Introduction
Ischemic stroke remains the one of the leading
neurological diseases, with high morbidity and mortality worldwide
(1). The main pathological processes
of ischemic stroke include excitotoxicity, inflammatory response
and oxidative stress, which ultimately lead to irreversible
neuronal injury (2). Among these,
oxidative stress is the most widespread process during
ischemia/reperfusion (I/R) injury and describes a major difficulty
for clinical treatment (3,4). However, the detailed mechanism of
oxidative stress has not yet been fully elucidated. Previous
studies have demonstrated that nicotinamide adenine dinucleotide
phosphate (NADPH) oxidases (NOX) are the primary enzymes
responsible for reactive oxygen species (ROS) generation and seven
NOXs have been identified in mammals, including NOX1-5, dual
oxidase (DUOX)-1 and DUOX-2 (5). A
previous study identified that NOX2 and NOX4 are the major NOXs
responsible for brain tissue ROS production in a rat ischemic
stroke model (6). Numerous factors
are reported to be associated with the regulation of NOX
expression, including the transcription factor, nuclear factor
(NF)-κB and myosin light chain (7).
However, the detailed mechanism by which NOX expression is
regulated has not yet been fully elucidated.
Transforming growth factor-β (TGF-β) is associated
with numerous biological processes, including cell growth,
differentiation, migration, survival, adhesion and apoptosis
(8), in addition to serving an
important role in tumor suppression (9). Through interacting with TGF-β receptors
(serine/threonine kinases), TGF-β triggers a cascade of events,
including activation of the extracellular signal-regulated kinase
signaling pathway and mechanistic target of rapamycin-mediated
protein synthesis (10). These
processes usually involve phosphorylation of SMAD2/3 and activin
receptor-like kinase (ALK)5 (10).
Emerging evidence has demonstrated that TGF-β signaling is crucial
in the pathogenesis of several central nervous system (CNS)
disorders, including neurodegenerative disorders (11). Previous studies have demonstrated
that increased TGF-β levels are associated with cerebral ischemic
injury (12,13). These results suggest that TGF-β
signaling may serve an important function in ischemic stroke.
Although previous studies have demonstrated that TGF-β signaling is
associated with ROS production (14), the association between ALK5/SMAD2/3
signaling and oxidative stress in ischemic stroke remains
unclear.
The present study aimed to investigate the role of
ALK5/SMAD2/3 signaling in oxidative stress. Using an I/R injury rat
model, PC-12 cell hypoxia/reperfusion (H/R) model and ALK5
silencing, it was identified that ALK5/SMAD2/3 signaling was
associated with the regulation of NOX2/4 expression. To the best of
our knowledge, this is the first study to explore the role of
ALK5/SMAD2/3 signaling in NOX-mediated oxidative stress in I/R
injury, which may provide a novel target for ischemic stroke
therapy.
Materials and methods
Animal experiments
A total of 16 male Sprague Dawley rats (age, 9
weeks; weight, 250–300 g) were purchased from Hunan SJA Laboratory
Animal Co., Ltd. (Changsha, China). Animals were allowed to
accommodate to 27°C, regular atmosphere, 12-h light/dark cycle,
with free to food and water for 1 week. Prior to experiments, rats
fasted for 24 h, with free access to water only. The study was
performed according to the Guide for the Care and Use of Laboratory
Animals, published by the National Institutes of Health (15) and experiments were approved by the
Hunan Normal University Veterinary Medicine Animal Care and Use
Committee (Changsha, China).
The rat model of I/R injury was established as
previously described (16). Briefly,
the middle cerebral artery of the rats was occluded for 2 h with
subsequent reperfusion for 24 h. The body temperature of the rats
was maintained at ~37°C throughout the procedure. Animals from the
sham group, without I/R injury, underwent the same procedure,
except that the occluding filament was inserted 7 mm above the
carotid bifurcation instead.
The animals were randomly allocated to two groups
(n=8 per group): The sham group, which underwent surgical
procedures without occlusion of the middle cerebral artery and the
I/R group, which was subjected to 2 h of ischemia followed by 24 h
of reperfusion. At the end of reperfusion, all animals were
sacrificed prior to obtaining brain tissues, which were collected
to assess cell apoptosis, mRNA and protein expression.
Terminal
deoxynucleotidyl-transferase-mediated dUTP nick end labeling
(TUNEL) staining
Cellular apoptosis in the brain tissue from
different treatment groups was analyzed by TUNEL assay according to
the manufacturer's instructions of a commercial kit (Colorimetric
TUNEL Apoptosis Assay kit; C1091; Beyotime Institute of
Biotechnology, Shanghai, China). Briefly, brains were embedded into
paraffin and fixed with 10% formaldehyde at 27°C for 20 min.
Following, brains were cut into 5-µm sections and incubated with 1×
biotin-labeled deoxyuridine triphosphate (Beyotime Institute of
Biotechnology, Haimen, China) at 37°C for 1 h. Samples were
incubated with streptavidin-horseradish peroxidase (HRP) at 37°C
for 30 min and 0.05% 3′-diaminobenzidine development solutions at
25°C for 10 min in sequential order. Brain slides were examined
microscopically (light microscope; magnification, ×200) and imaged
with a high-resolution digital camera (Nikon Eclipse 80i; Nikon
Corporation, Tokyo, Japan).
Cell culture
PC-12 cell line was purchased from the Type Culture
Collection of the Chinese Academy of Sciences (Shanghai, China).
Cells were cultured in Dulbecco's modified Eagle's medium (DMEM;
HyClone; GE Healthcare, Chicago, IL, USA) supplemented with 10%
fetal bovine serum (Thermo Fisher Scientific, Inc., Waltham, MA,
USA) and penicillin (100 U/ml) and streptomycin (0.1 mg/ml) and
maintained in 95% air and 5% CO2 at 37°C. Cells were
subcultured and seeded into 6- or 24-well plates at 105
cells/well for transfection experiments. Following transfection,
cells were digested with 0.2% trypsinogen and collected for mRNA or
protein analysis.
