Introduction
Radiation therapy is an important treatment modality
for thoracic cancers. However, radiation-induced pneumonia and
subsequent pulmonary fibrosis can be serious and fatal
complications, and they are major dose-limiting factors of
radiotherapy. Radiation-induced pulmonary fibrosis, characterized
by inflammatory cell infiltration, fibroblast proliferation, and
excessive deposition of extracellular matrix (ECM) proteins such as
collagen in the lung parenchyma (1–3),
presents 6 to 24 months after irradiation and continues to progress
over a period of years (4). The
pathological mechanisms of radiation-induced pulmonary fibrosis are
complex and involve numerous cell types. A large number of studies
have presented evidence showing the involvement of several
mediators in the pathogenesis of this disease (1,5).
Nonetheless, there is not an established treatment protocol for
radiation-induced pulmonary fibrosis. Therefore, the development of
therapeutic strategies for this disease is urgently needed.
Plasminogen activator inhibitor-1 (PAI-1) is the
main inhibitor of the plasminogen activator system, which blocks
fibrinolysis and promotes ECM accumulation in tissues (6–8). There
is a growing body of evidence demonstrating that PAI-1 plays a
crucial role in pulmonary fibrosis. Indeed, it has been shown that
overexpression of PAI-1 enhances bleomycin (BLM)-induced
pulmonary fibrosis (9). Moreover,
the inhibition of PAI-1 expression via gene deletion
(9,10), intrapulmonary administration of small
interfering RNAs (11), or a
specific PAI-1 inhibitor (12)
attenuates the development of BLM-induced pulmonary fibrosis in
mouse models. In vitro, PAI-1 is reported to contribute to
the development of pulmonary fibrosis by promoting ECM accumulation
and is associated with the epithelial-mesenchymal transition (EMT)
of lung epithelial cells and differentiation of fibroblasts to
myofibroblasts (7,12).
Several studies have demonstrated the involvement of
PAI-1 in radiation-induced tissue injury and fibrosis (13,14). The
overexpression of PAI-1 has been reported to lead to the
development of radiation-induced nephrosclerosis (15,16) and
enteritis (17). These observations
suggest that PAI-1 plays an important role in radiation-induced
tissue injury. However, the association between PAI-1 and
radiation-induced pulmonary fibrosis has not been fully elucidated.
In this study, we investigated the effect of PAI-1 gene
deficiency on radiation-induced lung fibrosis by analyzing the
degree of lung fibrosis induced by irradiation in PAI-1
knockout and wild-type (WT) mice.
Materials and methods
Animal model of radiation-induced lung
fibrosis
PAI-1-deficient (PAI-1−/−) C57BL/6
mice were purchased from Jackson Laboratory, (Bar Harbor, ME, USA)
and bred according to methods approved by the Institutional Animal
Care and Use Committee of Hiroshima University (Hiroshima, Japan).
All animal experiments were approved by the animal ethics committee
of Hiroshima University (permit no: A13-98). Age-, sex-, and body
weight-matched WT C57BL/6 mice were purchased from Charles River
Laboratories (Kanagawa, Japan). PAI-1−/− mice and WT
mice were anesthetized with intraperitoneal injection of
pentobarbital sodium (30 mg/kg). For euthanasia of mice, the dose
of 100–120 mg/kg pentobarbital sodium was used. A single dose of 15
Gy irradiation was delivered to the thorax of each mouse with an
X-ray irradiator (MBR-1520R-3; Hitachi, Ltd., Tokyo, Japan).
Irradiation characteristics were as follows: Beam energy, 150 kV;
X-ray dose rate, 1.3 Gy/min; source-surface distance, 50 cm;
diameter of the radiation field, 25 cm; filter, 0.5 mm Cu and 0.1
mm Al.
Analysis of bronchoalveolar lavage
(BAL) fluid
BAL fluids were collected 0, 4, 12, 18, and 24 weeks
after irradiation as previously described (11). Briefly, mice were sacrificed with a
lethal dose of pentobarbital, the tracheas were cannulated with an
18-gauge needle, and the lungs were lavaged three times with 0.5 ml
of phosphate-buffered saline (PBS). Lavage fluids were pooled and
were cleared of cells via centrifugation. Cells in BAL fluids were
counted with a standard hemocytometer. Differential cell counts
were obtained by Diff-Quik (Kokusai Shiyaku, Kobe, Japan) using
cytospin preparations (Shandon, Pittsburgh, PA, USA).
Measurements of concentrations of
PAI-1, TGF-β, and albumin in BAL fluid
PAI-1 and TGF-β levels in BAL fluids were measured
at 0, 4, 12, 18, and 24 weeks after irradiation using ELISA kits
for PAI-1 (Innovative Research, Novi, MI, USA) and TGF-β (R&D
Systems, Minneapolis, MN, USA), according to the manufacturers'
instructions. Mouse albumin concentrations in BAL fluids were
measured with an ELISA kit according to the manufacturers'
instructions (Shibayagi, Gunma, Japan).
Hydroxyproline assay
To quantify lung collagen contents, the
hydroxyproline contents of the left lungs were measured in each
group at 24 weeks after irradiation as described previously
(10).
Histology
After BAL and lung perfusion, mouse lungs were fixed
by inflation with a buffered 4% formalin solution. Lung tissue
specimens were embedded in paraffin and cut into 5-µm sections,
which were stained with hematoxylin and eosin or Masson's
trichrome.
Statistical analysis
Results are expressed as means ± SEM. The Wilcoxon
rank sum test was used to evaluate differences between two groups.
Comparisons of the multiple groups were evaluated by using
Kruskal-Wallis test followed by Steel-Dwass test or Steel test.
Correlations were analyzed with Pearson's correlation coefficient
test. The Kaplan-Meier method was used to analyze mouse survival
curves. Differences in survival between two groups were analyzed
using the log-rank test. All statistical analyses were performed
using JMP Genomics v.7.0 software (SAS Institute Inc., Cary, NC,
USA). P<0.05 was considered to indicate a statistically
significant difference.
