Introduction
Juvenile idiopathic arthritis (JIA) is one of the
most common immune-related diseases. We are still uncertain about
the etiology of this disease, which usually presents before the age
of 16, continues for at least 6 weeks, and has other potential
medical causes excluded (1). The
disease most commonly occurs in children between the ages of 7 and
12 years, but it may also present in adolescents up to 15 years of
age and in infants. It is a subset of childhood arthritis that may
be either transient and self-limiting, or chronic. The potential
causes of JIA are considered to involve genetics, environmental
factors, inflammatory cytokines and immune dysfunction. The
clinical features include fever, joint pain (often accompanied by a
skin rash), enlargement of the liver, spleen and lymph nodes, and
persistent inflammation that can cause joint deformity. The younger
the patient is, the more severe the symptoms are likely to be.
Older patients mainly experience joint-related symptoms only
(2–5).
The diagnosis of JIA is mainly based on the clinical
features and the results of laboratory tests for human leukocyte
antigen (HLA-B27), rheumatoid factor (RF) and matrix
metalloproteinase-3 (MMP-3) (6), but
hematological disorders have been seldom reported in JIA. During
examination of the bone marrow of patients with JIA, we noticed a
number of myelodysplastic changes. Additionally, we summarized the
features of the bone marrow cells from patients with JIA, providing
reference for the future clinical diagnosis and treatment of
JIA.
Materials and methods
Patients
The 107 patients included in this study were
initially diagnosed with JIA between May 2013 and October 2015. The
age range was 2–16 years old and median age was 11 years old,
including 67 boys and 40 girls. All 107 patients were diagnosed
with JIA from department of pediatrics. Our study was approved by
the Institutional Review Board at Ren Ji Hospital Affiliated to
Shanghai Jiao Tong University in Shanghai [IRB Approval no. RJKLS
(2016) 023].
Clinical features
