Introduction
Sepsis-related acute respiratory distress syndrome
(ARDS) is characterized by aggravated oxidative stress and cell
apoptosis with high morbidity and mortality (1,2).
Recently, a study demonstrated that mitochondrial dysfunction
contributed to the progression of experimental sepsis (3). Abnormal mitochondrial dynamics, namely
accelerated fission, served a crucial role in mitochondrial
dysfunction and cell death (4). In
mammalian cells, the process of mitochondrial fission is regulated
by the dynamin-related protein 1 (Drp1) and mitochondrial fission 1
protein (Fis1) (5). Fis1, the
mitochondrial outer membrane receptor for Drp1, is necessary for
the removal of damaged mitochondria by mitophagy (6). Previously studies demonstrated that
extensive mitochondrial Fis1 accelerated the emission of reactive
oxygen species (ROS) and marked oxidative stress promoted aberrant
fragmented mitochondria during programmed cell death (7,8).
Heme oxygenase-1 (HO-1)/carbon monoxide (CO), as the
endogenous antioxidant system of organisms, exhibits
anti-inflammatory, antioxidant and anti-apoptotic functions in
sepsis-related acute lung injury (9,10). CO,
whether produced endogenously as a product of heme degradation
catalyzed by HO-1 or released exogenously by CO donor compounds
named CO releasing molecule-2 (CORM2), has been reported to exhibit
cytoprotective actions against several experimental models
including sepsis or ischemia and reperfusion (11,12). The
authors' previous studies demonstrated that CO could suppress the
expression of Fis1 and alleviate lipopolysaccharide (LPS)-induced
oxidative stress damage in alveolar macrophages (3,13).
However, the possible mechanism by which HO-1/CO exerted a
cytoprotective effect has not completely elucidated. In addition,
as a major survival signaling pathway, activation of
phosphatidylinositol 3-kinase (PI3K)/protein kinase B (Akt)
resulted in the nuclear translocation of NF-E2 related factor 2
(Nrf2) and subsequently increased the expression of the downstream
target gene, HO-1 (14,15). Additionally, phosphorylation of Akt
at Ser473 via a PI3K-dependent pathway served a crucial role in
regulating mitochondrial oxidative stress-mediated tissue injury
(16,17). As described by Wang et al
(18), impaired PI3K/Akt signaling
may be associated with retinol binding protein 4 (RBP4)-induced
imbalance of the fusion and fission cycles in mitochondria.
As guardians of the alveolar-blood interface against
airborne particles and microbes, alveolar macrophages serve pivotal
roles in the pathogenesis of acute lung injury/ARDS (19). In this regards, the present study
aimed to elucidate whether PI3K/Akt pathway-mediated HO-1/CO
represses Fis1 levels and alleviates LPS-induced oxidative injury
in alveolar macrophages. The present study aimed to lay a
foundation for potential future clinical applications of CO or
CORM2 for sepsis-induced acute lung injury.
Materials and methods
Cell culture
Rat alveolar macrophage NR8383 cells (American Type
Culture Collection, Manassas, VA, USA) were cultured in Dulbecco's
modified Eagle medium, supplemented with 10% heat-inactivated fetal
bovine serum, 100 U/ml penicillin A, 0.1 mg/ml streptomycin and 2
mmol/l L-glutamine (All Invitrogen; Thermo Fisher Scientific, Inc.)
and maintained at 37°C in a humidified atmosphere of 5%
CO2 and 95% air. Cells were grown to 80–90% confluence
and seeded in 96-well plates for the subsequent experiments.
Experimental set-up
Cells were seeded at a density of 1×104
cells/ml in 96-well plates, and then stimulated with 10 µg/ml LPS
from Escherichia coli O111: B4 (Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany) for 24 h as described in previous studies
(13,20). To evaluate the role of CO on
LPS-induced oxidative injury and the possible mechanism, cells were
pretreated with 100 µM CORM2 (Sigma-Aldrich; Merck KGaA) or 25 µM
LY294002 (Merck KGaA), a specific inhibitor of PI3K, 1 h prior to
LPS administration and then sequentially incubated for 24 h, each
at 37°C (3,21). In the case of CORM2, the inactive
form (iCORM2; 100 µM) served as a negative control, a molecule
where the carbonyl groups were replaced with dimethyl sulfoxide
(DMSO) and finally bubbled with N2 gas to remove the
residual solubilized CO (22). The
final concentration of the solvent DMSO was <0.5%.
Cell viability
Cell viability was analyzed with an MTT assay. Prior
to being treated with different manipulations, NR8383 cells were
seeded in 96-well plates at a density of 1×104 cells/ml
and cultured overnight at 37°C. Briefly, 10 µl MTT (2.5 mg/ml in 1M
PBS) was added into each well at the end of culture. After a 3-h
incubation at 37°C and 5% CO2, the MTT solution was
discarded and DMSO was added to dissolve the blue formazan
crystals. The amount of formazan was measured
spectrophotometrically by determining the absorbance at a
wavelength of 540 nm using a microplate reader.
Biochemical measurements
Measurements of tumor necrosis factor-α (TNF-α; cat.
no. H052), interleukin-10 (IL-10) levels (cat. no. H009),
malondialdehyde (MDA) contents (cat. no. A003-1) and superoxide
dismutase (SOD) activities (cat. no. A001-3) in the NR8383 cell
supernatant were respectively performed with the relevant
commercially available ELISA kits supplied by Nanjing Jiancheng
Bioengineering Institute (Nanjing, China) according to the
manufacturer's protocols.
Transmission electron microscopy
Briefly, cultured cells were harvested by scraping
following exposure to CORM2 or LY294002 pretreatment. Cells were
fixed in 2.5% glutaradehyde/2% paraformaldehyde overnight at 4°C
and subsequently post-fixed in 1% osmium tetroxide for 1 h on ice,
dehydrated in gradient series of ethanol (70–100%), embedded with
Epon 812 resin blocks and cultured at 90°C for 72 h based on the
routine protocol described previously (13). Ultrathin sections (1-µm-thick) were
cut with a microtome (Leica, Microsystems GmbH, Wetzlar, Germany).
Finally, the images were observed using transmission electron
microscopy (JEOL, Ltd. Tokyo, Japan) at ×5,000 magnification by a
blinded observer. The mitochondrial morphology of NR8383 cells was
divided into three categories. Normal cells presented an intact
network of tubular mitochondria, fragmented cells displayed
predominantly spherical or rod-like mitochondria, and hyperfused
cells exhibited considerably interconnected, elongated mitochondria
(23).