Results
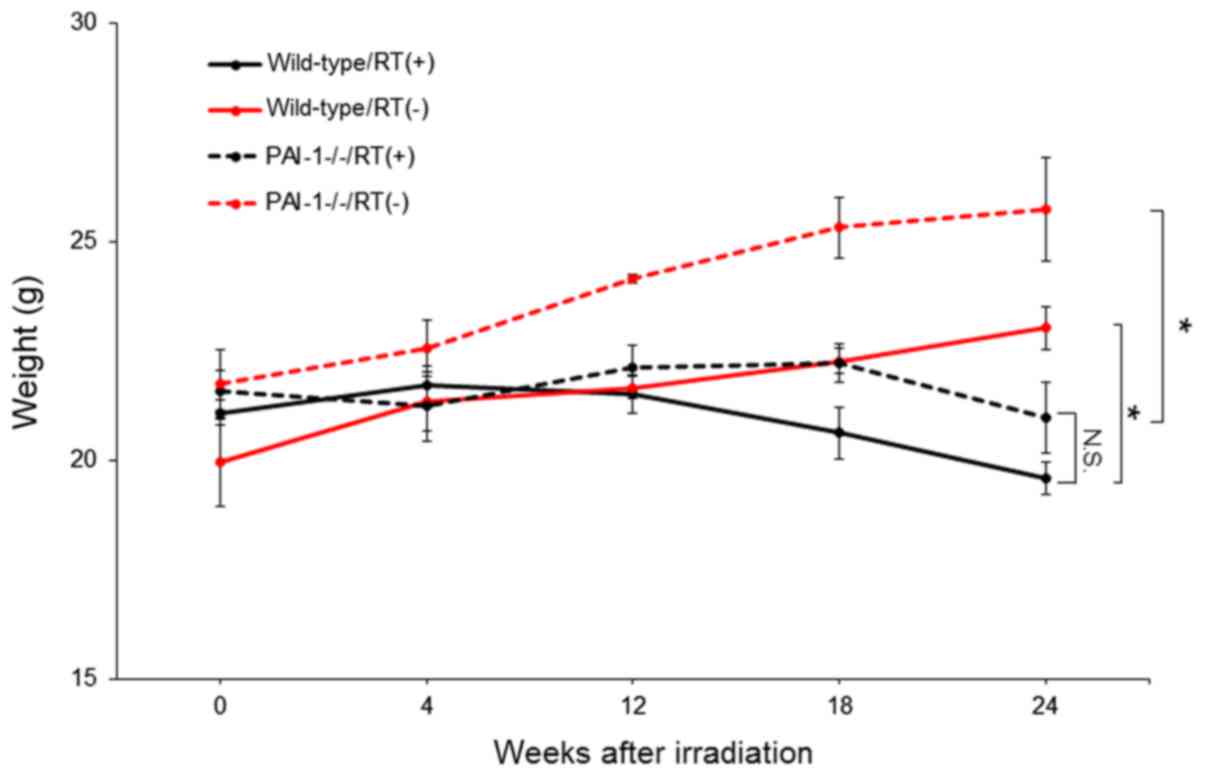
Effect of thoracic irradiation on body
weights of mice
The body weights of mice were measured over time to
assess the effect of thoracic irradiation on body weight changes.
Thoracic radiation reduced mouse body weights significantly in both
WT and PAI-1−/− mice, with no difference observed
between the two groups at 24 weeks after irradiation (Fig. 1).
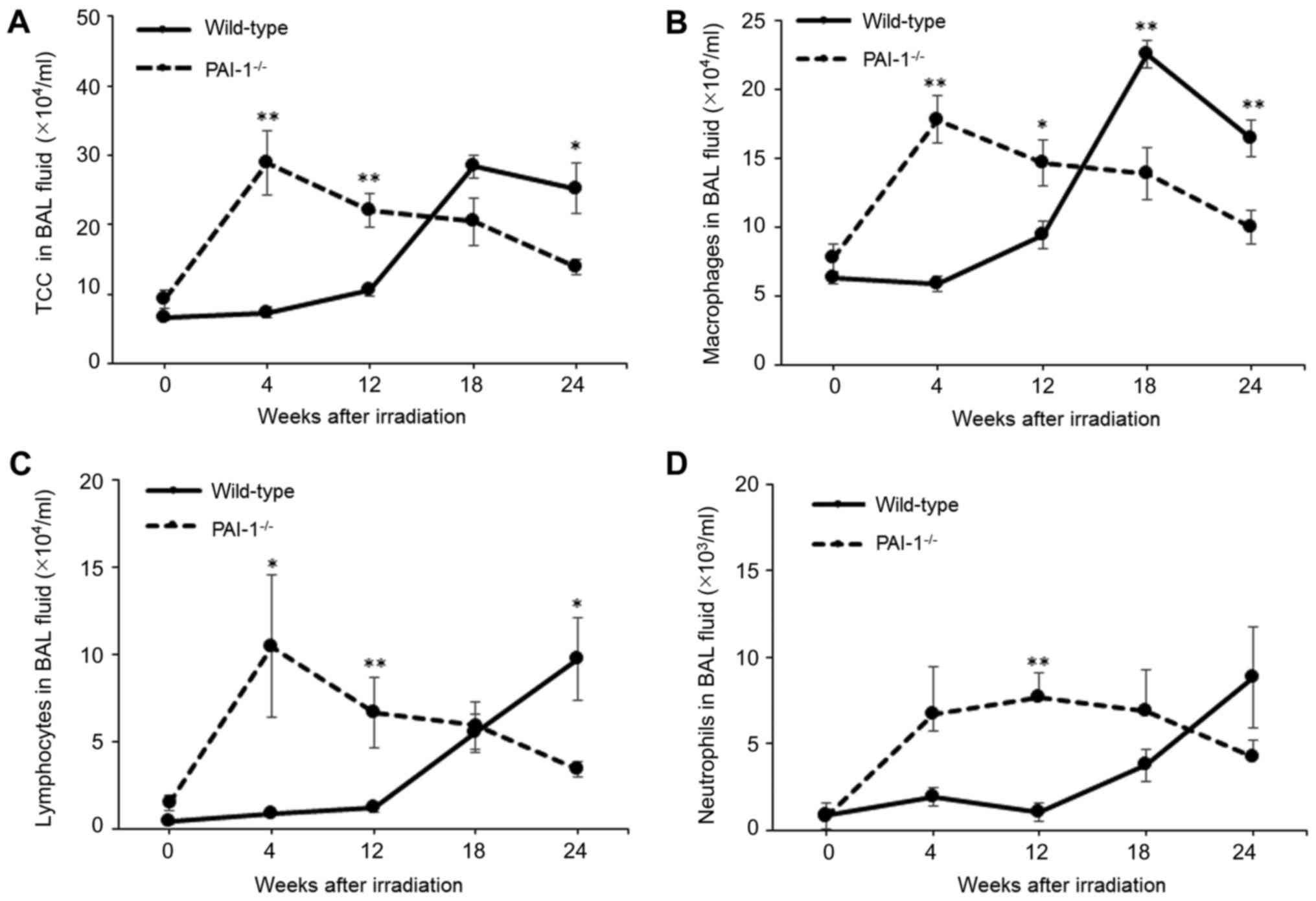
Assessment of inflammatory cells in
BAL fluid after thoracic irradiation
To assess the inflammatory response to thoracic
irradiation, inflammatory cells in BAL fluids were analyzed at
baseline and 4, 12, 18, and 24 weeks after irradiation. As shown in
Fig. 2A, total cell counts in BAL
fluids from PAI-1−/− mice peaked at 4 weeks after
irradiation and subsequently declined. In contrast, total cell
counts in BAL fluids from WT mice gradually increased to a peak at
18 weeks after irradiation. Total cell counts in BAL fluids were
significantly higher at 4 and 12 weeks and significantly lower at
24 weeks after irradiation in PAI-1−/− mice than in WT
mice. Based on differential cell counts in BAL fluids, numbers of
macrophages were significantly higher at 4 and 12 weeks and
significantly lower at 18 and 24 weeks after irradiation in