Twenty-eight patients (26.17%) experienced a fever
of variable duration, from 3 days to 3 months. Forty-eight patients
(39.3%) exhibited joint swelling, which lasted between 1 month and
4 years, especially in the knee and hip joints.
Materials
Bone marrow specimens were collected from the 107
patients who were initially diagnosed and treated at the Department
of Pediatrics at Renji Hospital (School of Medicine, Shanghai Jiao
Tong University, Shanghai, China) from May 2013 to October
2015.
Methods
Bone marrow aspirates were obtained from the
posterior iliac crest or the anterior iliac crest (7). Bone marrow slides containing particles
were prepared according to ICSH guidelines, using a Wright-Giemsa
Stain kit (by Baso Diagnostic Inc., Zhuhai, China), and then
examined under a microscope (7). The
marrow cellularity was observed under low power magnification
(×100), and described as follows: Acellular, reduced, normal,
increased, or markedly increased. The same magnification was used
to determine the numbers of megakaryocytes and hemophagocytes. A
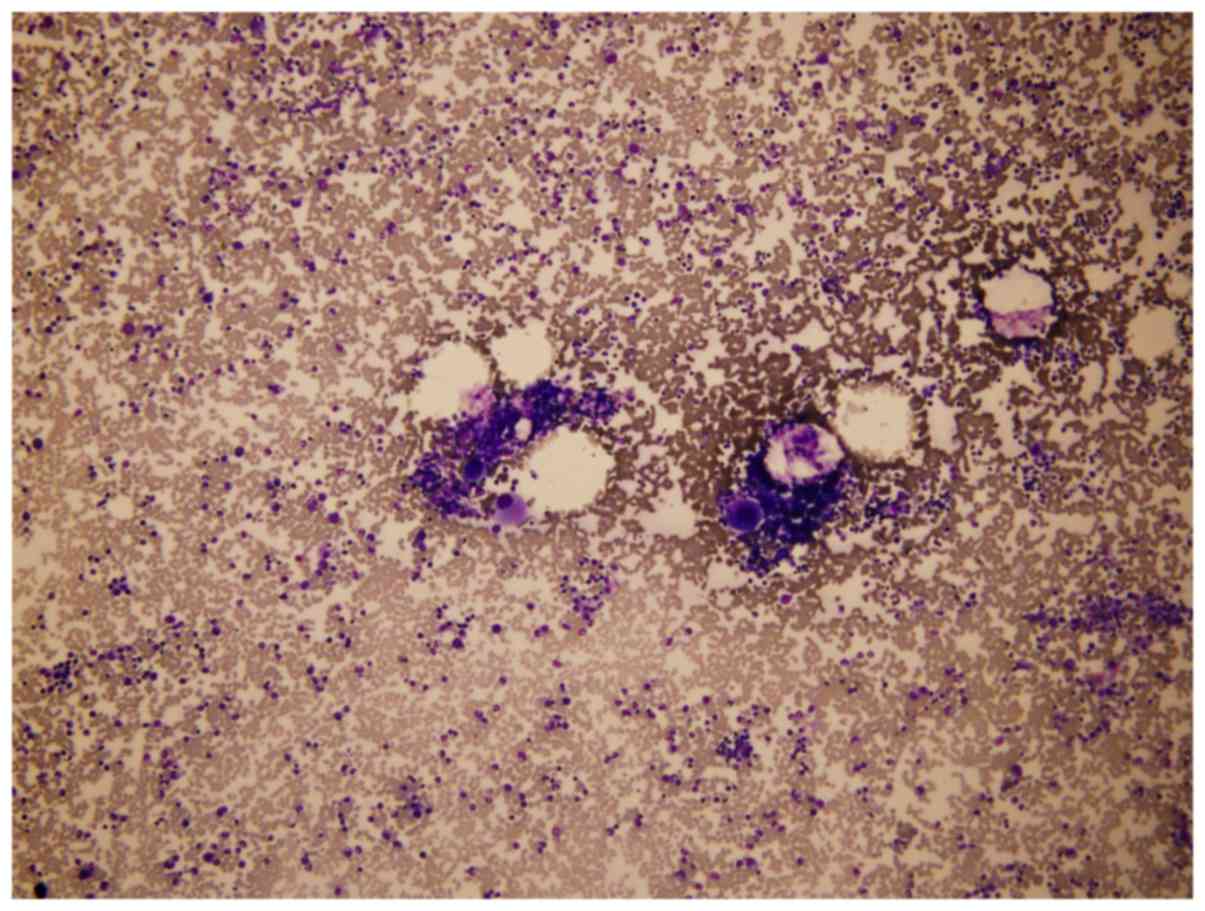
differential count of nucleated cells was then performed in
selected areas (Fig. 1), with ≥200
cells counted. Quantitative assessments of particular cell
lineages, and of particles within hemophagocytes, were performed
under ×1,000 magnification using oil lens. All microscopic
examinations were performed by the same researcher, in order to
avoid any bias in the assessments, in a blinded manner.
Statistical analysis
The Kruskal-Wallis test was used to test whether
numbers of hemophagocytes has the same distribution among the
subtypes. Software SAS version 9.13 (SAS institute Inc) was used to
perform the statistical analysis. And P<0.05 was considered to
indicate a statistically significant difference.
Results
Bone marrow examination
In 107 patients with JIA, twenty-seven (25.23%) had
normal cellularity of the bone marrow, fifty-four (50.47%) had
increased cellularity, and twenty-six (24.30%) had markedly
increased cellularity (Table I).
