Introduction
Hepatic fibrosis is a wound-repairing response to
frequent and repeated liver injuries that may lead to cirrhosis and
even liver cancer (1). Liver
fibrosis represents the consequence of an imbalance between
accumulation and dissolution of excessive extracellular matrix
(ECM) (2,3), and the inhibition of collagen
generation is an effective method to treat liver fibrosis (4,5).
Activated hepatic stellate cells (HSCs) are well known for their
potential role in increasing deposition of ECM and elevated
proliferation in liver fibrosis (6,7). During
liver injury, quiescent HSCs that store vitamin A in lipid droplets
(LDs) and reside in the spaces of Disse convert to an activated
phenotype and are depleted of vitamin A (6). The activation process of quiescent HSCs
can be driven by various stimuli, including lipopolysaccharide
(LPS) (8). LPS, the classic ligand
for Toll-like receptor 4 (TLR4) (9),
has been found to be associated with hepatic fibrogenesis through
direct interactions with HSCs (8).
LPS-induced nuclear factor-κB (NF-κB) activation and release of
inflammatory cytokines were also observed in activated HSCs
(10). However, the molecular
mechanisms underlying the effects of LPS on HSC activation are
poorly understood.
Fibroblast growth factor receptor 1 (FGFR1) is a
receptor tyrosine kinase that mediates a broad spectrum of cellular
and developmental processes, including apoptosis, proliferation,
and angiogenesis (11). Moreover,
substances targeting FGFR1 have been shown to have promise for
treatment in animal cancer models (12). Recently, the FGFR1 signalling system
has also been identified as a key player in the process of liver
injury (13–15). Selective blockade of FGFR1 inhibited
HSC activation by measuring the production of ECM (14). In addition, administration of NP603,
a novel inhibitor of FGFR1, significantly decreased hepatic
fibrosis in carbon tetrachloride (CCl4)-treated rats (14). These findings suggest that FGFR1
inhibition may be an ideal therapeutic approach to HSC activation
and liver fibrosis. Nevertheless, it is unclear whether FGFR1 is a
potential regulator in LPS-related inflammatory responses and
activation of HSCs.
The goal of this study was to determine the role of
FGFR1 in LPS-induced inflammation and HSCs activation.
Specifically, we used pharmacological and genetic means to inhibit
FGFR1 in HSCs. We showed that inhibition of FGFR1 ameliorated LPS
induced-NF-κB activation, inflammatory responses, fibrosis and cell
viability in HSCs. Furthermore, we found that the LPS/TLR4/c-Src
signalling axis appeared to mediate downstream FGFR1
phosphorylation.
Materials and methods
Reagents
AZD4547 were purchased from Shanghai Kai Yu
Pharmatech Technology Co., Ltd. (Shanghai China). LPS, PP2, and
TAK-242 were purchased from Sigma-Aldrich (St. Louis, MO, USA).
AZD4547, PP2, and TAK-242 were dissolved in DMSO for in
vitro experiments. Antibodies against TGF-β, collagen 1, α-SMA,
p-c-Src, c-Src, lamin B, and GAPDH were purchased from Santa Cruz
Biotechnology, Inc. (Dallas, TX, USA). Antibodies against p-FGFR1,
FGFR1, TLR4, TNF-α, IL-6, IκB-α and NF-κB P65 were from Cell
Signaling Technology, Inc. (Danvers, MA, USA).
Cell culture and treatment
HSCs were isolated from male Sprague-Dawley rats
(450–500 g) as described previously (16). Animal care and experimental protocols
were approved by the Committee on Animal Care of Zhuji People's
Hospital of Zhejiang Province (Zhuji, China; approval no.
zjdw2017-008). Briefly, after in situ perfusion of the liver
with 2-step pronase-collagenase digestion, HSCs were separated from
other nonparenchymal cells by density-gradient centrifugation using
OptiPrep (Axis-Shield, 1114542). HSCs were maintained in DMEM
containing 10% FBS, 100 U/ml penicillin, and 100 mg/ml streptomycin
in a humidified atmosphere of 5% CO2 at 37°C. All treatments were
initiated 12 h after isolation unless otherwise indicated. All
experiments were repeated at least 3 times.