Western blot analysis
Following pretreatment with 10 µg/ml LPS, 100 µM
CORM2, 100 µM iCORM2 and 25 µM LY294002, NR8383 cells were
sequentially incubated for 24 h at 37°C and lysed using 200 µl
radioimmunoprecipitation assay lysis buffer [containing 25 mM
Tris·HCl (pH 7.6), 150 mM NaCl, 1% Nonidet P-40, 1% sodium
deoxycholate, and 0.1% SDS (cat. no. 89901, Invitrogen, Thermo
Fisher Scientific, Inc.)] on ice for 40 min to extract the proteins
at 4°C. The lysates were centrifuged at 10,000 × g for 15 min at
4°C. The protein concentrations of the supernatants were determined
by Bradford assay. Protein samples (50 µg/lane) were separated by
10% SDS-PAGE for 2 h, transferred onto polyvinylidene fluoride
membranes and blocked using 5% non-fat powdered milk for 2 h at
37°C. Samples were then incubated at 4°C overnight with the
following primary antibodies: Anti-Fis1 (1:500; cat. no. SC-30122;
Santa Cruz Biotechnology, USA), anti-HO-1 (1:800; cat. no. ab13248;
Abcam, Cambridge, UK), anti-total (t)-Akt (1:500; cat. no.
ab176657), anti-phospho (p)-Akt (1:1,000; cat. no. ab38449; Abcam)
and anti-β-actin (1:1,000; cat. no. A1978; Sigma-Aldrich; Merck
KGaA). The blots were subsequently incubated with horseradish
peroxidase-conjugated goat anti-rabbit secondary immunoglobulin G
antibody (1:3,000; cat. no. A0545; Sigma-Aldrich; Merck KGaA) at
room temperature for 1 h. The immunoblots were visualized by the
enhanced chemiluminescence reagent (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA) and exposed to film. The image densities of
specific protein bands were normalized to target protein of β-actin
and determined by a densitometry (ImageLab™ Software 170–9690,
version 5-2-1; Bio-Rad Laboratories, Inc.).
Reverse transcription quantitative
polymerase chain reaction (RT-qPCR)
Following incubation with various chemicals for 24
h, NR8383 cells (1×104/well) were lysed for RNA
extraction and RT-qPCR. In brief, total RNA was extracted from rat
alveolar macrophage NR8383 cells using a Total RNAiso™ kit (Takara
Bio, Inc., Otsu, Japan; cat. no. D9108B) based on the
manufacturer's protocol. cDNA was synthesized using 1 µg total RNA
with a Revert First Strands cDNA Synthesis kit for 60 min at 42°C
(Takara Bio, Inc., Otsu, Japan; cat. no. DRRO47A) and RT-qPCR was
performed via a SYBR Premix Ex Taq™ kit (Takara Bio, Inc., Otsu,
Japan; cat. no. DRR420A). Initial denaturation of the PCR mix was
at 95°C for 10 min, followed by 40 cycles of denaturation at 95°C
for 30 sec, annealing at 95°C for 5 sec and extension at 60°C for
34 sec. The primers used were as follows: Fis1 forward,
5′-TACCCCGAGGCTGTCCTAAG-3′ and reverse, 5′-CAGGACATTAGGCCCAGAGC-3′,
147 bp; HO-1 forward, 5′-GAATCGAGCAGAACCAGCCT-3′ and reverse,
5′-CTCAGCATTCTCGCTTGGA-3′, 135 bp; β-actin forward,
5′-TGTGTCCGTCGTGGATCTGA-3′ and reverse,
5′-TTGCTGTTGAAGTCGCAGGAG-3′, 149 bp. β-actin served as the
reference gene, and fold changes were calculated using the
2−ΔΔCt method as described by Livak and Schmittgen
(24).
Statistical analysis
Statistical analysis was performed using SPSS
software version 16.0 (SPSS, Inc., Chicago, IL, USA) and GraphPad
Prism software version 6.0 (GraphPad Software, Inc., La Jolla, CA,
USA). All data were expressed as the mean ± the standard deviation
except for cell viability, for which the Kruskal-Wallis test
followed by Dunns post-hoc test was used as appropriate. The
statistical significance of the differences between groups was
determined by using one-way analysis of variance followed by
Bonferroni's post-hoc correction test. P<0.05 was considered to
indicate a statistically significant difference.
Results
PI3K/Akt pathway-mediated HO-1/CO
improves the viability of NR8383 cells
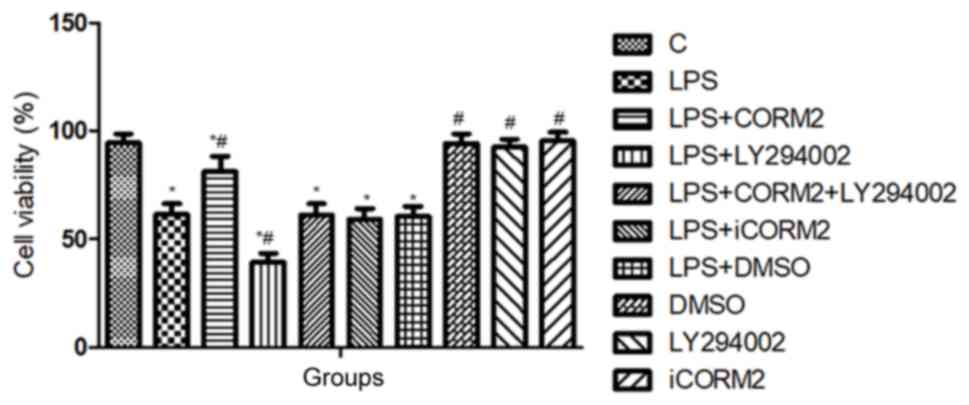
As shown in Fig. 1,
the viability of NR8383 cells was reduced to 61.4±4.92% by LPS,
which indicated that 10 µl/ml LPS initiated oxidative injury in
cells. Pretreatment with CORM2 significantly increased the
viability of NR8383 cells to 81±7.18% (P<0.05). However,
inclusion of LY294002, as the specific inhibitor of PI3K, further
decreased the numbers of viable cells treated with LPS, and
counteracted the improved cell viability offered by CORM2. No
significant differences in cell viability were observed in the
solvent DMSO and iCORM2 groups (P>0.05).