With respect to the megakaryocyte count, we noted (0–5) per slide
in 1 case (0.93%), (5–25) in 25 cases (23.36%), and (>25) in 81
cases (75.70%), which is a mild-moderate increase. Regarding
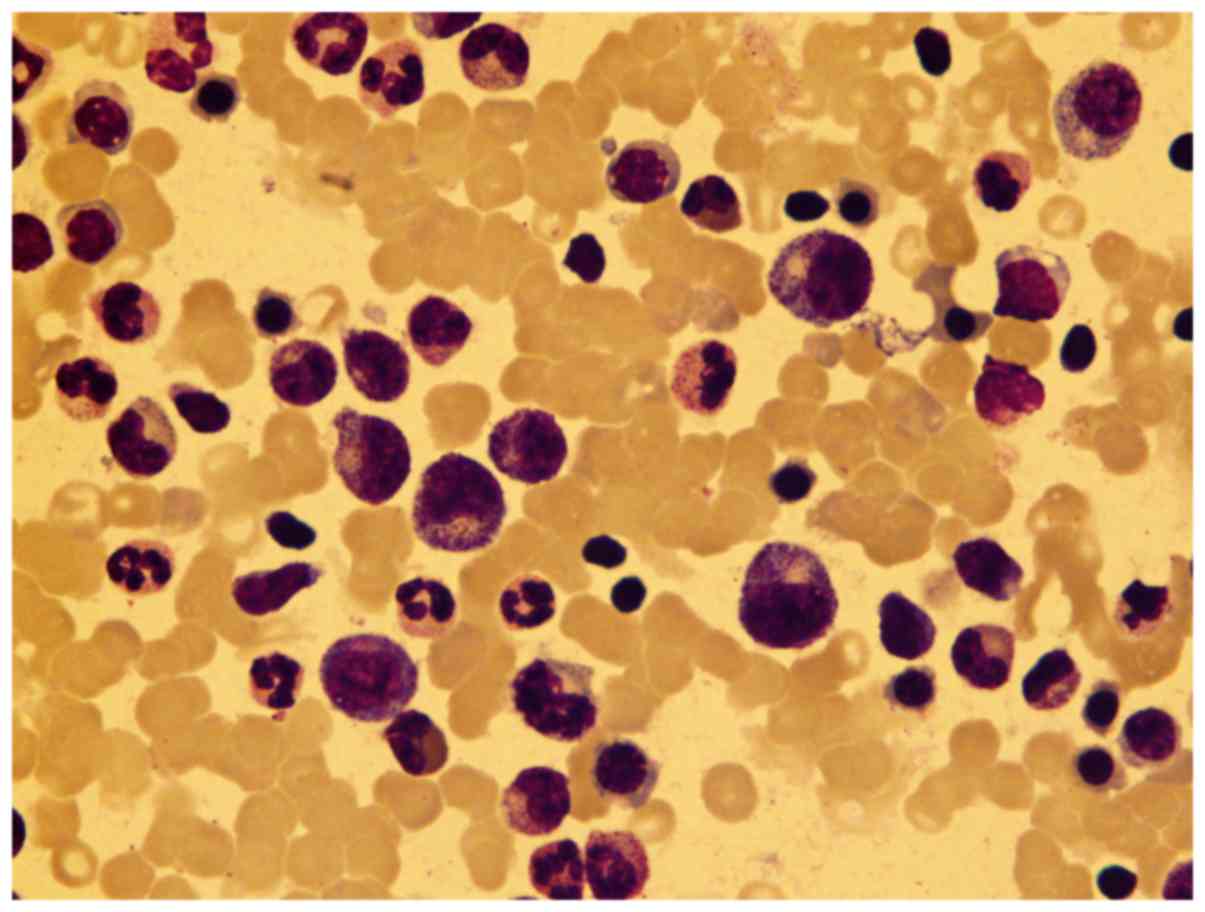
classification results, the myeloid series accounted for
(30.5–85%), including 25 cases >60% (23.36%), with a medium
percentage of 53%, and in 39 (36.45%) cases toxic granulation can
be seen (Fig. 2). The erythroid
series accounted for (6–44.0%), with a medium percentage of 22.5%.
Two patients had mild erythro-dysplastic hematopoiesis, including
megaloblastoid changes and nuclear dysmorphism. The lymphatic
series accounted for (3–42.5%). No obvious morphological changes
were observed.
 | Table I.Cellularity of bone marrow. |
Table I.
Cellularity of bone marrow.
