invasion in esophageal squamous
cell carcinoma. Int J Oncol. 49:2255–2264. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zuo J, Wang D, Shen H, Liu F, Han J and
Zhang X: MicroRNA-153 inhibits tumor progression in esophageal
squamous cell carcinoma by targeting SNAI1. Tumour Biol. Oct
13–2016.(Epub ahead of print). View Article : Google Scholar
|
|
8
|
Jing C, Ma G, LiX, Wu X, Huang F, Liu K
and Liu Z: MicroRNA-17/20a impedes migration and invasion via
TGF-β/ITGB6 pathway in esophageal squamous cell carcinoma. Am J
Cancer Res. 6:1549–1562. 2016.PubMed/NCBI
|
|
9
|
Hu N, Wang C, Clifford RJ, Yang HH, Su H,
Wang L, Wang Y, Xu Y, Tang ZZ, Ding T, et al: Integrative genomics
analysis of genes with biallelic loss and its relation to the
expression of mRNA and micro-RNA in esophageal squamous cell
carcinoma. BMC Genomics. 16:7322015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hu N, Clifford RJ, Yang HH, Wang C,
Goldstein AM, Ding T, Taylor PR and Lee MP: Genome wide analysis of
DNA copy number neutral loss of heterozygosity (CNNLOH) and its
relation to gene expression in esophageal squamous cell carcinoma.
BMC Genomics. 11:5762010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Garner E, Wallace JS, Argoty GA, Wilkinson
C, Fahrenfeld N, Heath LS, Zhang L, Arabi M, Aga DS and Pruden A:
Metagenomic profiling of historic Colorado Front Range flood impact
on distribution of riverine antibiotic resistance genes. Sci Rep.
6:384322016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Dennis G Jr, Sherman BT, Hosack DA, Yang
J, Gao W, Lane HC and Lempicki RA: DAVID: Database for annotation,
visualization, and integrated discovery. GenomeBiol. 4:P32003.
|
|
13
|
Torto-Alalibo T, Purwantini E, Lomax J,
Setubal JC, Mukhopadhyay B and Tyler BM: Genetic resources for
advanced biofuel production described with the Gene Ontology. Front
Microbiol. 5:5282014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ashburner M, Ball CA, Blake JA, Botstein
D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT,
et al: Gene ontology: Tool for the unification of biology. The gene
ontology consortium. Nat Genet. 25:25–29. 2000.
|
|
15
|
Du J, Yuan Z, Ma Z, Song J, Xie X and Chen
Y: KEGG-PATH: Kyotoencydopedia of genes and genomes-based pathway
analysis using a path analysis model. MolBiosyst. 10:2441–14447.
2014.
|
|
16
|
Kanehisa M and Goto S: KEGG:
Kyotoencyclopedia of genes and genomes. Nucleic Acids Res.
28:27–30. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Maere S, Heymans K and Kuiper M: BiNGO: A
Cytoscape plugin to assess overrepresentation of gene ontology
categories in biological networks. Bioinformatics. 21:3448–9344.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ren K, Li Y, Lu H, Li Z, Li Z, Wu K, Li Z
and Han X: Long Noncoding RNA HOTAIR Controls Cell Cycle by
Functioning as a Competing Endogenous RNA in Esophageal Squamous
Cell Carcinoma. Transl Oncol. 9:489–497. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang HF, Alshareef A, Wu C, Jiao JW,
Sorensen PH, Lai R, Xu LY and Li EM: miR-200b induces cell cycle
arrest and represses cell growth in esophageal squamous cell
carcinoma. Carcinogenesis. 37:858–869. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cao J, Ge MH and Ling ZQ: Fbxw7 tumor
suppressor: A vital regulator contributes to human tumorigenesis.
Medicine (Baltimore). 95:e24962016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Costa C, Santos M, Martínez-Fernández M,
Lorz C, Lázaro S and Paramio JM: Deregulation of the pRb-E2F4 axis
alters epidermal homeostasis and favors tumor development.