PAI-1−/− mice than in WT mice, and numbers of
lymphocytes were significantly higher at 4 and 12 weeks and
significantly lower at 24 weeks after irradiation in
PAI-1−/− mice than in WT mice.
PAI-1 and TGF-β levels in BAL
fluid
To determine the effect of thoracic irradiation on
the expression of PAI-1 and TGF-β in the lung, PAI-1 and TGF-β
levels in BAL fluid were measured using ELISA kits. In WT mice,
PAI-1 levels in BAL fluids were unchanged from baseline until 12
weeks after irradiation but started increasing after 18 weeks and
reached their highest levels at 24 weeks after irradiation. PAI-1
levels at 24 weeks were significantly higher compared with those
before irradiation (Fig. 3A). TGF-β
levels in BAL fluids from WT mice showed the same trend (Fig. 3B). In contrast, TGF-β levels in BAL
fluids from PAI-1−/− mice were almost unchanged until 24
weeks after irradiation, at which point they were significantly
lower than those in WT mice (31.51±7.18 vs. 152.95±70.05 pg/ml;
P=0.002; Fig. 3C).
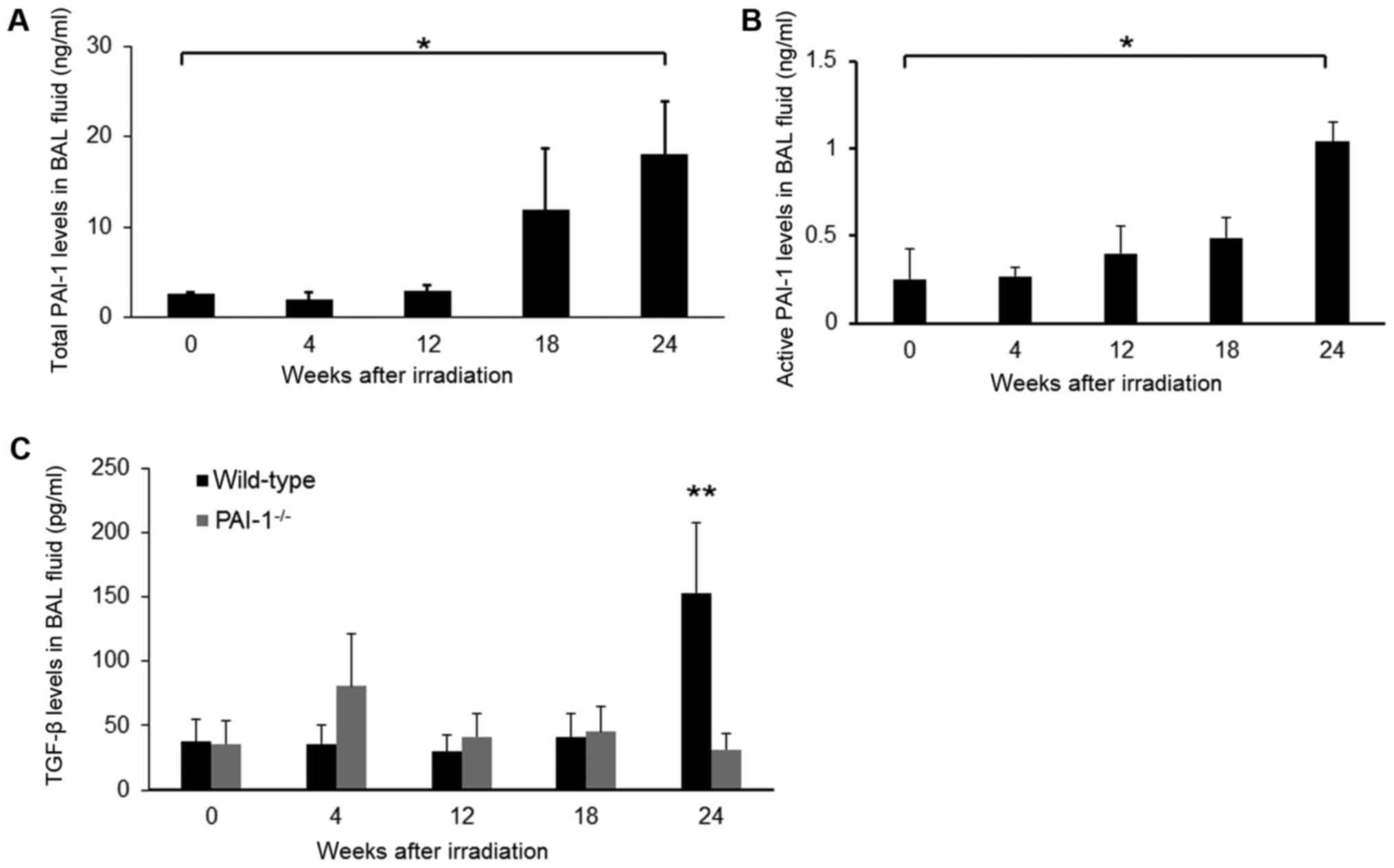 | Figure 3.(A) Total PAI-1 and (B) active PAI-1
levels in BAL fluids of irradiated WT mice. Data are shown as mean
± SEM for 5–8 mice at 0, 4, 12, 18, and 24 weeks after irradiation.