Cell model of H/R
To establish an H/R model, PC-12 cells were
subjected to 5 h of hypoxia (N2/CO2, 95:5) in
preconditioned hypoxic medium (serum-free DMEM without glucose or
sodium pyruvate, which was incubated at 37°C under hypoxic
conditions for 2 h), followed by 20 h of reoxygenation (5%
CO2). Hypoxic medium was replaced with fresh medium upon
switching to reoxygenation.
Cells were divided into three groups (n=6
wells/group): H/R group, which was subjected to 5 h of hypoxia
followed by 20 h of rexoygenation; +negative control (NC) small
interfering (si)RNA group, where cells were transfected with
negative control siRNA and +ALK5 siRNA group, where cells were
transfected with siRNA against ALK5 prior to H/R. At the end of
reoxygenation, cells were collected to assess apoptosis, ROS
production and mRNA and protein expression.
siRNA knockdown experiments
siRNA against ALK5 (sense,
5′-CAUAUUGCUGCAACCAGGATT-3′; antisense,
5′-UCCUGGUUGCAGCAAUAUGTT-3′) was designed and synthesized by Sangon
Biotech Co., Ltd., (Shanghai, China). Prior to the experiment,
mixtures of Lipofectamine 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.) and 80 nM ALK5 or NC siRNA (sense,
5′-UUCUCCGAACGUGUCACGUTT-3′; antisense,
5′-ACGUGACACGUUCGGAGAATT-3′) were prepared. Mixtures were added
into 24-well cell plates for 6 h for transfection at 30°C.
Following, cells were incubated in a CO2 incubator at
37°C for 24 h prior to gene expression measurement, according to
the manufacturer's protocol.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
RT-qPCR was used to analyze ALK5, NOX2 and NOX4 mRNA
levels in brain tissue. Total RNA was extracted using TRIzol
reagent (Takara Biotechnology Co., Ltd., Dalian, China) and
concentration and purity of RNA were determined
spectrophotometrically. A total of 200 ng RNA from each sample was
used for RT, which was performed according to the manufacturer's
protocol of the RT kit (DRR037A; Takara Biotechnology Co., Ltd.).
qPCR was performed to determine ALK5, NOX2 and NOX4 mRNA expression
levels, using SYBR Premix Ex Taq (Takara Biotechnology Co., Ltd.)
and an ABI 7300 system (Applied Biosystems; Thermo Fisher
Scientific, Inc.). qPCR primers for ALK5, NOX2, NOX4 and β-actin
are presented in Table I. Data
analysis was performed using the 2−ΔΔCq method using ABI
7300 software (v2.4; Thermo Fisher Scientific, Inc.) (17). Relative expression of ALK5, NOX2 and
NOX4 mRNA was normalized against β-actin mRNA.
 | Table I.Primers for reverse
transcription-quantitative polymerase chain reactions. |
Table I.
Primers for reverse
transcription-quantitative polymerase chain reactions.
| Gene | Forward primer
(5′-3′) | Reverse primer
(5′-3′) | Product size
(bp) |
|---|
| NOX2 |
ACAAGGTTTATGACGATGAGCC |
TTGAGCAACACGCACTGGAA | 174 |
| NOX4 |
CTGACAGGTGTCTGCATGGT |
ACTTCAACAAGCCACCCGAA | 160 |
| ALK5 |
TTGTTGAGGAGAAGCTGAGGC |
CACTGTAATGCCTTCGCCCC | 154 |
| β-actin |
CCCATCTATGAGGGTTACGC |
TTTAATGTCACGCACGATTTC | 150 |
Hoechst staining and lactate
dehydrogenase (LDH) release
Cellular apoptosis in PC-12 cells of different
treatment groups was analyzed by Hoechst assay (Beyotime Institute
of Biotechnology). Briefly, cells were incubated at 25°C with
Hoechst 33258 solutions (Beyotime Institute of Biotechnology) for
20 min. Images were examined microscopically (light microscope;
magnification, ×200) and photographed by a high-resolution digital
camera (Nikon Eclipse 80i; Nikon Corporation).
Cell toxicity of PC-12 was assessed by LDH release
using a microplate reader. The procedure was performed according to
the manufacturer's protocol of a commercial LDH release kit (C0016;
Beyotime Institute of Biotechnology, Shanghai, China).
Western blotting
Total protein from each sample was extracted using
the method described by Zhang et al (6). Proteins (40 µg) were separated on 10%
SDS-PAGE gels and transferred to polyvinylidene fluoride membranes.
Membranes were blocked with 5% milk at 25°C for 2 h and membranes
incubated with 1:2,000 diluted rabbit anti-phosphorylated
(p)-SMAD2/3 (sc-517575; Santa Cruz Biotechnology, Inc., Dallas, TX,
USA), NOX2 (sc-130543; Santa Cruz Biotechnology, Inc.), NOX4
(sc-30141; Santa Cruz Biotechnology, Inc.), ALK5 (sc-101574; Santa
Cruz Biotechnology, Inc.), caspase-3 (sc-7272; Santa Cruz
Biotechnology, Inc.) and mouse anti-β-actin (1:2,000; sc-47778;
Santa Cruz Biotechnology, Inc.) at 4°C for 16 h followed by
incubation with HRP-goat anti-mouse IgG (1:2,000; A0216; Beyotime
Institute of Biotechnology) or anti-rabbit IgG (1:2,000; A0208;
Beyotime Institute of Biotechnology). Bands were detected using an
enhanced chemiluminescence kit (GE Healthcare, Chicago, IL, USA)
and a Molecular Imager ChemiDoc XRS system (Bio-Rad Laboratories,
Inc., Hercules, CA, USA). Densitometric analysis was conducted with
ImageJ 1.43 (National Institutes of Health, Bethesda, MD, USA).