| Cellularity | Myeloid:erythroid
(M:E) ratio | Number of cases |
|---|
| Markedly
increased | 1:1 | 26 (24.30%) |
| Increased | 1:10 | 54 (50.47%) |
| Normal | 1:20 | 27 (25.23%) |
| Reduced | 1:50 | 0 |
| Acellular | 1:300 | 0 |
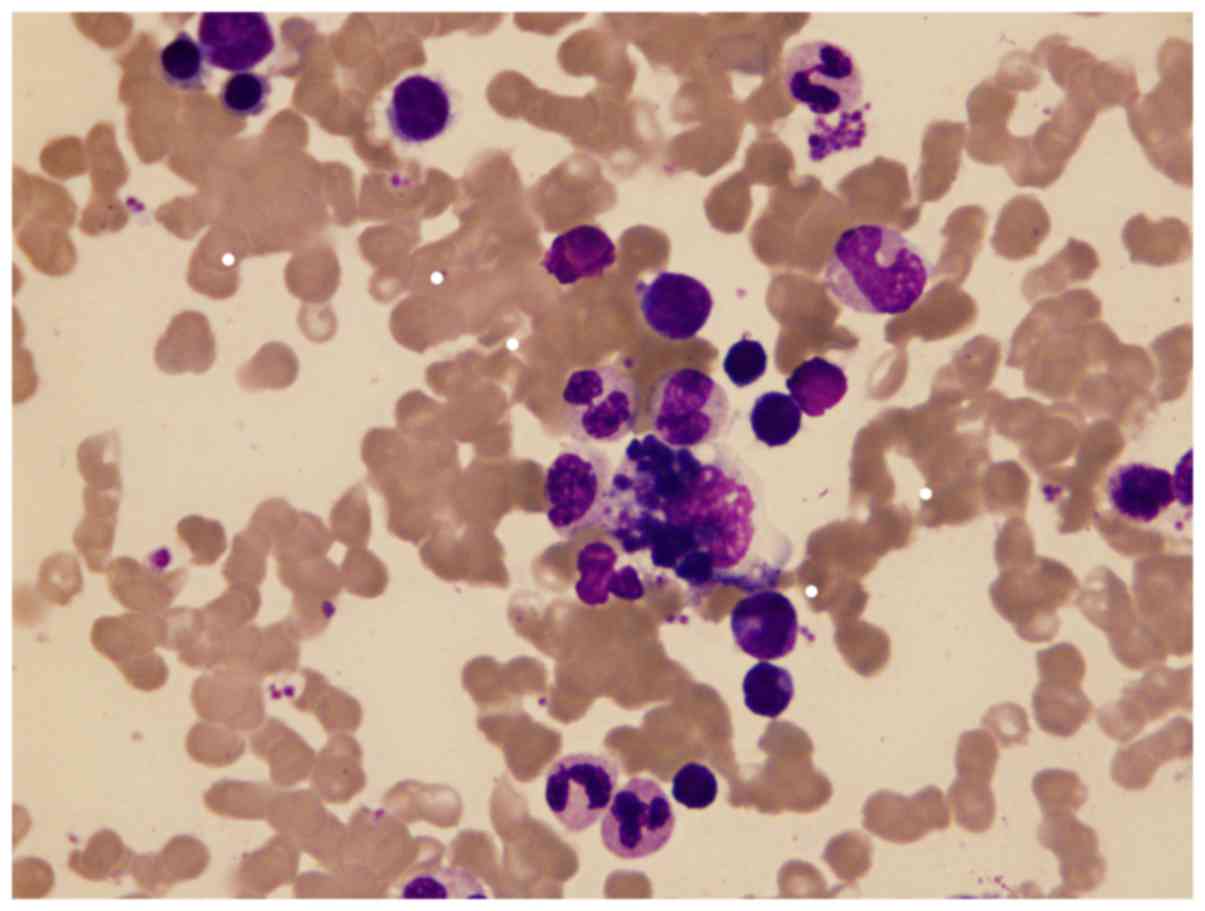
In all 107 patients with JIA, hemophagocytes were
observed (0–2/per slide) in 60 cases (56.07%), swallowing platelets
(Fig. 3) in 40 cases (66.67%),
erythrocytes in 22 cases (36.67%), neutrophil granulocytes in 4
cases (6.67%), lymphocytes in 1 case (1.67%), orthochromatic
normoblasts in 8 cases (13.33%), and impurities in 29 cases
(48.33%). Hemophagocytes were rarely seen (3–5/per slide) in 10
cases (9.35%), swallowing platelets in 8 cases (80.00%),
erythrocytes in 8 cases (80.00%), neutrophil granulocytes in 3
cases (30.00%), orthochromatic normoblasts in 2 cases (1.87%), and
impurities in 4 cases (3.74%). Hemophagocytes were clearly seen
(6–15/per slide) in 13 cases (12.15%), swallowing platelets in 10
cases (9.35%), erythrocytes in 8 cases (7.48%), neutrophil
granulocytes in 3 cases (2.80%), orthochromatic normoblasts in 3
cases (2.80%), and impurities in 9 cases (8.41%). Additionally, we
listed the number of hemophagocytes in the samples from each of the
107 patients with JIA (Table II),
but found no significant differences among the subtypes
(P>0.05).
| Table II.Number of hemophagocytes in each
category of juvenile idiopathic arthritis. |
Table II.