Measurement of cell viability by MTT
assay
Cell viability was assessed by MTT assay. HSCs were
plated in 96-well plates at 5,000 cells per well and were then
treated with or without LPS for 24 h. After incubation with MTT for
3 h, the reduction of MTT to purple formazan was detected by a
microplate reader at 540 nm. Cell viability was calculated as
follows: Cell viability = Atreated / Acontrol × 100%.
siRNA-induced gene silencing
FGFR1 gene silencing in cells was achieved by
transfecting cells with siRNA (5′-GCAGCGAUACCACCUACUUTT-3′) using
LipofectAMINE™ 2000 (Invitrogen; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA). Knockdown was verified by western blotting
(WB).
WB and co-immunoprecipitation
HSCs were lysed, and protein amounts were determined
by the Bradford assay (Bio-Rad). Nuclear and cytoplasmic proteins
were extracted from HSCs using nuclear and cytoplasmic protein
extraction kits (Beyotime Biotech, Nantong, China). Proteins were
separated by 10% SDS-PAGE and were electrotransferred to PVDF
membranes. Each membrane was blocked for 1.5 h with Tris-buffered
saline containing 0.05% Tween-20 and 5% non-fat milk. PVDF
membranes were then incubated with specific primary antibodies.
Immunoreactive bands were detected by incubating membranes with
horseradish peroxidase-conjugated secondary antibodies and
visualisation using enhanced chemiluminescence (Bio-Rad). The
amounts of the proteins were analysed using ImageJ analysis
software version 1.38e and were normalised to their respective
controls.
For immunoprecipitation studies, extracts were
incubated with anti-c-Src-antibody for 4 h and were then
precipitated with protein G-Sepharose beads at 4°C overnight. c-Src
and FGFR1 levels were detected by immunoblotting using specific
antibodies.
RT-qPCR
Total RNA was isolated from HSCs using TRIzol
(Invitrogen; Thermo Fisher Scientific, Inc.). Reverse transcription
and quantitative PCR were carried out using a two-step Platinum
SYBR Green qPCR SuperMix-UDG kit (Invitrogen; Thermo Fisher
Scientific, Inc.). An Eppendorf Mastercycler (Eppendorf, Hamburg,
Germany) was used for qPCR analysis. Primers for genes including
TNF-α, IL-6, collagen I, TGF-β, α-SMA, and β-actin were obtained
from Invitrogen; Thermo Fisher Scientific, Inc. (sequences are
listed in Table I). Target mRNA was
normalised to β-actin.
 | Table I.Sequences of primers for RT-qPCR
assay used in the study. |
Table I.
Sequences of primers for RT-qPCR
assay used in the study.
| Gene | Species | Forward primer | Reverse primer |
|---|
| TNF-α | Rat |
5′-TACTCCCAGGTTCTCTTCAAGG-3′ |
5′-GGAGGCTGACTTTCTCCTGGTA-3′ |
| IL-6 | Rat |
5′-GAGTTGTGCAATGGCAATTC-3′ |
5′-ACTCCAGAAGACCAGAGCAG-3′ |
| Collagen1 | Rat |
5′-CGAGTATGGAAGCGAAGGTT-3′ |
5′-ACGCTGTTCTTGCAGTGATA-3′ |
| TGF-β | Rat |
5′-AGGAGGAATTTGGCCAGGTG-3′ |
5′-GCTCACGAGGAGGCTAATCC-3′ |
| α-SMA | Rat |
5′-TGACCCAGATTATGTTTGAG-3′ |
5′-AGATAGGCACGTTGTGAGTC-3′ |
| β-actin | Rat |
5′-AAGTCCCTCACCCTCCCAAAAG-3′ |
5′-AAGCAATGCTGTCACCTTCCC-3′ |
Immunofluorescence cell staining
Cells were fixed with 4% paraformaldehyde,
permeabilised with 0.1% Triton X-100 and stained. Col-1 and α-SMA
staining were performed by incubating slides with anti-Col-1 or
anti-α-SMA antibody at 1:200 dilution overnight at 4°C.