Oncotarget. 7:75712–75728. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lambot MA, Peny MO, Fayt I, Haot J and
Noël JC: Overexpression of 27-kDa heat shock protein relates to
poor histological differentiation in human oesophageal squamous
cell carcinoma. Histopathology. 36:326–330. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Xia W, Qiu M, Chen R, Wang S, Leng X, Wang
J, Xu Y, Hu J, Dong G, Xu PL, et al: CCircular RNA has_circ_0067934
is upregulated in esophageal squamous cell carcinoma and promoted
proliferation. Sci Rep. 6:355762016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Xu Y, Qiu M, Chen Y, Wang J, Xia W, Mao Q,
Yang L, Li M, Jiang F, Xu L and Yin R: Long noncoding RNA, tissue
differentiation-inducing nonprotein coding RNA is upregulated and
promotes development of esophageal squamous cell carcinoma. Dis
Esophagus. 29:950–958. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhi H, Zhang J, Hu G, Lu J, Wang X, Zhou
C, Wu M and Liu Z: The deregulation of arachidonic acid
metabolism-related genes in human esophageal squamous cell
carcinoma. Int J Cancer. 106:327–333. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Mao Y, Li L, Liu J, Wang L and Zhou Y:
MiR-495 inhibits esophageal squamous cell carcinoma progression by
targeting Akt1. Oncotarget. 7:51223–51236. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zeng LS, Yang XZ, Wen YF, Mail SJ, Wang
MH, Zhang MY, Zheng XF and Wang HY: Overexpressed HDAC4 is
associated with poor survival and promotes tumor progression in
esophageal carcinoma. Aging (Albany NY). 8:1236–1249. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Luo J, Zhang C, Wang C, Li L, Li C, Li Q,
Zhang M and Wu Q: Miz-1 promotes the proliferation of esophageal
cancer cells via suppression of p21 and release of p21-arrested
cyclin D1. Oncol Rep. 35:3532–3540. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lin DC, Shi ZZ, Xue LY, Chen W, Xu X, Han
YL, Lv N and Wang MR: Expression of cell cycle related proteins
cyclin D1, p53 and p21WAF1/Cip1 in esophageal squamous cell
carcinoma. Yi Chuan. 32:455–460. 2010.(In Chinese). View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lang E, Zelenak C, Eberhard M, Bissinger
R, Rotte A, Ghashghaeinia M, Lupescu A, Lang F and Qadri SM: Impact
of cyclin-dependent kinase CDK4 inhibition on eryptosis. Cell
Physiol Biochem. 37:1178–1186. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Breit C, Bange T, Petrovic A, Weir JR,
Müller F, Vogt D and Musacchio A: Role of intrinsic and extrinsic
factors in the regulation of the mitotic checkpoint kinase Bub1.
PLoS One. 10:e01446732015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Takashima S, Saito H, Takahashi N, Imai K,
Kudo S, Atari M, Saito Y, Motoyama S and Minamiya Y: Strong
expression of cyclin B2 mRNA correlates with a poor prognosis in
patients with non-small cell lung cancer. Tumour Biol.
35:4257–4265. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chen JT, Younusi A, Cao L, Tian Z, Zhou YJ
and Song XH: Potential role of heat-shock proteins in giant cell
tumors. Genet Mol Res. 14:19144–19154. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kim SR, Kim KB, Chae YC, Park JW and Seo
SB: H3S10 phosphorylation-mediated transcriptional regulation by
Aurora kinase A. Biochem Biophys Res Commun. 469:22–28. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kusakabe M, Oku H, Matsuda R, Hori T, Muto
A, Igarashi K, Fukagawa T and Harata M: Genetic complementation
analysis showed distinct contributions of the N-terminal tail of
H2A.Z to epigenetic regulations. Genes Cells. 21:122–135. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Arai M, Kondoh N, Imazeki N, Hada A,
Hatsuse K, Matsubara O and Yamamoto M: The knockdown of endogenous
replication factor C4 decreases the growth and enhances the
chemosensitivity of hepatocellular carcinoma cells. Liver Int.
29:55–62. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ishibashi Y, Kinugasa T, Akagi Y, Ohchi T,
Gotanda Y, Tanaka N, Fujino S, Yuge K, Kibe S, Yoshida N, et al:
Minichromosome maintenance protein 7 is a risk factor for
recurrence in patients with Dukes C colorectal cancer. Anticancer
Res. 34:4569–4575. 2014.PubMed/NCBI
|
|
39
|
Watanabe E, Ohara R and Ishimi Y: Effect
of an MCM4 mutation that causes tumours in mouse on human MCM4/6/7
complex formation. J Biochem. 152:191–198. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Razavi SM, Jafari M, Heidarpoor M and
Khalesi S: Minichromosome maintenance-2 (MCM2) expression
differentiates oral squamous cell carcinoma from pre-cancerous
lesions. Malays J Pathol. 37:253–258. 2015.PubMed/NCBI
|
|
41
|
Su P, Wen S, Zhang Y, Li Y, Xu Y, Zhu Y,
Lv H, Zhang F, Wang M and Tian Z: Identification of the key genes
and pathways in esophageal carcinoma. Gastroenterol Res Pract.