Comparisons of the various time points were analyzed by
Kruskal-Wallis test followed by Steel test. *P<0.05 vs. the
control (time 0). (C) TGF-β levels in BAL fluids of irradiated WT
and PAI-1−/− mice. Data are shown as mean ± SEM for 5–8
mice per group at 0, 4, 12, 18, and 24 weeks after irradiation.
**P<0.01 vs. PAI-1-/- at the same time point. WT, wild-type;
PAI-1, plasminogen activator inhibitor-1; TGF-β, transforming
growth factor-β; BAL, bronchoalveolar lavage. |
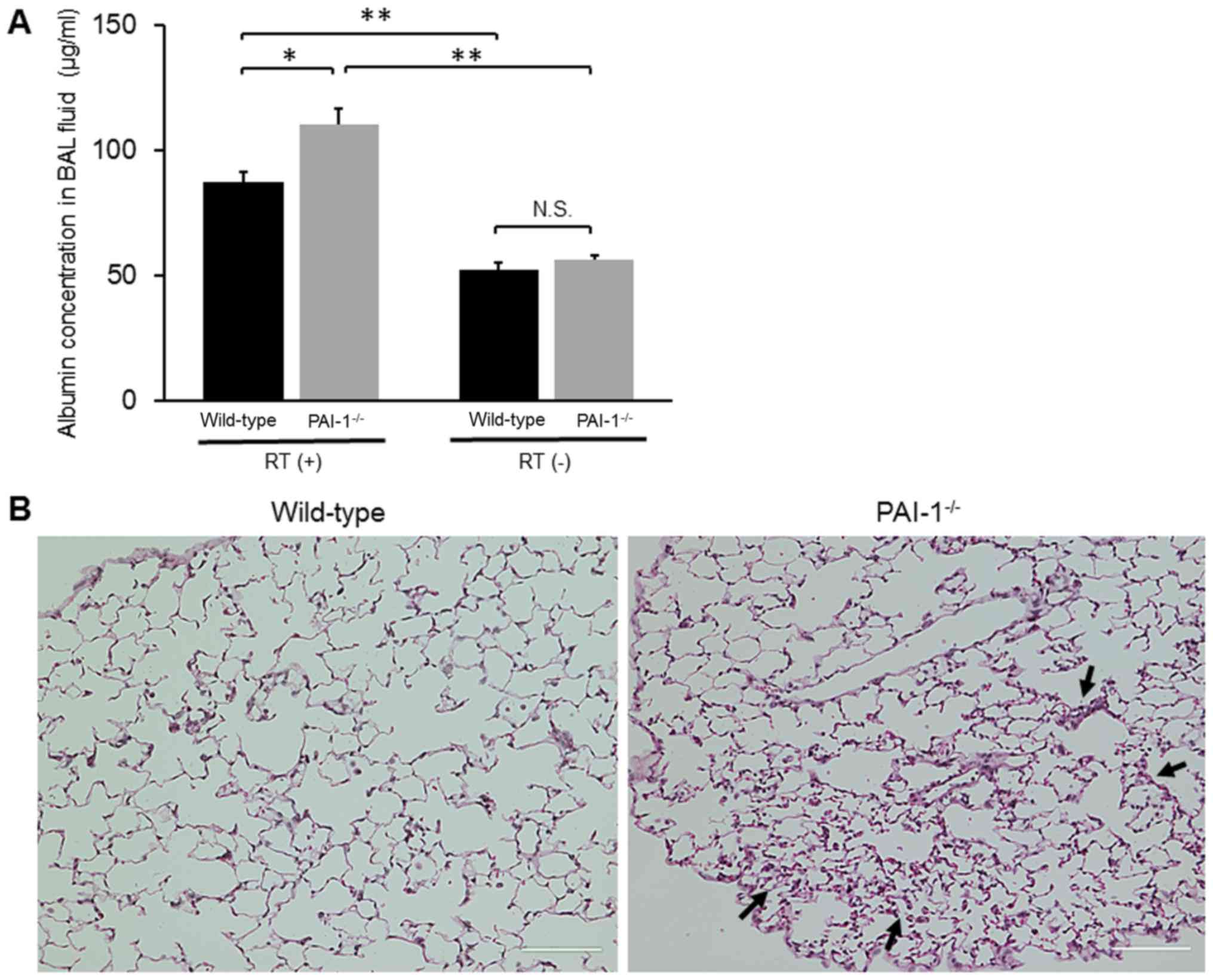
Effects of PAI-1 deficiency on
early-phase lung injury after irradiation
As mentioned above, we found that the number of
inflammatory cells increased 4 weeks after irradiation. Therefore,
to investigate the degree of inflammation in the lungs, we analyzed
the concentration of albumin in BAL fluids and assessed
histological changes in the lungs 4 weeks after irradiation.
Albumin concentrations in BAL fluids were significantly higher in
PAI-1−/− mice than in WT mice (110.62±5.98 vs.