PI3K/Akt pathway-mediated HO-1/CO
alleviates LPS-induced oxidative injury in NR8383 cells
To estimate the anti-oxidative and anti-inflammatory
effects of HO-1/CO mediated by the PI3K/Akt pathway, the levels of
MDA, SOD, TNF-α and IL-10 in the media of NR8383 cells were
detected (Fig. 2). The results
demonstrated that TNF-α levels and MDA contents were increased,
while IL-10 levels and SOD activities were decreased in LPS-exposed
cells, compared with the respective controls. Furthermore, CORM-2
pretreatment resulted in a 43.8 and 22.6% decrease of TNF-α levels
and MDA contents, respectively, and a 28.5 and 18.6% increase of
IL-10 levels and SOD activities, respectively. Conversely, the
above protective effects were counteracted by pretreatment with
LY294002, which indicated that the PI3K/Akt pathway participated in
the effects of HO-1/CO in alleviating LPS-induced oxidative injury
in NR8383 cells. As a comparison, iCORM2 or DMSO did not have any
significant influences on the levels of MDA, SOD, TNF-α and IL-10
(P>0.05).
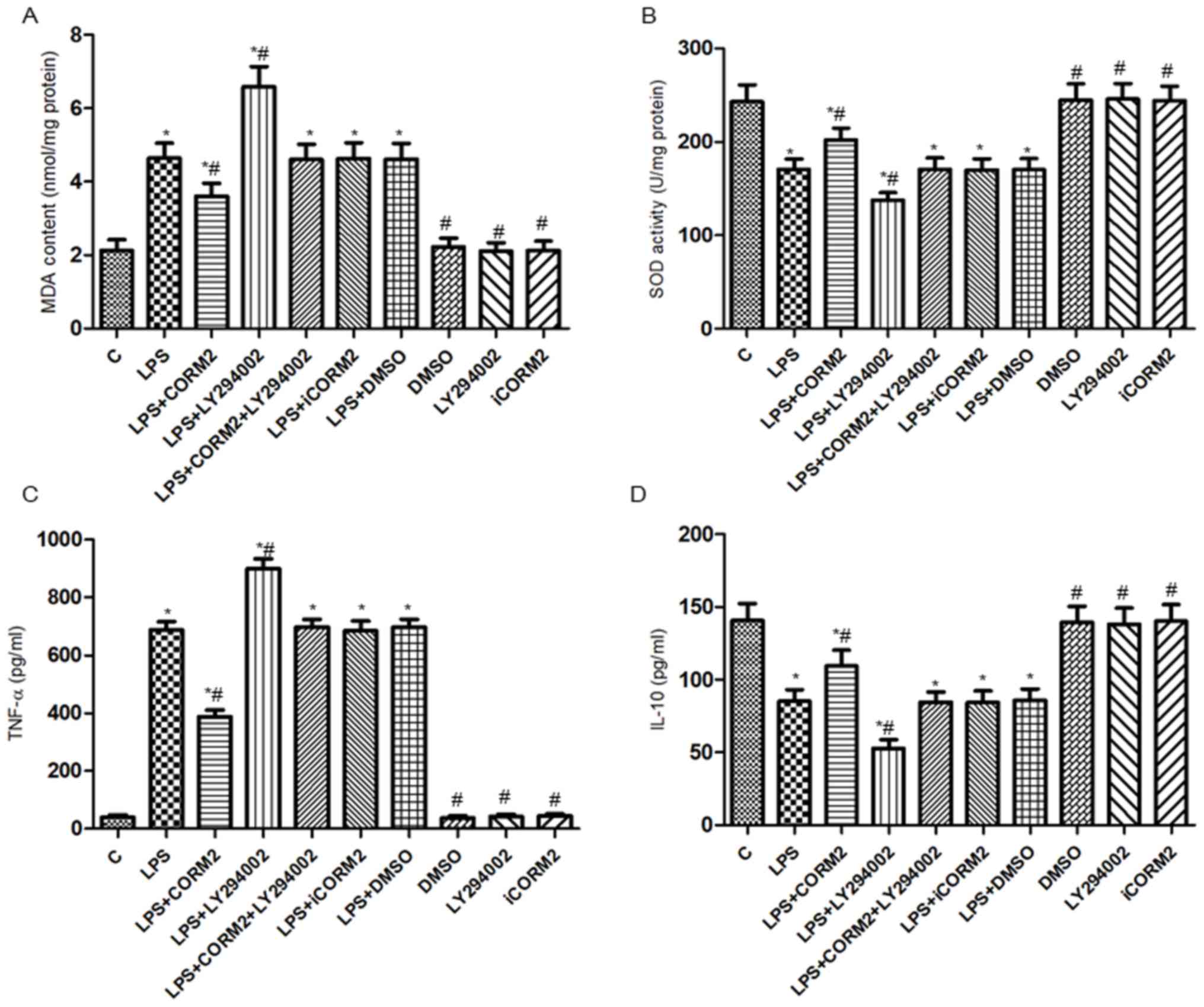 | Figure 2.PI3K/Akt pathway-mediated HO-1/CO
alleviated LPS-induced oxidative injury in NR8383 macrophages.
Cells were pre-incubated with 100 µM CORM2 or 25 µM LY294002 for 1
h prior to incubation with 10 µl/ml LPS for 24 h. The (A) MDA
content, (B) SOD activity, and (C) levels of TNF-α and (D) IL-10 in
the media of NR8383 cells were determined by the relevant
commercially available enzyme-linked immunosorbent assay kits. Data
were expressed as mean ± standard deviation of at least five
independent experiments using one-way analysis of variance and the
Bonferroni test for multiple comparisons. *P<0.05 vs. C,
#P<0.05 vs. LPS. LPS, lipopolysaccharide; HO-1, heme
oxygenase-1; CORM2, CO releasing molecule-2; iCORM2, inactive
CORM2; LY294002, the specific inhibitor of PI3K; TNF-α, tumor
necrosis factor-alpha; IL-10, interleukin-10; MDA, malondialdehyde;
SOD, superoxide dismutase; PI3K, phosphoinositide 3 kinase; Akt,
protein kinase B; CO, carbon monoxide; C, control. |
PI3K/Akt pathway-mediated HO-1/CO
suppresses the expression of Fis1 in NR8383 cells exposed to
LPS
To investigate whether the PI3K/Akt pathway
participated in the effects of HO-1/CO on the expressions of
mitochondrial Fis1 in NR8383 cells subjected to LPS, the
mitochondrial morphology (Fig. 3) as
well as the levels of t-Akt, p-Akt, HO-1 and Fis1 protein and mRNA
were detected by western blotting and RT-qPCR (Figs. 4 and 5). Consistent with our previous study
(3), exposure to LPS significantly
increased mitochondrial fragmentation, upregulated the expressions
of the Fis1 gene and protein (P<0.05), along with mild elevation
of the levels of the HO-1 and p-Akt proteins (P<0.05).