2016:29681062016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Li L, Wang W, Li X and Gao T: Association
of ECRG4 with PLK1, CDK4, PLOD1 and PLOD2 in esophageal squamous
cell carcinoma. Am J Transl Res. 9:3741–3748. 2017.PubMed/NCBI
|
|
43
|
Wang J, Li Q, Yuan J, Wang J, Chen Z, Liu
Z, Li Z, Lai Y, Gao J and Shen L: CDK4/6 inhibitor-SHR6390 exerts
potent antitumor activity in esophageal squamous cell carcinoma by
inhibiting phosphorylated Rb and inducing G1 cell cycle arrest. J
Transl Med. 15:1272017. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wang X, Li X, Li C, He C, Ren B, Deng Q,
Gao W and Wang B: Aurora-A modulates MMP-2 expression via AKT/NF-κB
pathway in esophageal squamous cell carcinoma cells. Acta Biochim
Biophys Sin (Shanghai). 48:520–527. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Qiu YT, Wang WJ, Zhang B, Mei LL and Shi
ZZ: MCM7 amplification and overexpression promote cell
proliferation, colony formation and migration in esophageal
squamous cell carcinoma by activating the AKT1/mTOR signaling
pathway. Oncol Rep. 37:3590–3596. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Choy B, LaLonde A, Que J, Wu T and Zhou Z:
MCM4 and MCM7, potential novel proliferation markers, significantly
correlated with Ki-67, Bmi1, and cyclin E expression in esophageal
adenocarcinoma, squamous cell carcinoma, and precancerous lesions.
Hum Pathol. 57:126–135. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Tanaka K, Mohri Y, Ohi M, Yokoe T, Koike
Y, Morimoto Y, Miki C, Tonouchi H and Kusunoki M: Mitotic
checkpoint genes, hsMAD2 and BubR1, in oesophageal squamous cancer
cells and their association with 5-fluorouracil and cisplatin-based
radiochemotherapy. Clin Oncol (R Coll Radiol). 20:639–646. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Huang T, Chen S, Han H, Li H, Huang Z,
Zhang J, Yin Q, Wang X, Ma X, Dai P, et al: Expression of Hsp90α
and cyclin B1 were related to prognosis of esophageal squamous cell
carcinoma and keratin pearl formation. Int J Clin Exp Pathol.
7:1544–1552. 2014.PubMed/NCBI
|
|
49
|
Zhang K, Li L, Zhu M, Wang G, Xie J, Zhao
Y, Fan E, Xu L and Li E: Comparative analysis of histone H3 and H4
post-translational modifications of esophageal squamous cell
carcinoma with different invasive capabilities. J Proteomics.
112:180–189. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Li WQ, Hu N, Hyland PL, Gao Y, Wang ZM, Yu
K, Su H, Wang CY, Wang LM, Chanock SJ, et al: Genetic variants in
DNA repair pathway genes and risk of esophageal squamous cell
carcinoma and gastric adenocarcinoma in a Chinese population.
Carcinogenesis. 34:1536–1542. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Roncalli M, Bosari S, Marchetti A,
Buttitta F, Bossi P, Graziani D, Peracchia A, Bonavina L, Viale G
and Coggi G: Cell cycle-related gene abnormalities and product
expression in esophageal carcinoma. Lab Invest. 78:1049–1057.
1998.PubMed/NCBI
|
|
52
|
Pettersson A, Nylund G, Khorram-Manesh A,
Nordgren S and Delbro DS: Nicotine induced modulation of SLURP-1
expression in human colon cancer cells. Auton Neurosci. 148:97–100.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Fu JH, Wang LQ, Li T and Ma GJ:
RNA-sequencing based identification of crucial genes for esophageal
squamous cell carcinoma. J Cancer Res Ther. 11:240–425. 2015.
|
|
54
|
Cancer Genome Atlas Network; Comprehensive
genomic characterization of head and neck squamous cell carcinomas.
Nature. 517:576–582. 2015. View Article : Google Scholar : PubMed/NCBI
|