87.34±4.31 µg/ml; P=0.01; Fig. 4A).
In addition, histological examination of lungs showed that the
degree of inflammatory cell infiltration into the alveoli was
higher in PAI-1−/− mice than in WT mice (Fig. 4B).
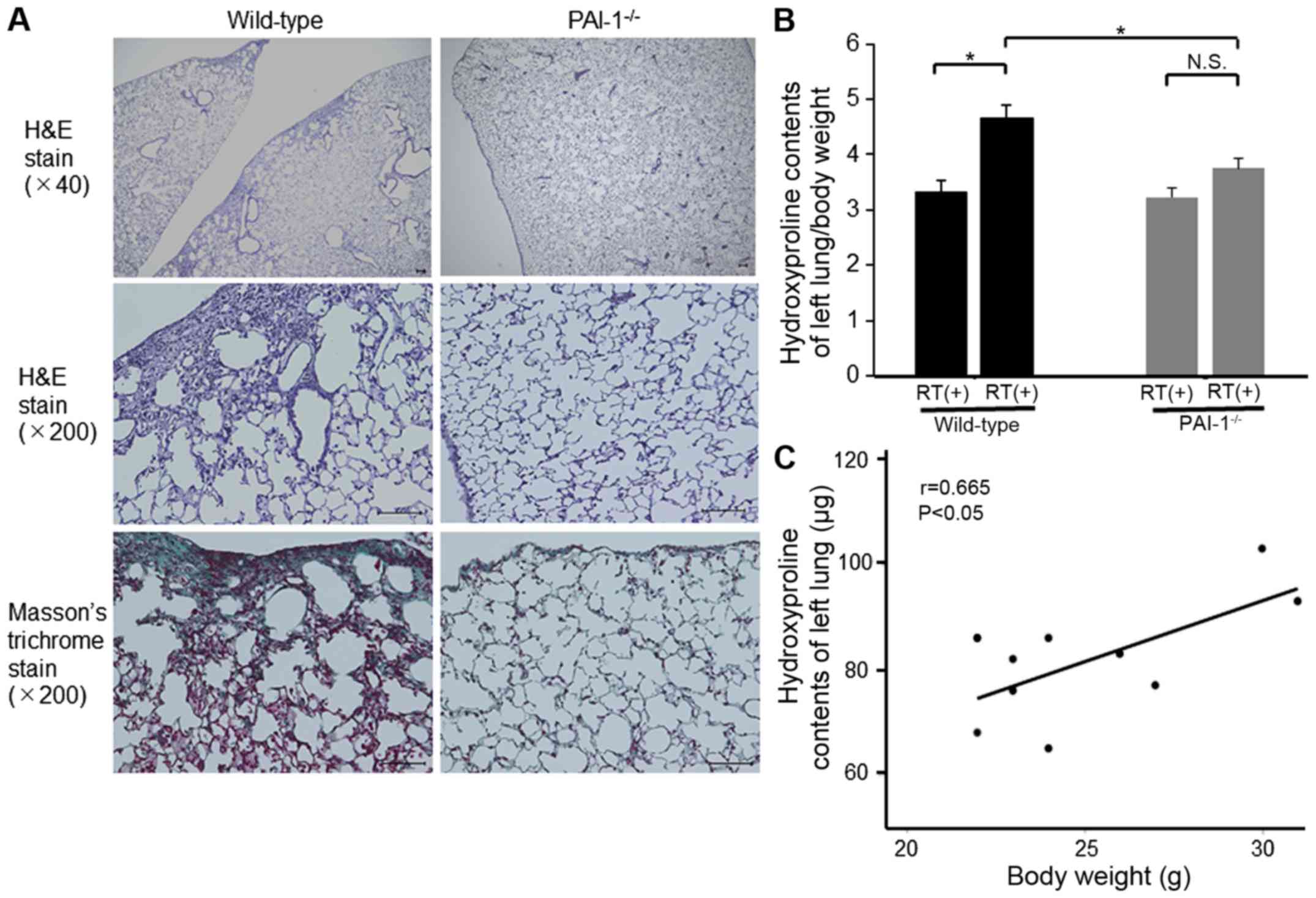
Effect of PAI-1 deficiency on
radiation-induced pulmonary fibrosis
To assess the degree of radiation-induced pulmonary
fibrosis, right lungs were histologically examined, and the
hydroxyproline contents of left lungs were measured at 24 weeks
after irradiation. Representative histological images of lungs
revealed that the extent of pulmonary fibrosis was apparently
attenuated in PAI-1−/− mice compared with that in WT
mice (Fig. 5A). In addition, the
hydroxyproline contents of the left lung per mg body weight were
found to be significantly lower in PAI-1−/− mice than in
WT mice at 24 weeks after irradiation (3.75±0.45 vs. 4.66±0.57 µg/g
body weight; P=0.031; Fig. 5B). The
hydroxyproline content was adjusted for mouse body weight as
previously reported (18). Actually,
we confirmed that there was a correlation between hydroxyproline
content and body weight (r=0.665, P=0.036; Fig. 5C).