NOX and caspase-3 activity
NOX activity was measured using a commercial NADPH
oxidase activity quantification kit (GMS50096.1, Shanghai Genmed
Pharmaceutical Technology Co., Ltd., Shanghai, China) following the
manufacturer's protocol. Briefly, cells were centrifuged at 12,000
× g for 30 min at 4°C and the supernatant of the cell lysates was
incubated with oxidized cytochrome c in a quartz cuvette at 30°C
for 3 min, NOX substrate (NADPH) was added to the reaction mixture
and incubated at 30°C for a further 15 min. The change of
absorbance at 550 nm was monitored. NOX activity was estimated by
calculating the cytochrome c reduction per min.
Measurements of caspase-3 activity were performed
according to the manufacturer's protocol of a commercial Caspase-3
Activity Assay kit (Beyotime Institute of Biotechnology). Briefly,
cell lysate (10 µl) was mixed with working solution containing
caspase-3 substrate (90 µl, Ac-DEVD-pNA) and the mixture was
incubated at 37°C for 60 min. The absorbance was recorded at 405
nm. Enzyme activity was recorded as U/g protein, where 1 U was
defined as the amount of enzyme required to react with 1 nmol of
Ac-DEVD-pNA per h at 37°C.
Determination of ROS levels
Intracellular ROS levels were determined with
2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA), a
cell-permeable indicator of ROS (Beyotime Institute of
Biotechnology). DCFH-DA is non-fluorescent until cleavage of the
acetate groups by intracellular ROS. Briefly, PC-12 cells
(107 cells/well) were washed with PBS and incubated with
DCFH-DA (10 µM) at 37°C for 20 min. ROS-mediated fluorescence was
observed under a fluorescence microscope, with excitation set to
502 nm and emission at 523 nm. Results are expressed using
arbitrary units.
Statistical analysis
SPSS software (version 11; SPSS, Inc., Chicago, IL,
USA) was used for statistical analysis. Data are presented as mean
± standard error of the mean. Differences among multiple groups
were analyzed by analysis of variance with Bonferroni's multiple
comparison test. Student's t-test was used to compare two groups.
P<0.05 was considered to indicate a statistically significant
difference.
Results
I/R injury induces ROS production and
apoptosis in brain tissues
To explore the mechanism for clinical I/R injury, a
rat I/R model was established that simulated ischemic stroke. ROS
level, caspase-3 enzyme activity and expression were determined and
apoptosis was detected by TUNEL staining, in order to evaluate
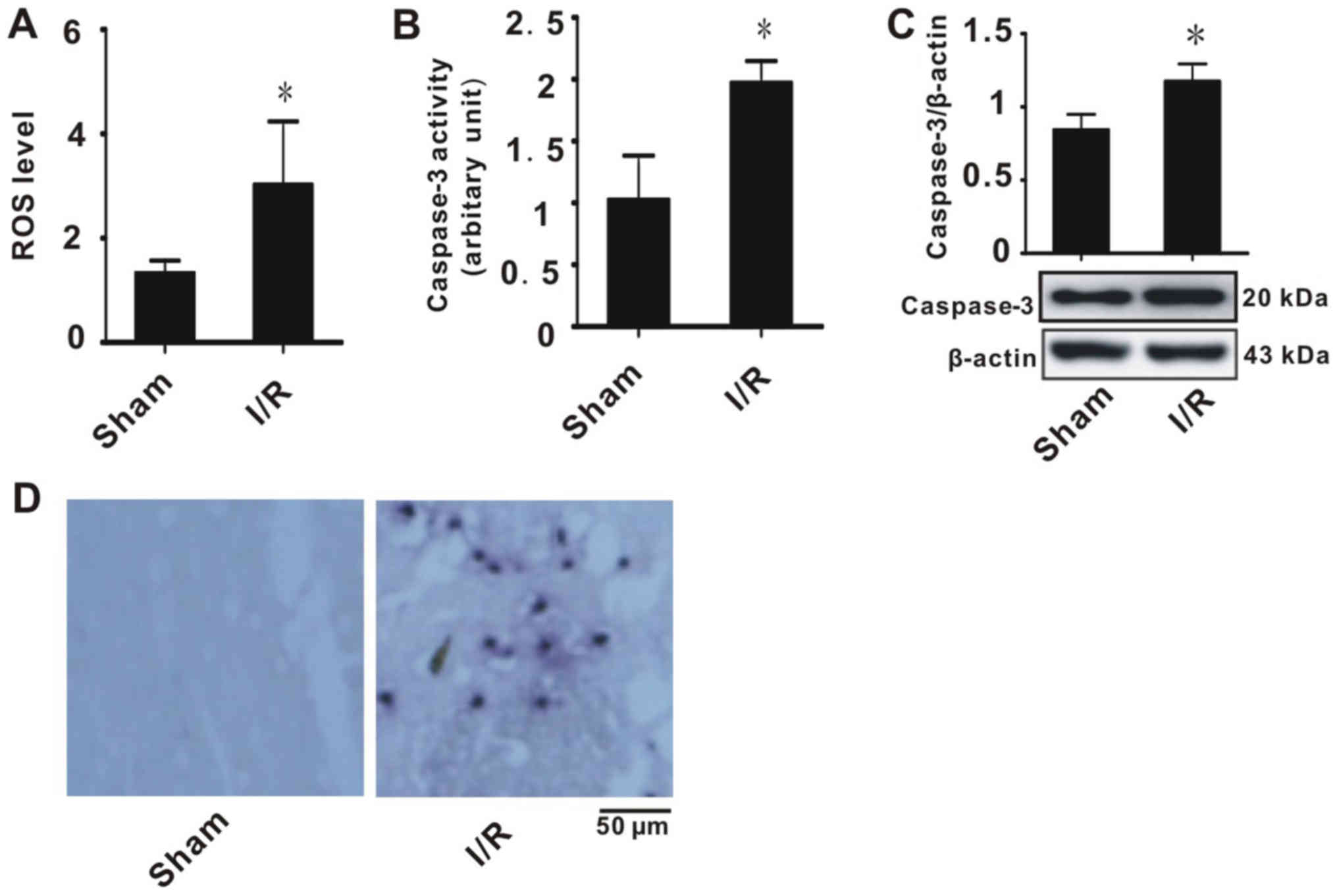
oxidative stress injury. As indicated in Fig. 1A, tissues with I/R injury exhibited
higher ROS production compared with the sham group. This suggested
that these tissues suffered from oxidative stress. In addition, it
was indicated that I/R significantly increased caspase-3 expression
and activity compared with the sham group (Fig. 1B and C). TUNEL staining indicated
that tissues with I/R had significantly increased apoptosis
compared with the sham group (Fig.