Additionally, increased levels of mRNA and proteins of HO-1 and
p-Akt, but decreased levels of Fis1 mRNA and protein were observed
in CORM2-pretreated cells stimulated by LPS, accompanied by
increased mitochondrial elongation. However, pre-incubation with
LY294002 of cells exhibited a 19.5–27.9% reduction in the
expressions of the p-Akt protein, HO-1 mRNA and proteins, while a
14.2–28.8% elevation in the levels of Fis1 mRNA and proteins was
demonstrated. Concordantly, more spherical or rod-like fragmented
mitochondria were observed in LY294002-pretreated NR8383 cells
subjected to LPS. There were no significant translational
differences in the expression of t-Akt protein following
stimulation with various reagents (P>0.05). iCORM2 or DMSO
induced no effects on the mRNA and protein levels of
above-mentioned indicators.
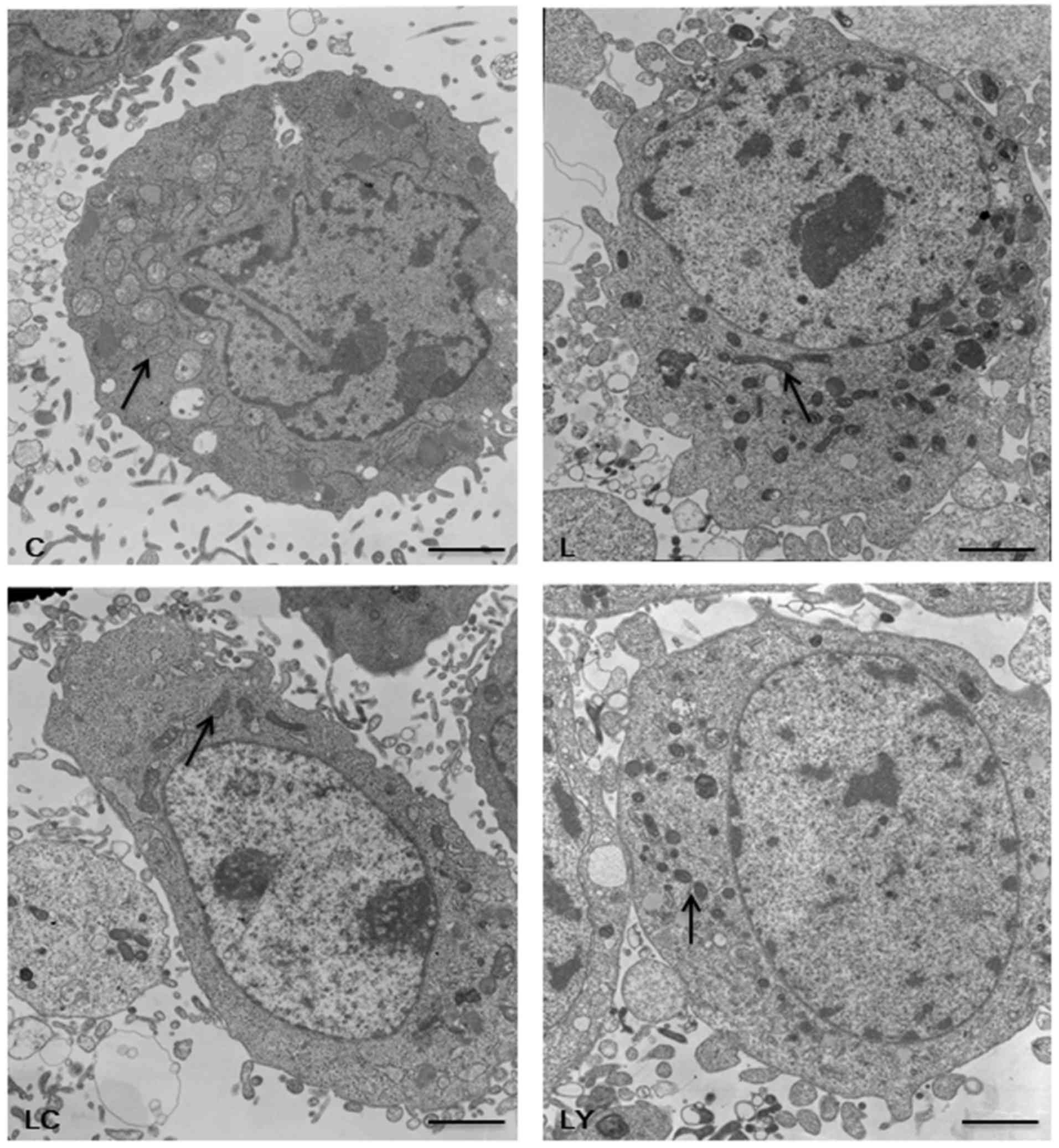 | Figure 3.PI3K/Akt pathway-mediated HO-1/CO
suppresses the fission of mitochondria in NR8383 cells. Images of
the morphological structures of mitochondria in NR8383 cells were
observed by a transmission electron microscopy under ×5000
magnification. The Control group (group C) showed the normal shape
of mitochondria. LPS exposure resulted in more swollen and
fragmented mitochondria (group L). Pretreatment with LY294002
aggravated the mitochondrial fragmentation in LPS-treated NR8383
cells (group LY). Conversely, inclusion of CORM2 revealed more
branched, elongated mitochondria in NR8383 cells exposed to LPS
(group LC). Arrows indicated the normal, fragmented or elongated
mitochondria (Scale bar, 2 µm; magnification ×5,000). LPS,
lipopolysaccharide; HO-1, heme oxygenase-1; CORM2, CO releasing
molecule-2; LY294002, the specific inhibitor of PI3K; PI3K,
phosphoinositide 3 kinase; Akt, protein kinase B; CO, carbon
monoxid; group C, the control group; group L, the LPS group; group
LY, the LPS+LY294002 group; group LC, the LPS+CORM2 group. |
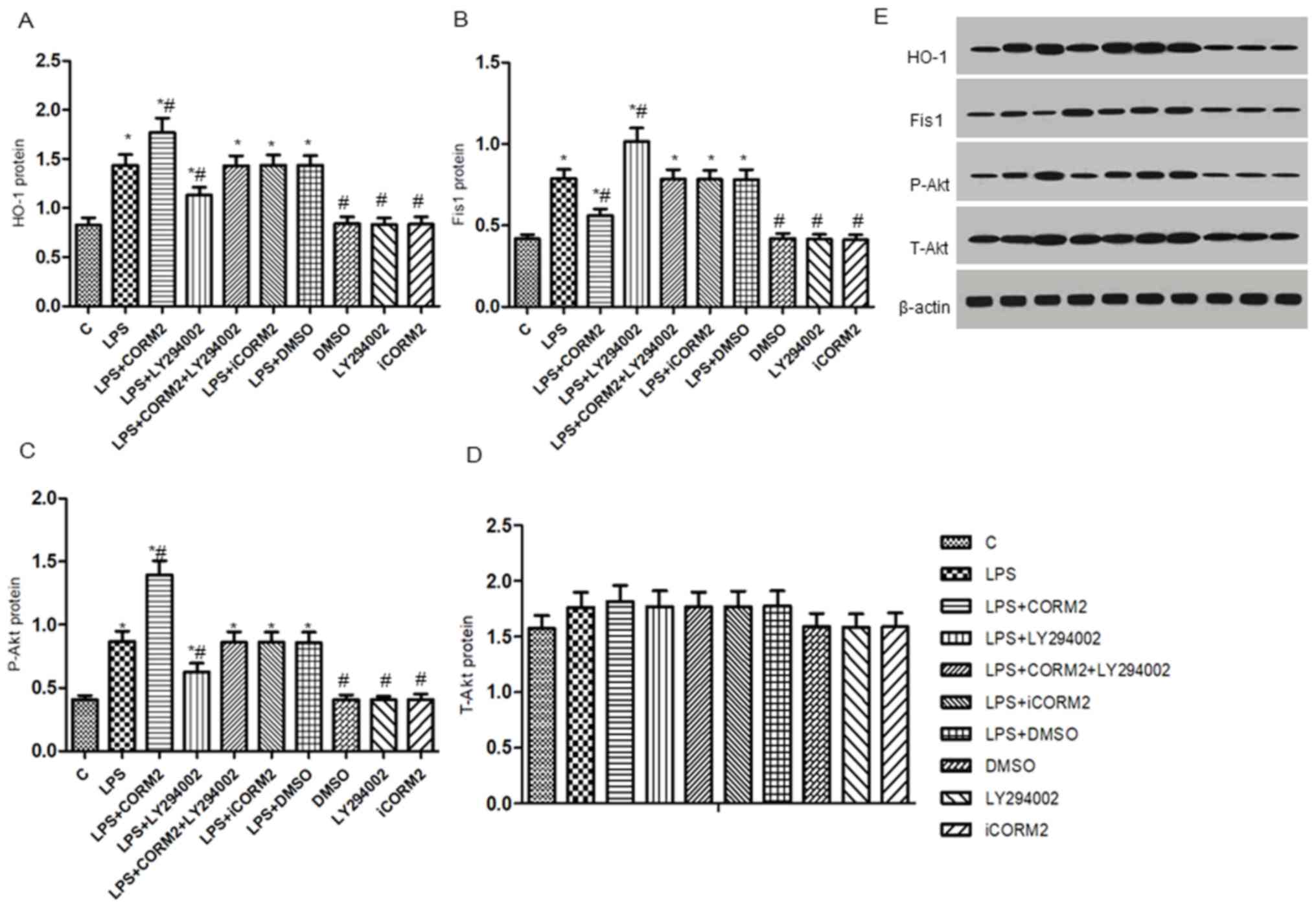 | Figure 4.PI3K/Akt pathway-mediated HO-1/CO
represses the levels of Fis1 protein in NR8383 cells subjected to
LPS. Relative expression levels of the (A) HO-1, (B) Fis1, (C)
p-Akt and (D) t-Akt proteins by western blotting in cells presented
in histograms. (E) The image densities of specific protein bands
were normalized to β-actin and semi-quantified by densitometry.
Data were expressed as the mean ± standard deviation of at least
five independent experiments using a one-way analysis of variance