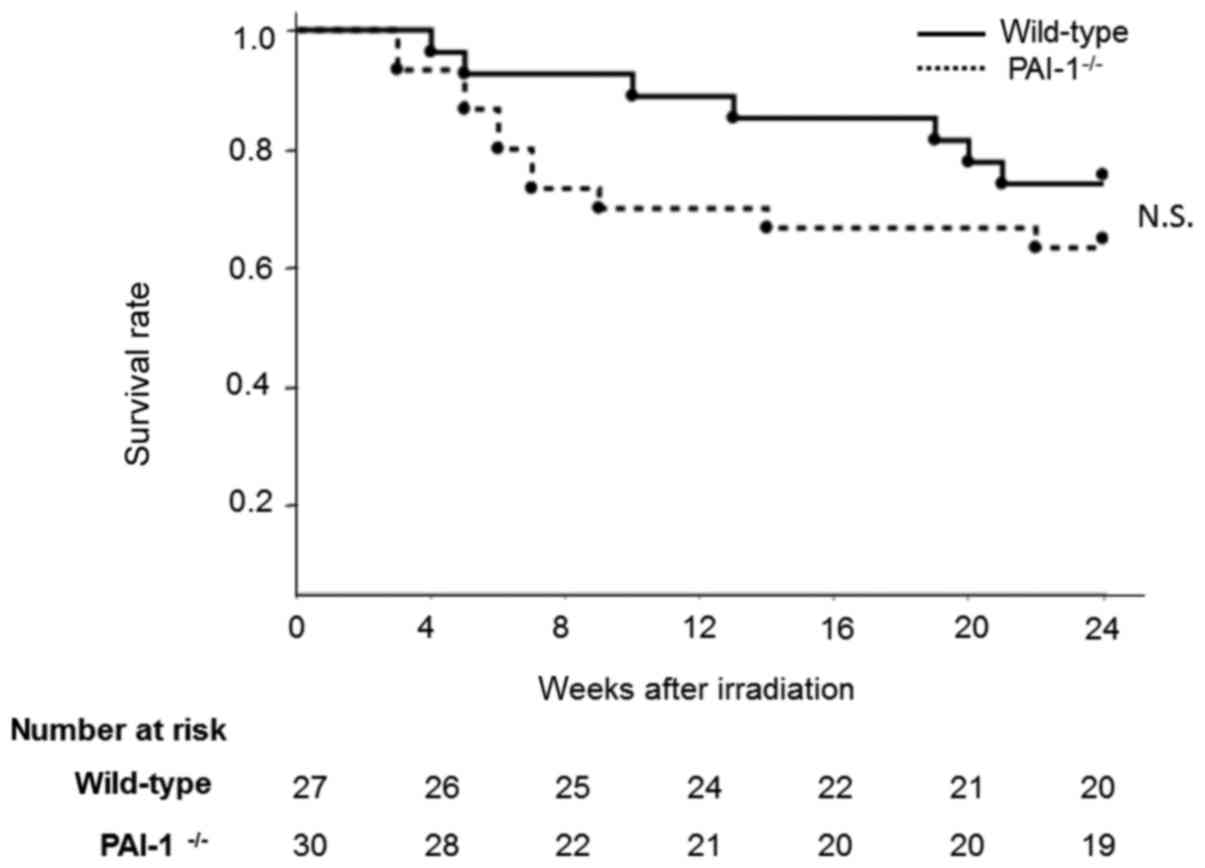
Effect of thoracic irradiation on
mouse mortality
In PAI-1−/− mice, a high mortality rate
was observed from 4 to 12 months after irradiation due to
inflammation in the lungs. In contrast, in WT mice, increased
mortality was observed 18 weeks after irradiation due to lung
fibrosis. However, there was no significant difference in the
survival rates of the two groups at 24 weeks after irradiation
(P=0.322; Fig. 6).
Discussion
In the present study, we showed that pulmonary
fibrosis developed at 24 weeks after thoracic irradiation in WT
mice with accompanying elevations in both PAI-1 and TGF-β in BAL
fluid; however, in PAI-1−/− mice, the degree of
pulmonary fibrosis was limited, and there was no elevation in TGF-β
in BAL fluid. These data indicate the direct involvement of PAI-1
in the development of radiation-induced pulmonary fibrosis.
We demonstrated that PAI-1 gene deficiency reduced
the progression of radiation-induced fibrosis in this study.
Previous studies showed that PAI-1 deletion or suppression using
siRNA or a specific PAI-1 inhibitor limited the development of
BLM-induced pulmonary fibrosis in murine models (9–11). In
the field of radiation-induced pulmonary fibrosis, there are
several studies that have investigated whether PAI-1 is associated
with radiation-induced pulmonary fibrosis. In animal models, the
intra-abdominal administration of recombinant truncated PAI-1
protein or pentoxifylline, an antifibrotic agent, attenuates
radiation-induced fibrosis in the lungs by reducing PAI-1 activity
and promoting fibrin proteolysis (19,20). In
contrast, genetic deficiency of nuclear factor erythroid 2-related
factor 2 (NRF2) increases PAI-1 expression in irradiated murine
lungs, resulting in the development of late tissue injury in mice
(21). These results suggest that
PAI-1 may be promising as a therapeutic target for
radiation-induced pulmonary fibrosis. This is the first study to
investigate the effect of PAI-1 deficiency on the progression of
radiation-induced pulmonary fibrosis.
Several studies have shown that transforming growth
factor beta (TGF-β) is the major cytokine responsible for pulmonary
fibrosis after irradiation (1–3,22,23).
Furthermore, TGF-β is known to be a potent inducer of PAI-1
expression in various diseases associated with fibrosis (7,24). A
previous study showed that both TGF-β and PAI-1 expression levels
were increased in the skin of irradiated rats (25). In addition, the administration of
anti-TGF-β antibody limited radiation-induced PAI-1 expression and
the degree of skin fibrosis in this study. These results indicate
that PAI-1 is associated with the process of radiation-induced
tissue fibrosis as a downstream effector of TGF-β.
In the present study, we demonstrated that the
degree of pulmonary fibrosis and the levels of TGF-β in BAL fluid
significantly increased in WT mice after irradiation. In contrast,
these results were not observed in PAI-1−/− mice. In
addition, a previous report also showed that the inhibition of
PAI-1 activity via systemic delivery of recombinant truncated PAI-1
protein attenuated pulmonary fibrosis and the expression of TGF-β
in BAL fluids of irradiated mice (19). These results suggest that PAI-1 is
not only a downstream effector of TGF-β but is also involved in
radiation-induced pulmonary fibrosis through the regulation of
TGF-β expression. In fact, in mesangial cells, PAI-1 is reported to
activate the TGF-β1 gene promoter (26) and enhance the expression of TGF-β
(27). Furthermore, we previously
showed that exogenous TGF-β induced EMT and stimulated the
production of endogenous TGF-β in A549 cells. In contrast, a
PAI-1-specific inhibitor limited TGF-β-induced EMT and the
production of TGF-β in A549 cells (12). These observations imply that PAI-1
would be an effective treatment target for pulmonary fibrosis as a
regulator of TGF-β.