1D). These results suggested that the rats exhibited evident
brain injury.
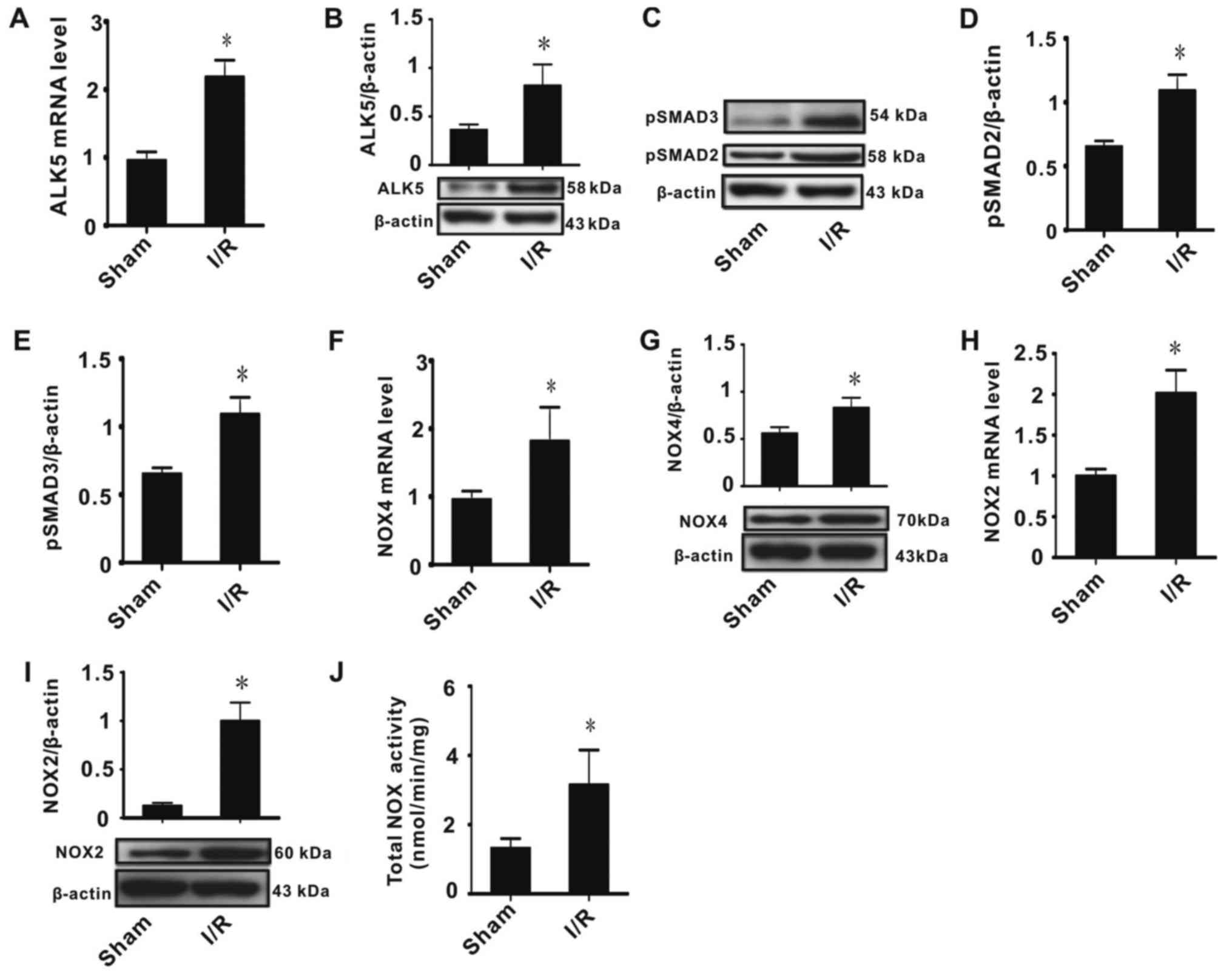
I/R injury affects ALK5, p-SMAD2/3,
NOX2 and NOX4 expression
As ALK5/p-SMAD2/3 signaling regulates gene
expression and NOX2/4 activity is the main source of cellular ROS,
effects of I/R injury on ALK5, p-SMAD2/3, NOX2 and NOX4 mRNA and
protein expression were determined, in addition to measuring the
total NOX activity. The results indicated that I/R injury
significantly increased ALK5 mRNA and protein expression (Fig. 2A and B) and phosphorylation of
SMAD2/3 (Fig. 2C-E). It was observed
that I/R injury significantly increased NOX2/4 expression and
activity (Fig. 2F-J). These data
indicated that NOX2/4 expression was associated with ALK5/p-SMAD2/3
expression.