and the Bonferroni test for multiple comparisons. *P<0.05 vs. C,
#P<0.05 vs. LPS. T-Akt, total protein kinase B (Akt);
P-Akt, phosphorylated Akt; HO-1, heme oxygenase-1; Fis1,
mitochondrial fission 1 protein; LPS, lipopolysaccharide; CORM2, CO
releasing molecule-2; iCORM2, inactive CORM2; LY294002, the
specific inhibitor of PI3K; CO, carbon monoxide; C, control. |
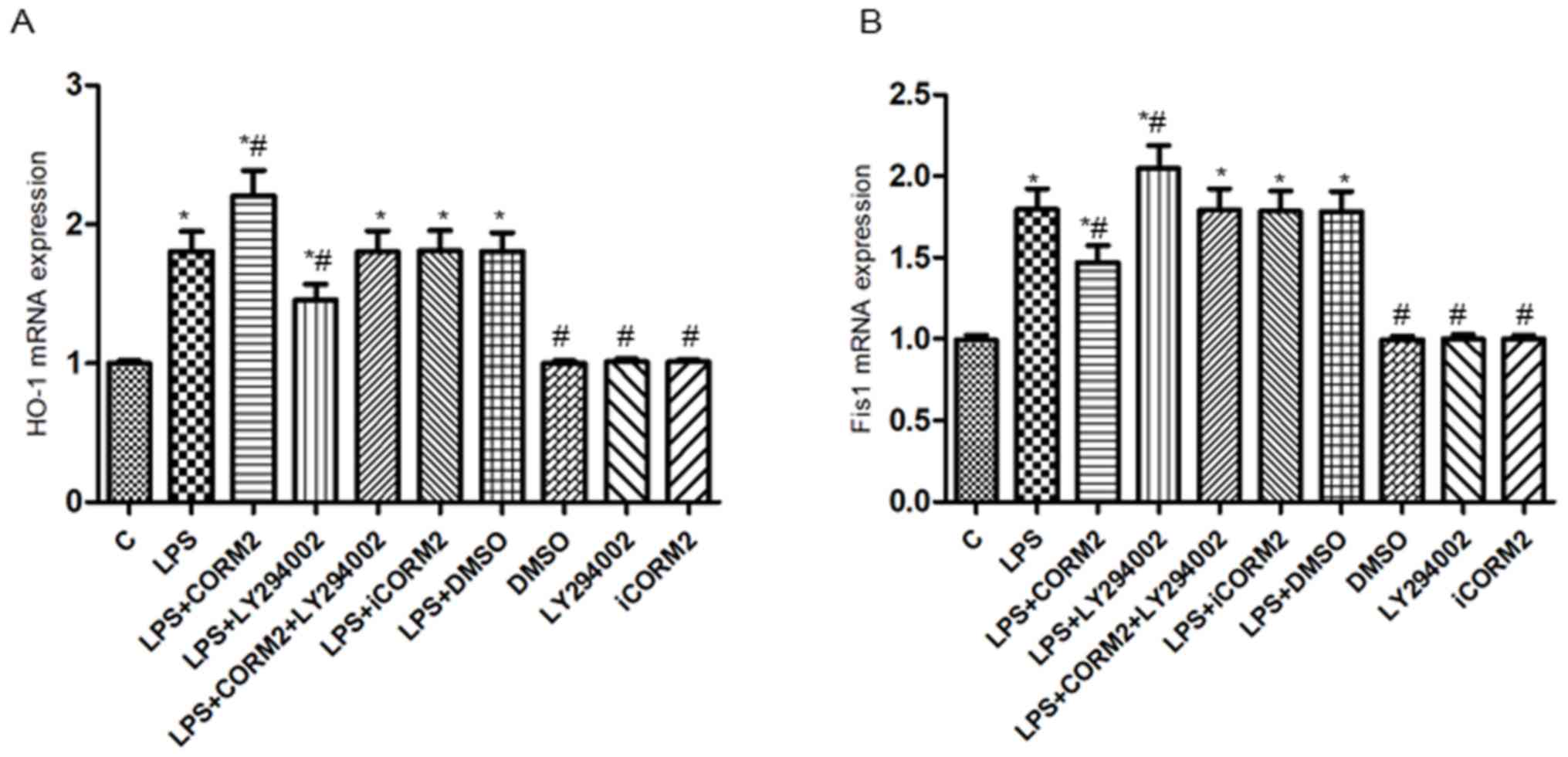 | Figure 5.PI3K/Akt pathway-mediated HO-1/CO
represses the levels of Fis1 mRNA in NR8383 cells subjected to LPS.
Relative expression levels of (A) HO-1mRNA and (B) Fis1 mRNA
detected by reverse transcription quantitative polymerase chain
reaction. β-actin served as the reference gene. Data were expressed
as the mean ± standard deviation of at least five independent
experiments using a one-way analysis of variance and the Bonferroni
test for multiple comparisons. *P<0.05 vs. C,
#P<0.05 vs. LPS. Fis1, mitochondrial fission 1
protein; LPS, lipopolysaccharide; CORM2, CO releasing molecule-2;
iCORM2, inactive CORM2; LY294002, the specific inhibitor of PI3K;
DMSO, dimethyl sulfoxide; PI3K, phosphoinositide 3 kinase; Akt,
protein kinase B; CO, carbon monoxide; HO-1, Heme oxygenase-1; C,
control. |
Discussion
Alveolar macrophages have been used to remove
inhaled particles including dusts, bacteria, and viruses from the
airways (25). In addition,
macrophages serve a role in the modulation of host defenses and
could be activated by LPS to initiate an inflammatory response and
generate, and release inflammatory cytokines and ROS (26). The rat alveolar macrophage NR8383
cells behave similarly to primary rat alveolar macrophages with
respect to the respiratory burst, phagocytosis and cytokine
responses (27). Herein, the
well-characterized NR8383 macrophages were used to investigate the
effects of the HO-1/CO system on the levels of Fis1 and LPS-induced
oxidative injury of cells and attempted to elucidate the possible
mechanism of action. A number of studies indicated that abnormal
mitochondrial fission was a determinant in the pathogenesis of
sepsis (2,28,29).
Fis1, a small molecule uniformly located in the mitochondrial outer
membrane, was necessary for mitochondrial fission and removal of
dysfunctional mitochondria (30,31). Lee
et al (32) reported that
downregulated human Fis1 powerfully inhibited cell death to a
significantly greater degree compared with the downregulation of
Drp1. In addition, the activity of SOD used in the present study