Number of hemophagocytes in each
category of juvenile idiopathic arthritis.
| Number of
Hemophagocytes/per slide | Systemic
arthritis | Enthesitis-related
arthritis | Polyarthritis
RF-positive | Polyarthritis
RF-negative | Oligoarthrtitis | Undifferentiated
arthritis | Total |
|---|
| 0 | 4 | 9 | 0 | 1 | 1 | 15 | 30 |
| 0–2 | 9 | 13 | 4 | 0 | 3 | 29 | 58 |
| 3–5 | 0 | 3 | 0 | 0 | 0 | 4 | 7 |
| 6–15 | 1 | 3 | 1 | 0 | 1 | 6 | 12 |
| Total | 14 | 28 | 5 | 1 | 5 | 54 | 107 |
In our study, 4 patients were received MAS in their
discharge diagnosis, with 3 sJIA and 1 undifferentiated JIA.
Hemophagocytes were occasionally seen (0–2/per slide) in the 3 sJIA
cases, and clearly seen (6–15/per slide) in the 1 case of
undifferentiated JIA.
Discussion
We found abnormal quantitative myelodysplastic
changes in our 107 patients with JIA. An increase in the
cellularity and a mild-moderate increase in the megakaryocytic
series had been found in most cases, in addition to other
qualitative changes. The most common morphological change was toxic
granulation in the myeloid series. For the erythroid series,
megaloblastic changes and nuclear dysmorphism were seen in 2/107
cases. Furthermore, 77.57% of the patients had hemophagocytes
present in bone marrow smears. As bone marrow aspiration is an
invasive examination, no healthy, age-matched volunteers were
included in this study.
None of the patients had any indication of certain
infections at the time of bone marrow examination, according to
their clinical history; thus, abnormal smear findings, such as
toxic granulation and the presence of hemophagocytes, may be
related to the disease itself. The pathophysiological mechanism
underlying the hematopoietic changes in JIA remains uncertain.
Cellular immune system dysfunction has been implicated, and may
lead to alterations in the microenvironment of the bone marrow,
through the effects of dysregulated cytokines and other local
intracellular messengers (8,9).
Mellins et al (8) reported that systemic JIA (sJIA),
currently classified as a subtype of JIA (1), appears to be driven by the continuous
activation of innate immune pathways with abnormally regulated
production of innate proinflammatory cytokines, suggesting that
sJIA is an autoinflammatory disorder. It is recognized that a
proportion (10–30%) of patients with sJIA develop macrophage
activation syndrome (MAS). MAS is a syndrome characterized as
overreactive inflammation driven by the excessive activation and
expansion of T cells (mainly CD8+), leading to the
activation of hemophagocytic macrophages (9,10); it is
also known as a complication of pediatric rheumatic disorders,
particularly JIA (11) and systemic
lupus erythematosus (SLE) (12,13). It
is recognized as a severe, potentially life-threatening
complication, the same disorder as the secondary or ‘reactive’ form
of hemophagocytic lymphohistiocytosis (HLH) (14). Both conditions have high mortality
rates (15). Even with appropriate
and timely treatment, one English study reported mortality rates
was 2/9 in 2001 (16), and one
Iranian study reported mortality rates was 2/5 in 2011 (17).
Abnormal quantitative and qualitative features in
bone marrow smears, especially the presence of hematopoietic cells,
and their phagocytosis by macrophages, is common. The frequent
presence of hemophagocytes indicates that bone marrow examinations
are one of the most important auxiliary examinations for the
diagnosis of JIA, in order to exclude or make the diagnose of
MAS.