ALK5 knockdown affects ALK5,
p-SMAD2/3, NOX2 and NOX4 expression in PC-12 cells
The association between ALK5/p-SMAD2/3 and NOX2/4
was assessed. An H/R model was constructed in vitro to
simulate I/R injury. Cells were treated with siRNA against ALK5 or
NC siRNA. The results indicated that ALK5 siRNA significantly
decreased ALK5 mRNA and protein expression, indicating that ALK5
was knocked down successfully (Fig. 3A
and B). Effects of ALK5 knockdown on phosphorylation of SMAD2/3
and expression of NOX2/4 were evaluated. Cells treated with ALK5
siRNA exhibited a significantly lower SMAD2/3 phosphorylation level
compared with cells treated with H/R alone (Fig. 3C-E). ALK5 knockdown significantly
decreased NOX2/4 mRNA and protein expression and total NOX enzyme
activity, when compared with the H/R group (Fig. 3F-J). These results suggested that
ALK5/p-SMAD2/3 signaling serves a key function in the regulation of
NOX2/4 expression and in oxidative stress injury.
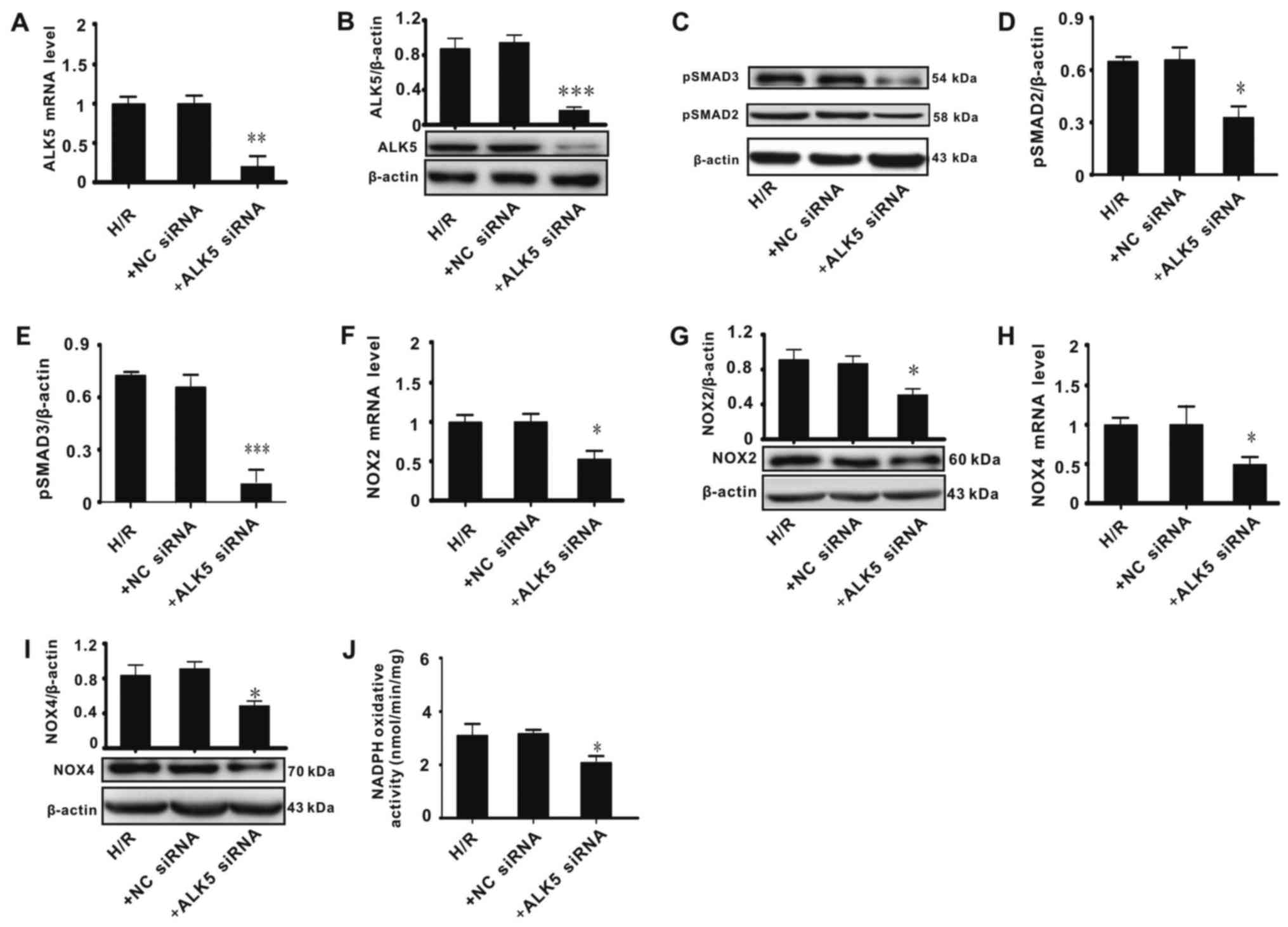 | Figure 3.ALK5 knockdown affects ALK5,
p-SMAD2/3, NOX2 and NOX4 expression in PC-12 cells. ALK5 (A) mRNA
and (B) protein levels. (C) Representative western blots for
p-SMAD2/3 expression. Ratio of (D) p-SMAD2 and (E) p-SMAD3 to
β-actin. NOX2 (F) mRNA and (G) protein levels. NOX4 (H) mRNA and
(I) protein levels. (J) Total NOX activity. Data were evaluated
using analysis of variance and Bonferroni's post hoc test.