In this study, the numbers of total cells,
lymphocytes and macrophages, as well as the albumin levels in BAL
fluids were significantly higher and the degree of infiltration of
inflammatory cells was histologically more prominent at 4 weeks
after irradiation in PAI-1−/− mice than in WT mice.
Furthermore, several PAI-1−/− mice died between 4 and 12
months after irradiation due to inflammation in the lungs. These
findings are compatible with those of a previous study that showed
that the numbers of total cells, neutrophils, and macrophages in
BAL fluids were significantly higher in PAI-1−/− mice
than in WT mice with acid-induced acute lung injury (28). Although in these studies the extent
of pulmonary fibrosis was lower in PAI-1−/− mice than in
WT mice, these findings suggest that PAI-1 inhibition should be
avoided in the early phase after irradiation. To elucidate the
mechanism by which an acute inflammatory response is induced in
lung injuries by PAI-1 deficiency, further examination is
needed.
Radiation-induced pulmonary fibrosis is a chronic
progressive condition resulting from high levels of reactive oxygen
species (ROS) induced by irradiation and the subsequent injury of
alveolar epithelial cells and vascular endothelial cells (29). The underlying mechanism of
radiation-induced pulmonary fibrosis is considered to be similar to
that of idiopathic pulmonary fibrosis (IPF). In the present study,
we examined the involvement of PAI-1 and TGF-β, which activate
fibroblasts, induce EMT in epithelial cells, and accelerate the
coagulation cascade, in the process of pulmonary fibrosis. However,
a large number of other factors have been reported to be associated
with pulmonary fibrosis. Previous studies have shown that several
mediators, such as platelet-derived growth factor, vascular
endothelial growth factor, and fibroblast growth factor, are also
involved in fibroblast activation in IPF progression (30,31).
Furthermore, it has been reported that endoplasmic reticulum stress
in alveolar type II cells (32) and
T lymphocytes, such as Th-2 and Th-17 T-cells, are also implicated
in pulmonary fibrosis (31). Further
studies are therefore required to elucidate the involvement of
these factors in radiation-induced pulmonary fibrosis.
In conclusion, our study shows that PAI-1
genetic deficiency attenuates the development of pulmonary fibrosis
after irradiation. These results suggest that PAI-1 may represent a
novel therapeutic target for radiation-induced pulmonary
fibrosis.
Acknowledgements
The authors would like to thank Mrs. Yukari Iyanaga
(Department of Molecular and Internal Medicine, Graduate School of
Biomedical & Health Sciences, Hiroshima University, Hiroshima,
Japan) for providing technical assistance. The abstract was
presented at the International Conference of the American Thoracic
Society May 13–18 2016 in San Francisco, and published as abstract
no. A2378 in American Journal of Respiratory and Critical Care
Medicine 2016, Volume 193.
Funding
The present study was supported by grants-in-aid for
Scientific Research from the Ministry of Education, Culture,
Sports, Science and Technology of Japan.
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
SS and NH designed the present study. SS performed
the experiments. SS, TM, TS, YH, SM, TN, HI, KF, HH and NH
contributed to analysis and interpretation of the data. SS, TM, TS,
YH, SM, TN, HI, KF, HH and NH drafted the manuscript. All authors
reviewed and approved the final manuscript.
Ethics approval and consent to
participate
All animal experiments were approved by the animal
ethics committee of Hiroshima University (permit no: A13-98) and
animals were maintained according to guidelines for the Ethical Use
of Animals in Research at Hiroshima University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ding NH, Li JJ and Sun LQ: Molecular
mechanisms and treatment of radiation-induced lung fibrosis. Curr
Drug Targets. 14:1347–1356. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Madani I, De Ruyck K, Goeminne H, De Neve
W, Thierens H and Van Meerbeeck J: Predicting risk of
radiation-induced lung injury. J Thorac Oncol. 2:864–874. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rübe CE, Uthe D, Schmid KW, Richter KD,
Wessel J, Schuck A, Willich N and Rube C: Dose-dependent induction
of transforming growth factor beta (TGF-beta) in the lung tissue of
fibrosis-prone mice after thoracic irradiation. Int J Radiat Oncol
Biol Phys. 47:1033–1042. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Carver JR, Shapiro CL, Ng A, Jacobs L,
Schwartz C, Virgo KS, Hagerty KL, Somerfield MR and Vaughn DJ: ASCO
Cancer Survivorship Expert Panel: American Society of Clinical
Oncology clinical evidence review on the ongoing care of adult
cancer survivors: Cardiac and pulmonary late effects. J Clin Oncol.
25:3991–4008. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rubin P, Johnston CJ, Williams JP,
McDonald S and Finkelstein JN: A perpetual cascade of cytokines
postirradiation leads to pulmonary fibrosis. Int J Radiat Oncol
Biol Phys. 33:99–109. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kohler HP and Grant PJ:
Plasminogen-activator inhibitor type 1 and coronary artery disease.
N Engl J Med. 342:1792–1801. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ghosh AK and Vaughan DE: PAI-1 in tissue
fibrosis. J Cell Physiol. 227:493–507. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Iwaki T, Urano T and Umemura K: PAI-1,
progress in understanding the clinical problem and its aetiology.
Br J Haematol. 157:291–298. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Eitzman DT, McCoy RD, Zheng X, Fay WP,
Shen T, Ginsburg D and Simon RH: Bleomycin-induced pulmonary
fibrosis in transgenic mice that either lack or overexpress the
murine plasminogen activator inhibitor-1 gene. J Clin Invest.
97:232–237. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hattori N, Degen JL, Sisson TH, Liu H,
Moore BB, Pandrangi RG, Simon RH and Drew AF: Bleomycin-induced
pulmonary fibrosis in fibrinogen-null mice. J Clin Invest.