PE-conjugated secondary antibody (1:200) was used for detection.
Cells were counterstained with DAPI nuclear stain. Images were
captured (original magnification 400; Nikon, Tokyo, Japan).
Statistical analysis
All data represented 3 independent experiments and
were expressed as the means ± SEM. Statistical analyses were
performed using GraphPad Pro. Prism 5.0 (GraphPad Software, Inc.,
La Jolla, CA, USA). Student t-tests or one-way ANOVAs followed by
multiple comparisons tests with Bonferroni corrections were
employed to analyse the differences between sets of data. A P-value
<0.05 was considered to indicate a statistically significant
difference.
Results
Small-molecule FGFR1 inhibitor
reversed LPS-induced HSC activation
We used a small-molecule inhibitor, AZD4547, that
specifically inhibits FGFR1 activity (Fig. 1A) (17). Freshly isolated HSCs were treated
with LPS (100 ng/ml for 15 min) and the effects on FGFR1 activation
were determined. LPS induced a robust increase in FGFR1
phosphorylation that was inhibited in a dose-dependent fashion by
AZD4547 pretreatment for 1 h (Fig.
1B). To evaluate the effect of AZD4547 on LPS-induced HSCs
activation, we determined cell viability of HSCs. In accordance
with previous studies (18), LPS
significantly stimulated HSC proliferation (Fig. 1C), indicating that LPS increased HSC
activation. Treatment with AZD4547 reduced LPS-induced cell
viability (Fig. 1C).
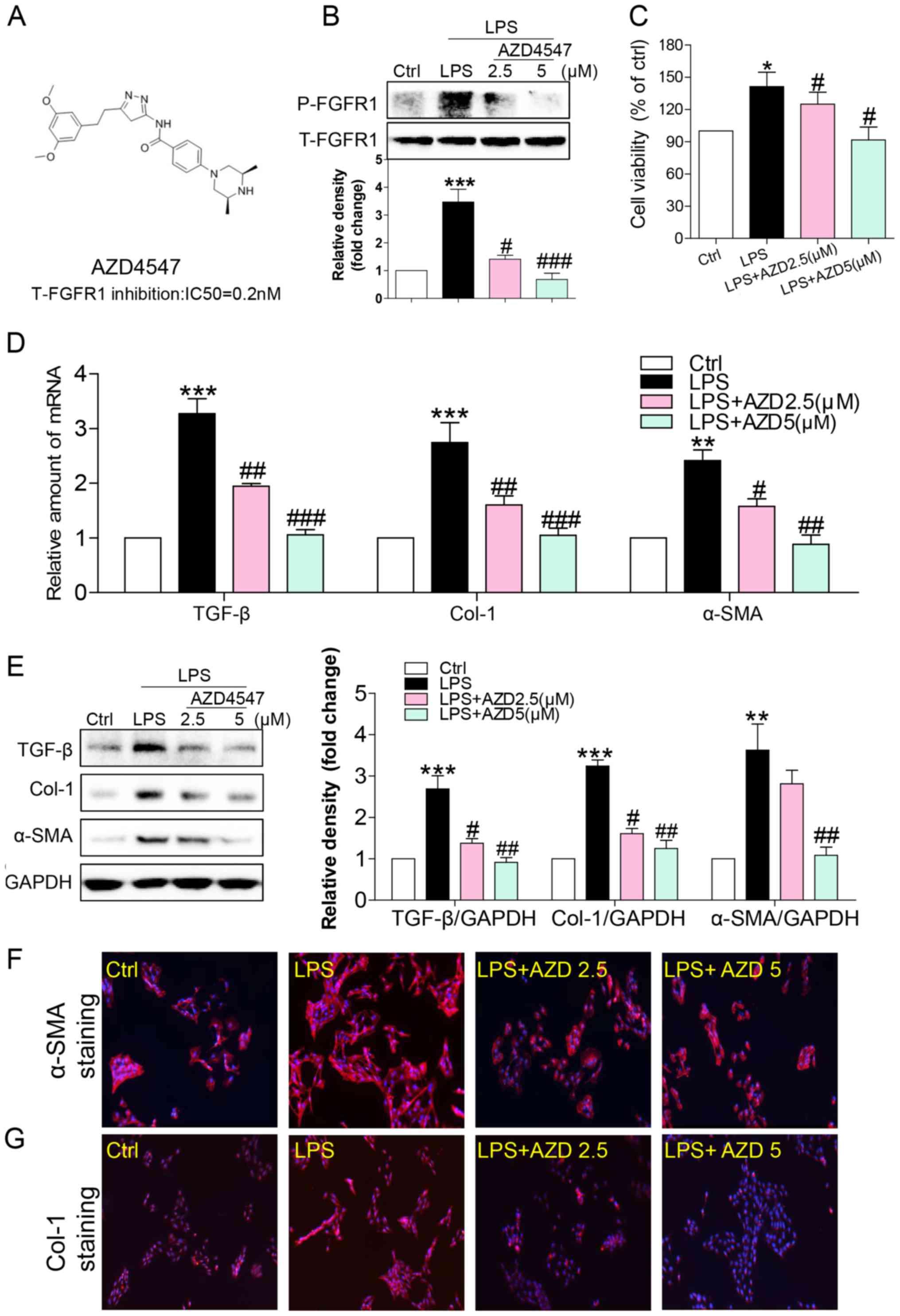 | Figure 1.FGFR1 inhibitor AZD4547 attenuates
LPS-induced HSCs activation. (A) Chemical structures and
FGFR1-inhibitory IC50 values of AZD4547. HSCs were pretreated with
AZD4547 (AZD, 2.5, 5 µM) for 1 h, and then exposed to LPS (100
ng/ml) for the indicated times. (B) Exposure to LPS for 15 min.
p-FGFR1 levels detected by western blotting (WB). (C) Exposure to
LPS for 24 h. The cell viability of HSCs detected by MTT assay. (D)