*P<0.05, **P<0.01, ***P<0.001 vs. H/R. ALK5, activin
receptor-like kinase 5; p-, phosphorylated; NOX, nicotinamide
adenine dinucleotide phosphate oxidase; H/R, 5 h hypoxia/20 h
reoxygenation; +NC siRNA, negative control small interfering RNA;
+ALK5 siRNA, siRNA against ALK5. |
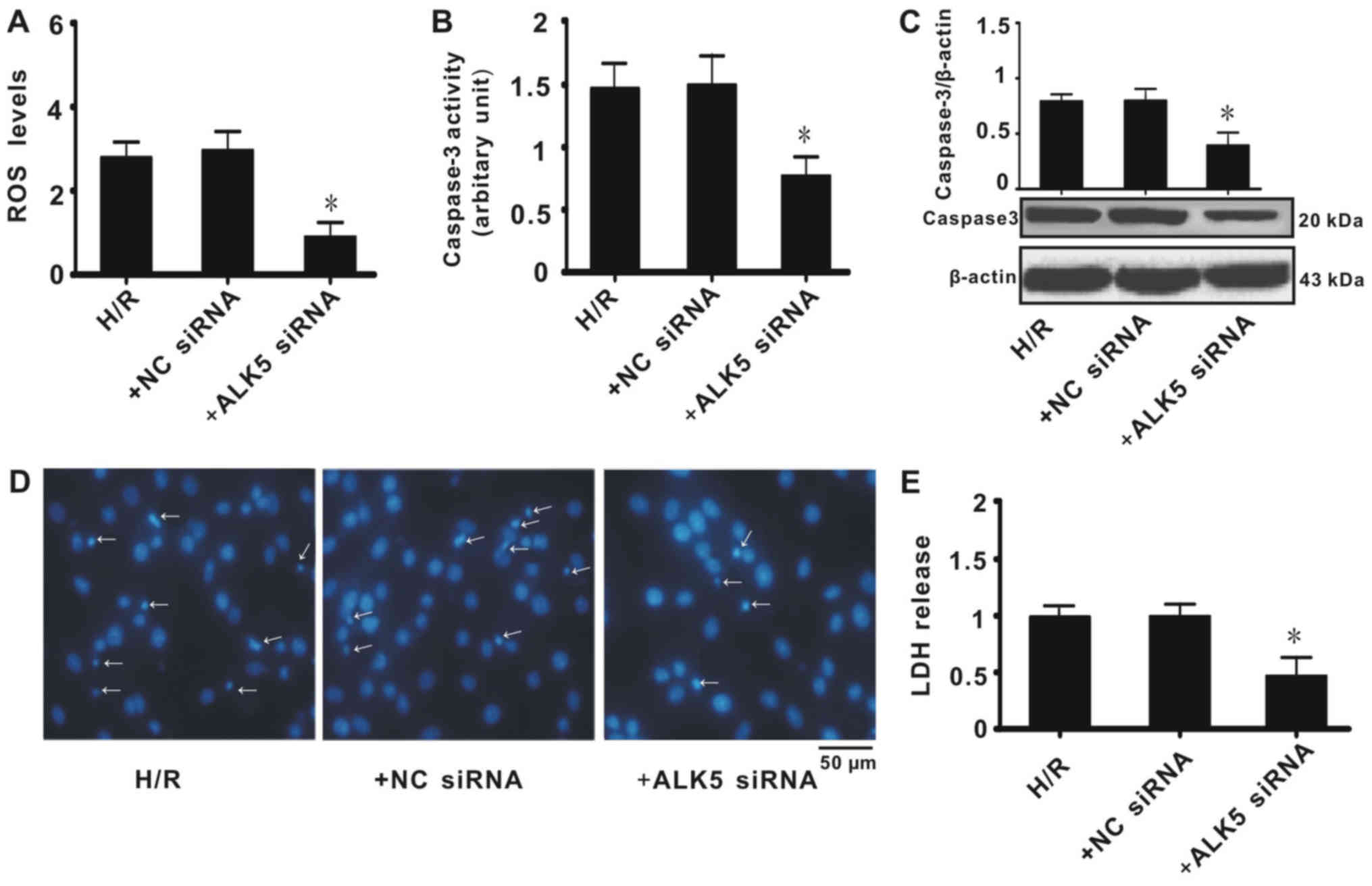
ALK5 knockdown affects ROS generation
and apoptosis in PC-12 cells
To further determine whether ALK5 knockdown affects
ROS generation in PC-12 cells, the total ROS level was measured. It
was revealed that cells treated with ALK5 siRNA had significantly
decreased ROS levels when compared with the H/R group (Fig. 4A). Expression and enzyme activity of
caspase-3, a mediator for apoptosis, were also measured. It was
identified that ALK5 knockdown significantly decreased caspase-3
expression and enzyme activity (Fig. 4B
and C). Hoechst staining indicated that ALK5 knockdown
decreased apoptosis in PC-12 cells when compared with the H/R or NC
siRNA groups (Fig. 4D). Consistent
with the results for ROS generation, caspase-3 activity and Hoechst
staining, LDH release was significantly decreased in PC-12 cells
treated with ALK5 siRNA when compared with the H/R group (Fig. 4E). These results indicated that ALK5
knockdown inhibited apoptosis of PC-12 cells and that the
underlying mechanism was associated with the inhibition of ROS
generation.
Discussion
The results of the present study demonstrated that
ALK5/p-SMAD2/3 signaling was associated with oxidative stress
following I/R injury. It was identified that increased oxidative
stress was associated with increased NOX2/4 and that NOX2/4
expression was associated with ALK5 expression and phosphorylation
of SMAD2/3. ALK5 knockdown significantly alleviated the damage to
PC-12 cells induced by H/R treatment. H/R contributed to increased
LDH release and ROS, which are primarily produced by NOX2/4 in
mammalian neuronal cells (18).
However, siRNA against ALK5 significantly suppressed NOX2/4
expression and decreased phosphorylation of SMAD2/3. The present
study indicated that activation of ALK5/p-SMAD2/3 signaling
exacerbates the extent of brain injury in I/R by upregulating
NOX2/4 expression.
Oxidative stress is associated with the pathogenesis
of a range of neurodegenerative disorders, including Alzheimer's
disease, Parkinson's disease, Huntington's disease, amyotrophic
lateral sclerosis, multiple sclerosis and ischemic stroke (5,18,19).