106:1341–1350. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Senoo T, Hattori N, Tanimoto T, Furonaka
M, Ishikawa N, Fujitaka K, Haruta Y, Murai H, Yokoyama A and Kohno
N: Suppression of plasminogen activator inhibitor-1 by RNA
interference attenuates pulmonary fibrosis. Thorax. 65:334–340.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Omori K, Hattori N, Senoo T, Takayama Y,
Masuda T, Nakashima T, Iwamoto H, Fujitaka K, Hamada H and Kohno N:
Inhibition of plasminogen activator inhibitor-1 attenuates
transforming growth factor-β-dependent epithelial mesenchymal
transition and differentiation of fibroblasts to myofibroblasts.
PLoS One. 11:e01489692016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hageman J, Eggen BJ, Rozema T, Damman K,
Kampinga HH and Coppes RP: Radiation and transforming growth
factor-beta cooperate in transcriptional activation of the
profibrotic plasminogen activator inhibitor-1 gene. Clin Cancer
Res. 11:5956–5964. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhao W, Spitz DR, Oberley LW and Robbins
ME: Redox modulation of the pro-fibrogenic mediator plasminogen
activator inhibitor-1 following ionizing radiation. Cancer Res.
61:5537–5543. 2001.PubMed/NCBI
|
|
15
|
Brown NJ, Nakamura S, Ma L, Nakamura I,
Donnert E, Freeman M, Vaughan DE and Fogo AB: Aldosterone modulates
plasminogen activator inhibitor-1 and glomerulosclerosis in vivo.
Kidney Int. 58:1219–1227. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Oikawa T, Freeman M, Lo W, Vaughan DE and
Fogo A: Modulation of plasminogen activator inhibitor-1 in vivo: A
new mechanism for the anti-fibrotic effect of renin-angiotensin
inhibition. Kidney Int. 51:164–172. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Vozenin-Brotons MC, Milliat F, Linard C,
Strup C, François A, Sabourin JC, Lasser P, Lusinchi A, Deutsch E,
Girinsky T, et al: Gene expression profile in human late radiation
enteritis obtained by high-density cDNA array hybridization. Radiat
Res. 161:299–311. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Suzuki N, Ohta K, Horiuchi T, Takizawa H,
Ueda T, Kuwabara M, Shiga J and Ito K: T lymphocytes and
silica-induced pulmonary inflammation and fibrosis in mice. Thorax.
51:1036–1042. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chung EJ, McKay-Corkum G, Chung S, White
A, Scroggins BT, Mitchell JB, Mulligan-Kehoe MJ and Citrin D:
Truncated plasminogen activator inhibitor-1 protein protects from
pulmonary fibrosis mediated by irradiation in a murine model. Int J
Radiat Oncol Biol Phys. 94:1163–1172. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lee JG, Shim S, Kim MJ, Myung JK, Jang WS,
Bae CH, Lee SJ, Kim KM, Jin YW, Lee SS and Park S: Pentoxifylline
regulates plasminogen activator inhibitor-1 expression and protein
kinase A phosphorylation in radiation-induced lung fibrosis. Biomed
Res Int. 2017:12792802017.PubMed/NCBI
|
|
21
|
Travis EL, Rachakonda G, Zhou X, Korhonen
K, Sekhar KR, Biswas S and Freeman ML: NRF2 deficiency reduces life
span of mice administered thoracic irradiation. Free Radic Biol
Med. 51:1175–1183. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Anscher MS, Thrasher B, Zgonjanin L,
Rabbani ZN, Corbley MJ, Fu K, Sun L, Lee WC, Ling LE and Vujaskovic
Z: Small molecular inhibitor of transforming growth factor-beta
protects against development of radiation-induced lung injury. Int
J Radiat Oncol Biol Phys. 71:829–837. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Flechsig P, Dadrich M, Bickelhaupt S,
Jenne J, Hauser K, Timke C, Peschke P, Hahn EW, Gröne HJ, Yingling
J, et al: LY2109761 attenuates radiation-induced pulmonary murine
fibrosis via reversal of TGF-β and BMP-associated proinflammatory
and proangiogenic signals. Clin Cancer Res. 18:3616–3627. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liu RM: Oxidative stress, plasminogen
activator inhibitor 1, and lung fibrosis. Antioxid Redox Signal.
10:303–319. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Schultze-Mosgau S, Kopp J, Thorwarth M,
Rödel F, Melnychenko I, Grabenbauer GG, Amann K and Wehrhan F:
Plasminogen activator inhibitor-I-related regulation of procollagen
I (alpha1 and alpha2) by antitransforming growth factor-beta1
treatment during radiation-impaired wound healing. Int J Radiat
Oncol Biol Phys. 64:280–288. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Seo JY, Park J, Yu MR, Kim YS, Ha H and
Lee HB: Positive feedback loop between plasminogen activator
inhibitor-1 and transforming growth factor-beta1 during renal
fibrosis in diabetes. Am J Nephrol. 30:481–490. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Nicholas SB, Aguiniga E, Ren Y, Kim J,
Wong J, Govindarajan N, Noda M, Wang W, Kawano Y, Collins A and
Hsueh WA: Plasminogen activator inhibitor-1 deficiency retards
diabetic nephropathy. Kidney Int. 67:1297–1307. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Allen GB, Cloutier ME, Larrabee YC,
Tetenev K, Smiley ST and Bates JH: Neither fibrin nor plasminogen
activator inhibitor-1 deficiency protects lung function in a mouse
model of acute lung injury. Am J Physiol Lung Cell Mol Physiol.
296:L277–L285. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Huang Y, Zhang W, Yu F and Gao F: The
cellular and molecular mechanism of radiation-induced lung injury.
Med Sci Monit. 23:3446–3450. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sgalla G, Iovene B, Calvello M, Ori M,
Varone F and Richeldi L: Idiopathic pulmonary fibrosis:
Pathogenesis and management. Respir Res. 19:322018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Betensley A, Sharif R and Karamichos D: A
systematic review of the role of dysfunctional wound healing in the
pathogenesis and treatment of idiopathic pulmonary fibrosis. J Clin
Med. 6:pii: E2. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Burman A, Tanjore H and Blackwell TS:
Endoplasmic reticulum stress in pulmonary fibrosis. Matrix Biol.
68–69:355–365. 2018. View Article : Google Scholar
|