reflected the ability of the cell to scavenge free radicals, while
MDA content reflected the severity of membrane damage and degrees
of oxidative stress (33).
Respectively, TNF-α and IL-10 are commonly used cytokines of
pro-inflammatory and anti-inflammatory responses. In the current
study, exposure to LPS resulted in increased levels of Fis1 mRNA
and proteins, accompanied by reduced cell viability, elevated MDA
contents and TNF-α levels, but declined SOD activities and IL-10
levels, which indicated that LPS upregulated the expressions of
Fis1 and gave rise to oxidative injury of NR8383 cells.
As an endogenous antioxidant, the HO-1 system
confers cytoprotection against the oxidative cellular injury
(9). CO, as one of the products of
heme degradation, whether it comes from CORM2 or HO-1 induction,
appears to induce antioxidative, anti-inflammatory and
anti-apoptotic effects (11).
Therefore, CORM2 was used as a potent donor of CO in the present
study and pretreatment with CORM2 significantly inhibited
mitochondrial fragmentation, upregulated the expressions of HO-1
mRNA and proteins, and downregulated levels of Fis1 mRNA and
proteins in NR8383 cells subjected to LPS. CORM2 alleviated
LPS-induced oxidative cellular injury characterized by elevated SOD
activities and IL-10 levels, while reducing MDA contents and TNF-α
levels, which was consistent with previous studies by the authors
of the present study (3,14). Furthermore, the PI3K/Akt signaling
pathway could be activated by the LPS-induced Toll-like
receptor-4-mediated pathway, which is upstream of phase II
detoxifying enzyme, HO-1, and serves critical roles in inflammatory
responses and oxidative stress-mediated tissue injury (34,35).
Consequently, upregulated p-Akt protein, and HO-1 mRNA and protein
levels were identified in CORM2-pretreated cells. However,
inclusion of LY294002, the specific inhibitor of PI3K, markedly
reversed the CORM2-mediated protective actions described above,
which resulted in marked mitochondrial fragmentation, decreased
phosphorylation of Akt, reduced levels of HO-1 mRNA and protein,
and elevated expression levels of Fis1 mRNA and protein. This was
followed by reduced cell viability, elevated MDA contents and TNF-α
levels, and decreased SOD activities and IL-10 levels. The results
of the current study clarified the role of the PI3K/Akt
pathway-mediated HO-1/CO in suppressing the expression of Fis1 and
alleviating LPS-induced oxidative injury in alveolar
macrophages.