Excessive ROS generation, including superoxide anions, hydrogen
peroxide and hydroxyl radicals, is one of the main causes for
mitochondrial DNA mutations, mitochondrial respiratory chain
damage, membrane permeability alteration and Ca2+
dyshomeostasis (20). Previous
studies have reported that ROS are generated endogenously from
molecular oxygen by cellular oxidases, including mono- and
dioxygenases of the mitochondrial electron chain transport system,
peroxidases, NOX, nitric oxide synthases, cytochrome 450,
cyclooxygenases, lipooxygenases and xanthine oxidases (5,21).
Although seven subtypes of NOXs have been identified in rat
tissues, including NOX1-5, DUOX1 and DUOX2, NOX2 and NOX4 were
demonstrated to be the major contributors to brain tissue ROS
production (6). In a previous study,
it has been identified that NOX2/4 expression is increased in brain
tissues following I/R injury (6).
Consistent with this, NOX inhibition or knockout has been
demonstrated to significantly alleviate oxidative stress and
protect against I/R injury, which demonstrates that NOX2/4 serve
key functions in cerebral ischemic stroke (22).
Numerous factors, including transcription factors
(NF-κB, activator protein-1, signal transducer and activator of
transcription 1/3 and CCAAT/enhancer binding protein), nuclear
receptors (peroxisome proliferator-activated receptor α) and
epigenetic regulators (histone acetylation and microRNAs), are
associated with the regulation of NOX expression (23). Previously, the tumor suppressor TGF-β
was also demonstrated to be a key factor in ROS generation
(24,25). It has previously been identified that
TGF-β may induce ROS generation through the TGF-β/ALK5/SMAD2/3,
mitogen-activated protein kinase or c-Jun N-terminal kinase
pathways (13). The canonical TGF-β
signaling pathway includes TGF-βs, TGF-β receptors (including ALK1
and ALK5) and SMAD1-8 (10). For
ALK5, the TGF-β signaling is as follows: TGF-β binds ALK5 to form a
complex exhibiting kinase activity; the activated kinase
phosphorylates downstream substrates, SMAD2/3, aided by SMAD4;
p-SMAD2/3 translocates to the nucleus and promotes gene expression
(8,10). Emerging evidence has demonstrated
that TGF-β signaling is critical in the pathogenesis of several CNS
disorders, including neurodegenerative disorders and ischemic
stroke (11,12). In the present study, it was
identified that ALK5/SMAD2/3 signaling was associated with NOX2/4
expression and ROS generation following I/R injury. Similarly,
previous studies have identified that increased TGF-β levels were
associated with cerebral ischemic injury (12). It is speculated that ALK5/SMAD2/3
signaling serves an important role in ischemic stroke by regulating
NOX2/4 expression and ROS generation.
Excessive ROS are harmful to cells and body due to
their damage to biological macromolecules, including alcohol
dehydrogenase 2 (26,27). Therefore, inhibiting the production
of ROS is an effective method for treatment of oxidative stress
injuries and explains the neuroprotective role of NOX inhibitors
(28), including gp91ds-tat and
apocynin (29,30). These results suggest that NOX2/4 are
potential targets for antioxidant therapy. Considering the role of
the ALK5/SMAD2/3 signaling pathway in the regulation of NOX2/4
expression, inhibiting ALK5/SMAD2/3 signaling may be an effective
therapy for reducing oxidative stress. In summary, the present
study demonstrated that the ALK5/SMAD2/3/NOX pathway serves a key
function in I/R injury and provides a potential novel target for
ischemic stroke antioxidant treatment.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81603107 and
81600040), the Education Department of Hunan Province (grant no.
15C0161).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
ZL and APW contributed to the acquisition of data.
XMD, GHH, MLZ and ZBY contributed to the design of the experiments
and drafting of the manuscript. All authors have read and approved
the manuscript.
Ethics approval and consent to
participate
The use of animals in the present study was approved
by the Hunan Normal University Veterinary Medicine Animal Care and
Use Committee.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
I/R
|
ischemia/reperfusion
|
|
NOX
|
nicotinamide adenine dinucleotide
phosphate oxidase
|
|
ROS
|
reactive oxygen species
|
|
H/R
|
hypoxia/reoxygenation
|
|
ALK5
|
activin receptor-like kinase 5
|
|
TGF-β
|
transformation growth factor-β
|
References
|
1
|
Pandya RS, Mao L, Zhou H, Zhou S, Zeng J,
Popp AJ and Wang X: Central nervous system agents for ischemic
stroke: Neuroprotection mechanisms. Cent Nerv Syst Agents Med Chem.
11:81–97. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jhelum P, Karisetty BC, Kumar A and
Chakravarty S: Implications of epigenetic mechanisms and their
targets in cerebral ischemia models. Curr Neuropharmacol.