In conclusion, we investigated the characteristics
of bone marrow cells in 107 patients with JIA. And we noticed
increased cellularity; increased megakaryocyte count and the
presence of hemophagocytes were in the majority of bone marrow
specimens. We propose that JIA is associated with specific
myelodysplastic changes, and that cellular immune system
dysfunction and overreactive inflammatory cytokines may contribute
to the development of these myelodysplastic changes in the bone
marrow. In order to exclude or make the diagnose of MAS, bone
marrow examination should be considered as an important auxiliary
examination.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The dataset used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
DZ wrote the manuscript. JZ and FC designed the
study. DZ and YZ analyzed and interpreted the patient data. FC gave
advice about the study and gave final approval of the version to be
published. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Institutional
Review Board at Ren Ji Hospital Affiliated to Shanghai Jiao Tong
University in Shanghai [IRB Approval no. RJKLS (2016) 023]. Patient
consent was waived by the ethics committee.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Petty RE, Southwood TR, Manners P, Baum J,
Glass DN, Goldenberg J, He X, Maldonado-Cocco J, Orozco-Alcala J,
Prieur AM, et al: International League of Associations for
Rheumatology classification of juvenile idiopathic arthritis:
Second revision, edmonton, 2001. J Rheumatol. 31:390–392.
2004.PubMed/NCBI
|
|
2
|
Foeldvari I and Bidde M: Validation of the
proposed ILAR classification criteria for juvenile idiopathic
arthritis. International League of associations for rheumatology. J
Rheumatol. 27:1069–1072. 2000.PubMed/NCBI
|
|
3
|
Ravelli A and Martini A: Juvenile
idiopathic arthritis. Lancet. 369:767–778. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Peixuan and Xiufen Cheng Hu: Juvenile
idiopathic arthritis and macrophage activation syndrome. J App Clin
Pediatr. 24:1631–1633. 2009.
|
|
5
|
Petty RE, Southwood TR, Baum J, Bhettay E,
Glass DN, Manners P, Maldonado-Cocco J, Suarez-Almazor M,
Orozco-Alcala J and Prieur AM: Revision of the proposed
classification criteria for juvenile idiopathisc arthritis: Durban,
1997. J Rheumatol. 25:1991–1994. 1998.PubMed/NCBI
|
|
6
|
Giancane G, Consolaro A, Lanni S, Davì S,
Schiappapietra B and Ravelli A: Juvenile idiopathic arthritis:
Diagnosis and treatment. Rheumatol Ther. 3:187–207. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lee SH, Erber WN, Porwit A, Tomonaga M and
Peterson LC: International Council for Standardization In
Hematology: ICSH guidelines for the standardization of bone marrow
specimens and reports. Int J Lab Hematol. 30:349–364. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mellins ED, Macubas C and Grom AA:
Pathogenesis of systemic juvenile idiopathic arthritis: Some
answers, more questions. Nat Rev Rheumatol. 7:416–426. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hadchouel M, Prieur AM and Griscelli C:
Acute hemorrhagic, hepatic, and neurologic manifestations in
juvenile rheumatoid arthritis: possible relationship to drugs or
infection. J Pediatr. 106:561–566. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Grom AA: Natural killer cell dysfunction:
A common pathway in systemic-onset juvenile rheumatoid arthritis,
macrophage activation syndrome, and hemophagocytic
lymphohistiocytosis? Arthritis Rheum. 50:689–698. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Stéphan J, Koné-Paut I, Galambrun C, Mouy
R, Bader-Meunier B and Prieur A: Reactive haemophagocytic syndrome
in children with inflammatory disorders. A retrospective study of
24 patients. Rheumatology (Oxford). 40:1285–1292. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pringe A, Trail L, Ruperto N, Buoncompagni
A, Loy A, Breda L, Martini A and Ravelli A: Macrophage activation
syndrome in juvenile systemic lupus erythematosus: An
under-recognized complication? Lupus. 16:587–592. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Parodi A, Davì S, Pringe AB, Pistorio A,
Ruperto N, Magni-Manzoni S, Miettunen P, Bader-Meunier B, Espada G,
Sterba G, et al: Macrophage activation syndrome in juvenile
systemic lupus erythematosus: A multinational multicenter study of
thirty-eight patients. Arthritis Rheum. 60:3388–3399. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Grom A: Macrophage activation syndrome and
reactive hemophagocytic lymphohistiocytosis: The same entities?
Curr Opin Rheumatol. 15:587–590. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Janka G: Hemophagocytic syndromes. Blood
Rev. 21:245–253. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sawhney S, Woo P and Murray KJ: Macrophage
activation syndrome: A potentially fatal complication of rheumatic
disorders. Arch Dis Child. 85:421–426. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Moradinejad MH and Ziaee V: The incidence
of macrophage activation syndrome in children with rheumatic
disorders. Minerva Pediatr. 63:459–466. 2011.PubMed/NCBI
|