There were certain limitations to the present study.
The recently published report by Monick et al (36) indicated that 100 mg/ml LPS-mediated
induction of ceramide resulted in PI3K activation and promoted
survival of human alveolar macrophages in the setting of pulmonary
sepsis. As a comparison, rat alveolar macrophage NR8383 cells
exposed to 10 µg/ml LPS for 24 h were used as the classical model
of LPS-induced cellular oxidative injury in the current study. In
this case, the endogenous cytoprotection of HO-1 on mitochondrial
Fis1, and the possible associated signaling pathway were
investigated. Increased expressions of p-Akt and HO-1 was
demonstrated in NR8383 cells treated with LPS. However, the
activation of the PI3K/Akt pathway-mediated mild increase of HO-1
level was not sufficient to inhibit mitochondrial fission in the
context of LPS-induced oxidative stress. The results were
consistent with previous studies (3,13).
Future studies should include mechanistic data with regard to the
interactions between PI3K and Fis1. In addition, as previous
studies reported, activation of the Nrf2/ARE pathway accounted for
the beneficial effects of HO-1 against sepsis-related multiple
organ dysfunctions (37,38). Furthermore, PI3K/Akt signaling was
involved in the nuclear translocation of Nrf2 (14). Therefore, further studies are
required to clarify the role of the Nrf2/ARE pathway in the HO-1/CO
system in regulating the balance of mitochondrial fusion/fission.
Ultimately, the P38 mitogen associated protein kinase,
extracellular signal regulated kinase and protein kinase C
signaling pathways had been reported to mediate the induction of
HO-1 (39). Hence, to identify which
signaling cascades other than the PI3K/Akt pathway facilitated the
cytoprotective effects of HO-1/CO system against oxidative cellular
injury requires further investigation.
In conclusion, the present study revealed that the
PI3K/Akt signaling pathway mediated the HO-1/CO system in
repressing the levels of mitochondrial fission protein, Fis1 and
alleviating LPS-induced oxidative injury in NR8383 alveolar
macrophages. This result may serve as supporting evidence for
further studies regarding the clinical applications of CO or CORM2
to relieve sepsis-related lung injury or oxidative cellular
injury.
Acknowledgements
The authors would like to thank Professor Holger K.
Eltzschig (The University of Texas Health Science Center at
Houston, McGovern Medical School) for his technical expertise and
valuable advice.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant no. 81772106).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JY conceived and designed this project. JS, YZ, ZL,
LG, SD and RM performed the key experiments and associated data
analyses. JS and ZL classified and interpreted data, and performed
statistical analysis. JS wrote the manuscript and all authors
contributed to revision of the manuscript. JY oversaw the entire
project.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Pierrakos C and Vincent JL: The changing
pattern of acute respiratory distress syndrome over time: A
comparison of two periods. Eur Respir J. 40:589–595. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Moussa MD, Santonocito C, Fagnoul D,
Donadello K, Pradier O, Gaussem P, De Backer D and Vincent JL:
Evaluation of endothelial damage in sepsis-related ARDS using
circulating endothelial cells. Intensive Care Med. 41:231–238.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yu JB, Shi J, Wang D, Dong SA, Zhang Y,
Wang M, Gong LR, Fu Q and Liu DQ: Heme oxygenase-1/carbon
monoxide-regulated mitochondrial dynamic equilibrium contributes to
the attenuation of endotoxin-induced acute lung injury in rats and
in lipopolysaccharide-activated macrophages. Anesthesiology.
125:1190–1201. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sebastián D, Palacín M and Zorzano A:
Mitochondrial dynamics: Coupling mitochondrial fitness with healthy
aging. Trends Mol Med. 23:201–215. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Westermann B: Mitochondrial fusion and
fission in cell life and death. Nat Rev Mol Cell Biol. 11:872–884.
2010. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chang CR and Blackstone C: Dynamic
regulation of mitochondrial fission through modification of the
dynamin-related protein Drp1. Ann N Y Acad Sci. 1201:34–39. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lee S, Park YY, Kim SH, Nguyen OT, Yoo YS,
Chan GK, Sun X and Cho H: Human mitochondrial Fis1 links to cell
cycle regulators at G2/M transition. Cell Mol Life Sci. 71:711–725.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mai S, Klinkenberg M, Auburger G,
Bereiter-Hahn J and Jendrach M: Decreased expression of Drp1 and
Fis1 mediates mitochondrial elongation in senescent cells and
enhances resistance to oxidative stress through PINK1. J Cell Sci.
123:917–926. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ryter SW and Choi AM: Targeting heme
oxygenase-1 and carbon monoxide for therapeutic modulation of
inflammation. Transl Res. 167:7–34. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Constantin M, Choi AJ, Cloonan SM and
Ryter SW: Therapeutic potential of heme oxygenase-1/carbon monoxide
in lung disease. Int J Hypertens. 2012:8592352012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Qin W, Zhang J, Lv W, Wang X and Sun B:
Effect of carbon monoxide-releasing molecules II-liberated CO on
suppressing inflammatory response in sepsis by interfering with
nuclear factor kappa B activation. PLoS One. 8:e758402013.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ryter SW and Choi AM: Cytoprotective and
anti-inflammatory actions of carbon monoxide in organ injury and
sepsis models. Novartis Found Symp. 280:165–175. 2007.PubMed/NCBI
|
|
13
|
Shi J, Yu JB, Liu W, Wang D, Zhang Y, Gong
LR, Dong SA and Liu DQ: Carbon monoxide alleviates
lipopolysaccharide-induced oxidative stress injury through
suppressing the expression of Fis1 in NR8383 cells. Exp Cell Res.