15:815–830. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jiang YF, Liu ZQ, Cui W, Zhang WT, Gong
JP, Wang XM, Zhang Y and Yang MJ: Antioxidant effect of salvianolic
acid B on hippocampal CA1 neurons in mice with cerebral ischemia
and reperfusion injury. Chin J Integr Med. 21:516–522. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chen H, Yoshioka H, Kim GS, Jung JE, Okami
N, Sakata H, Maier CM, Narasimhan P, Goeders CE and Chan PH:
Oxidative stress in ischemic brain damage: Mechanisms of cell death
and potential molecular targets for neuroprotection. Antioxid Redox
Signal. 14:1505–1517. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ma MW, Wang J, Zhang Q, Wang R, Dhandapani
KM, Vadlamudi RK and Brann DW: NADPH oxidase in brain injury and
neurodegenerative disorders. Mol Neurodegener. 12:72017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhang HF, Li TB, Liu B, Lou Z, Zhang JJ,
Peng JJ, Zhang XJ, Ma QL, Peng J and Luo XJ: Inhibition of myosin
light chain kinase reduces NADPH oxidase-mediated oxidative injury
in rat brain following cerebral ischemia/reperfusion. Naunyn
Schmiedebergs Arch Pharmacol. 388:953–963. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhang YS, Liu B, Luo XJ, Li TB, Zhang JJ,
Peng JJ, Zhang XJ, Ma QL, Hu CP, Li YJ, et al: Nuclear cardiac
myosin light chain 2 modulates NADPH oxidase 2 expression in
myocardium: A novel function beyond muscle contraction. Basic Res
Cardiol. 110:382015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Massagué J: How cells read TGF-beta
signals. Nat Rev Mol Cell Biol. 1:169–178. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Katz LH, Li Y, Chen JS, Muñoz NM, Majumdar
A, Chen J and Mishra L: Targeting TGF-β signaling in cancer. Expert
Opin Ther Targets. 17:743–760. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hata A and Chen YG: TGF-β signaling from
receptors to Smads. Cold Spring Harb Perspect Biol. 8:a0220612016.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gomes FC, Vde Sousa O and Romão L:
Emerging roles for TGF-beta1 in nervous system development. Int J
Dev Neurosci. 23:413–424. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Vivien D and Ali C: Transforming growth
factor-β signalling in brain disorders. Cytokine Growth Factor Rev.
17:121–128. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Docagne F, Ali C, Lesne S, Nicole O,
MacKenzie ET, Buisson A and Vivien D: Does transforming growth
factor-beta (TGF-beta) act as a neuroprotective agent in cerebral
ischemia? J Soc Biol. 197:145–150. 2003.(In French). View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hagler MA, Hadley TM, Zhang H, Mehra K,
Roos CM, Schaff HV, Suri RM and Miller JD: TGF-β signalling and
reactive oxygen species drive fibrosis and matrix remodelling in
myxomatous mitral valves. Cardiovasc Res. 99:175–184. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Clark JD, Gebhart GF, Gonder JC, Keeling
ME and Kohn DF: Special report: The 1996 guide for the care and use
of laboratory animals. ILAR J. 38:41–48. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Belarbi K, Cuvelier E, Destée A, Gressier
B and Chartier-Harlin MC: NADPH oxidases in Parkinson's disease: A
systematic review. Mol Neurodegener. 12:842017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yang ZB, Tan B, Li TB, Lou Z, Jiang JL,
Zhou YJ, Yang J, Luo XJ and Peng J: Protective effect of vitexin
compound B-1 against hypoxia/reoxygenation-induced injury in
differentiated PC12 cells via NADPH oxidase inhibition. Naunyn
Schmiedebergs Arch Pharmacol. 387:861–871. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Guo C, Sun L, Chen X and Zhang D:
Oxidative stress, mitochondrial damage and neurodegenerative
diseases. Neural Regen Res. 8:2003–2014. 2013.PubMed/NCBI
|
|
20
|
Grivennikova VG and Vinogradov AD:
Partitioning of superoxide and hydrogen peroxide production by
mitochondrial respiratory complex I. Biochim Biophys. 1827:446–454.
2013. View Article : Google Scholar
|
|
21
|
Di Meo S, Reed TT, Venditti P and Victor
VM: Role of ROS and RNS Sources in Physiological and Pathological
Conditions. Oxid Med Cell Longev. 2016:12450492016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kleinschnitz C, Grund H, Wingler K,
Armitage ME, Jones E, Mittal M, Barit D, Schwarz T, Geis C, Kraft
P, et al: Post-stroke inhibition of induced NADPH oxidase type 4
prevents oxidative stress and neurodegeneration. PLoS Biol.
8:e10004792010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Manea SA, Constantin A, Manda G, Sasson S
and Manea A: Regulation of Nox enzymes expression in vascular
pathophysiology: Focusing on transcription factors and epigenetic
mechanisms. Redox Biol. 5:358–366. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ghatak S, Hascall VC, Markwald RR,
Feghali-Bostwick C, Artlett CM, Gooz M, Bogatkevich GS,
Atanelishvili I, Silver RM, Wood J, et al: Transforming growth
factor β1 (TGFβ-1)-induced CD44V6-NOX4 signaling in pathogenesis of
idiopathic pulmonary fibrosis. J Biol Chem. 292:10490–10519. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hsieh HL, Wang HH, Wu WB, Chu PJ and Yang
CM: Transforming growth factor-β1 induces matrix
metalloproteinase-9 and cell migration in astrocytes: Roles of
ROS-dependent ERK-and JNK-NF-κB pathways. J Neuroinflammation.
7:882010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chen CH, Ferreira JC, Gross ER and
Mochly-Rosen D: Targeting aldehyde dehydrogenase 2: New therapeutic
opportunities. Physiol Rev. 94:1–34. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Glatt H, Rost K, Frank H, Seidel A and
Kollock R: Detoxification of promutagenic aldehydes derived from
methylpyrenes by human aldehyde dehydrogenases ALDH2 and ALDH3A1.
Arch Biochem Biophys. 477:196–205. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Rey FE, Cifuentes ME, Kiarash A, Quinn MT
and Pagano PJ: Novel competitive inhibitor of NAD(P)H oxidase
assembly attenuates vascular O(2)(−) and systolic blood pressure in
mice. Circ Res. 89:408–414. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chen H, Song YS and Chan PH: Inhibition of
NADPH oxidase is neuroprotective after ischemia-reperfusion. J
Cereb Blood Flow Metab. 29:1262–1272. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Simonyi A, Serfozo P, Lehmidi TM, Cui J,
Gu Z, Lubahn DB, Sun AY and Sun GY: The neuroprotective effects of
apocynin. Front Biosci (Elite Ed). 4:2183–2193. 2012. View Article : Google Scholar : PubMed/NCBI
|