349:162–167. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cantley LC: The phosphoinositide 3-kinase
pathway. Science. 296:1655–1657. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lin Y, Kuang W, Wu B, Xie C, Liu C and Tu
Z: IL-12 induces autophagy in human breast cancer cells through
AMPK and the PI3K/Akt pathway. Mol Med Rep. 16:4113–4118. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jiang YQ, Chang GL, Wang Y, Zhang DY, Cao
L and Liu J: Geniposide prevents hypoxia/reoxygenation-induced
apoptosis in H9c2 Cells: Improvement of mitochondrial dysfunction
and activation of GLP-1R and the PI3K/Akt signaling pathway. Cell
Physiol Biochem. 39:407–421. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zheng T, Yang X, Wu D, Xing S, Bian F, Li
W, Chi J, Bai X, Wu G, Chen X, et al: Salidroside ameliorates
insulin resistance through activation of a mitochondria-associated
AMPK/PI3K/Akt/GSK3β pathway. Br J Pharmacol. 172:3284–3301. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang J, Chen H, Liu Y, Zhou W, Sun R and
Xia M: Retinol binding protein 4 induces mitochondrial dysfunction
and vascular oxidative damage. Atherosclerosis. 240:335–344. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wu D, Pan P, Su X, Zhang L, Qin Q, Tan H,
Huang L and Li Y: Interferon regulatory factor-1 mediates alveolar
macrophages pyroptosis during LPS-induced acute lung injury in
mice. Shock. 46:329–338. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Williams JG, Bernstein S and Prager M:
Effect of melatonin on activated macrophage TNF, IL-6, and reactive
oxygen intermediates. Shock. 9:406–411. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ying Z, Xie X, Chen M, Yi K and
Rajagopalan S: Alpha-lipoic acid activates eNOS through activation
of PI3-kinase/Akt signaling pathway. Vascul Pharmacol. 64:28–35.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sawle P, Foresti R, Mann BE, Johnson TR,
Green CJ and Motterlini R: Carbon monoxide-releasing molecules
(CO-RMs) attenuate the inflammatory response elicitedby
lipopolysaccharide in RAW264.7 murine macrophages. Br J Pharmacol.
145:800–810. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ballweg K, Mutze K, Königshoff M,
Eickelberg O and Meiners S: Cigarette smoke extract affects
mitochondrial function in alveolar epithelial cells. Am J Physiol
Lung Cell Mol Physiol. 307:L895–L907. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C (T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Guo C, Yang L, Luo J, Zhang C, Xia Y, Ma T
and Kong L: Sophoraflavanone G from Sophora alopecuroides inhibits
lipopolysaccharide-inducedinflammation in RAW264.7 cells by
targeting PI3K/Akt, JAK/STAT and Nrf2/HO-1 pathways. Int
Immunopharmacol. 38:349–356. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Reidy MF and Wright JR: Surfactant protein
A enhances apoptotic cell uptake and TGF-beta1 release by
inflammatory alveolar macrophages. Am J Physiol Lung Cell Mol
Physiol. 285:L854–L861. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wu Y, Wang H, Zhang J, Ma X, Meng J, Li Y,
Hou Z and Luo X: Adenosine deaminase that acts on RNA 1p150 in
alveolar macrophage is involved in LPS-induced lung injury. Shock.
31:410–415. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Gonzalez AS, Elguero ME, Finocchietto P,
Holod S, Romorini L, Miriuka SG, Peralta JG, Poderoso JJ and
Carreras MC: Abnormal mitochondrial fusion-fission balance
contributes to the progression of experimental sepsis. Free Radic
Res. 48:769–783. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Preau S, Delguste F, Yu Y, Remy-Jouet I,
Richard V, Saulnier F, Boulanger E and Neviere R: Endotoxemia
engages the RhoA kinase pathway to impair cardiac function by
altering cytoskeleton, mitochondrial fission, and autophagy.
Antioxid Redox Signal. 24:529–542. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gomes LC and Scorrano L: High levels of
Fis1, a pro-fission mitochondrial protein, trigger autophagy.
Biochim Biophys Acta. 1777:860–866. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ikeda Y, Sciarretta S, Nagarajan N,
Rubattu S, Volpe M, Frati G and Sadoshima J: New insights into the
role of mitochondrial dynamics and autophagy during oxidative
stress and aging in the heart. Oxid Med Cell Longev.
2014:2109342014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lee YJ, Jeong SY, Karbowski M, Smith CL
and Youle RJ: Roles of the mammalian mitochon-drial fission and
fusion mediators Fis1, Drp1, and Opa1 in apoptosis. Mol Biol Cell.
15:5001–5011. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yu JB, Wang Y, Li Z, Dong SA, Wang D, Gong
LR, Shi J, Zhang Y, Liu DQ and Mu R: Effect of heme oxygenase-1 on
mitofusion-1 protein in LPS-induced ALI/ARDS in rats. Sci Rep.
6:365302016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lu CY, Yang YC, Li CC, Liu KL, Lii CK and
Chen HW: Andrographolide inhibits TNFα-induced ICAM-1 expression
via suppression of NADPH oxidase activation and induction of HO-1
and GCLM expression through the PI3K/Akt/Nrf2 and PI3K/Akt/AP-1
pathways in human endothelial cells. Biochem Pharmacol. 91:40–50.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kim KC, Kang KA, Zhang R, Piao MJ, Kim GY,
Kang MY, Lee SJ, Lee NH, Surh YJ and Hyun JW: Up-regulation of
Nrf2-mediated heme oxygenase-1 expression by eckol, a phlorotannin
compound, through activation of Erk and PI3K/Akt. Int J Biochem
Cell Biol. 42:297–305. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Monick MM, Mallampalli RK, Carter AB,
Flaherty DM, McCoy D, Robeff PK, Peterson MW and Hunninghake GW:
Ceramide regulates lipopolysaccharide-induced phosphatidylinositol
3-kinase and Akt activity in human alveolar macrophages. J Immunol.
167:5977–5985. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Chen H, Xie K, Han H, Li Y, Liu L, Yang T
and Yu Y: Molecular hydrogen protects mice against polymicrobial
sepsis by ameliorating endothelial dysfunction via an Nrf2/HO-1
signaling pathway. Int Immunopharmacol. 28:643–654. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yu JB, Shi J, Gong LR, Dong SA, Xu Y,
Zhang Y, Cao XS and Wu LL: Role of Nrf2/ARE pathway in protective
effect of electroacupuncture against endotoxic shock-induced acute
lung injury in rabbits. PLoS One. 9:e1049242014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Chen HH, Chen YT, Huang YW, Tsai HJ and
Kuo CC: 4-Ketopinoresinol, a novel naturally occurring ARE
activator, induces the Nrf2/HO-1 axis and protects against
oxidative stress-induced cell injury via activation of PI3K/AKT
signaling. Free Radic Biol Med. 52:1054–1066. 2012. View Article : Google Scholar : PubMed/NCBI
|