Exposure to LPS for 6 h, the mRNA levels of TGF-β, col-1, and α-SMA
were detected by RT-qPCR and normalised by β-actin. Incubation with
LPS for 24 h. (E) The levels of TGF-β, Col-1 and α-SMA in cell
lysates were detected by WB. (F and G) Immunofluorescence staining
of HSCs for Col-1 (red), and α-SMA (red) in LPS-treated cells
pretreated with AZD4547 prior to LPS exposure (blue=DAPI).
Representative micrographs are shown (*P<0.05, **P<0.01,
***P<0.001, vs. Ctrl group; #P<0.05, ##P<0.01,
###P<0.001, vs. LPS group). |
Increased production and/or activity of transforming
growth factor (TGF)-β was critical for sustaining HSC activation
and fibrosis (19). Upon sustained
LPS treatment for 6 h, mRNA levels of TGF-β increased (Fig. 1D). Pretreatment with AZD4547
decreased TGF-β mRNA levels in a dose-dependent manner (Fig. 1D). As shown in Fig. 1D, LPS stimulated mRNA expression of
ECM, including collagen I and α-smooth muscle actin (α-SMA), both
of which were reduced by AZD4547 pretreatment in a dose-dependent
manner. AZD4547 also dose-dependently reversed LPS-stimulated TGF-β
(Fig. 1E), Col-1 (Fig. 1E), and α-SMA (Fig. 1E) protein expression. These results
were also verified by staining cells for α-SMA (Fig. 1F), and Col-1 (Fig. 1G). These findings strongly suggested
that the FGFR1 small-molecule inhibitor attenuated LPS-related
fibrosis in HSCs, and that inhibition of liver fibrosis protein
expression by AZD4547 may be associated with decreased HSC
viability.
FGFR1 inhibitor AZD4547 decreased
LPS-induced inflammatory responses in HSCs
LPS caused inflammatory responses in the liver,
mediating the progression of HSC activation (8,10,20). We
evaluated whether AZD4547 altered the expression of
pro-inflammatory cytokines. Immunoblotting showed an increased
expression of inflammatory cytokines, including tumour necrosis
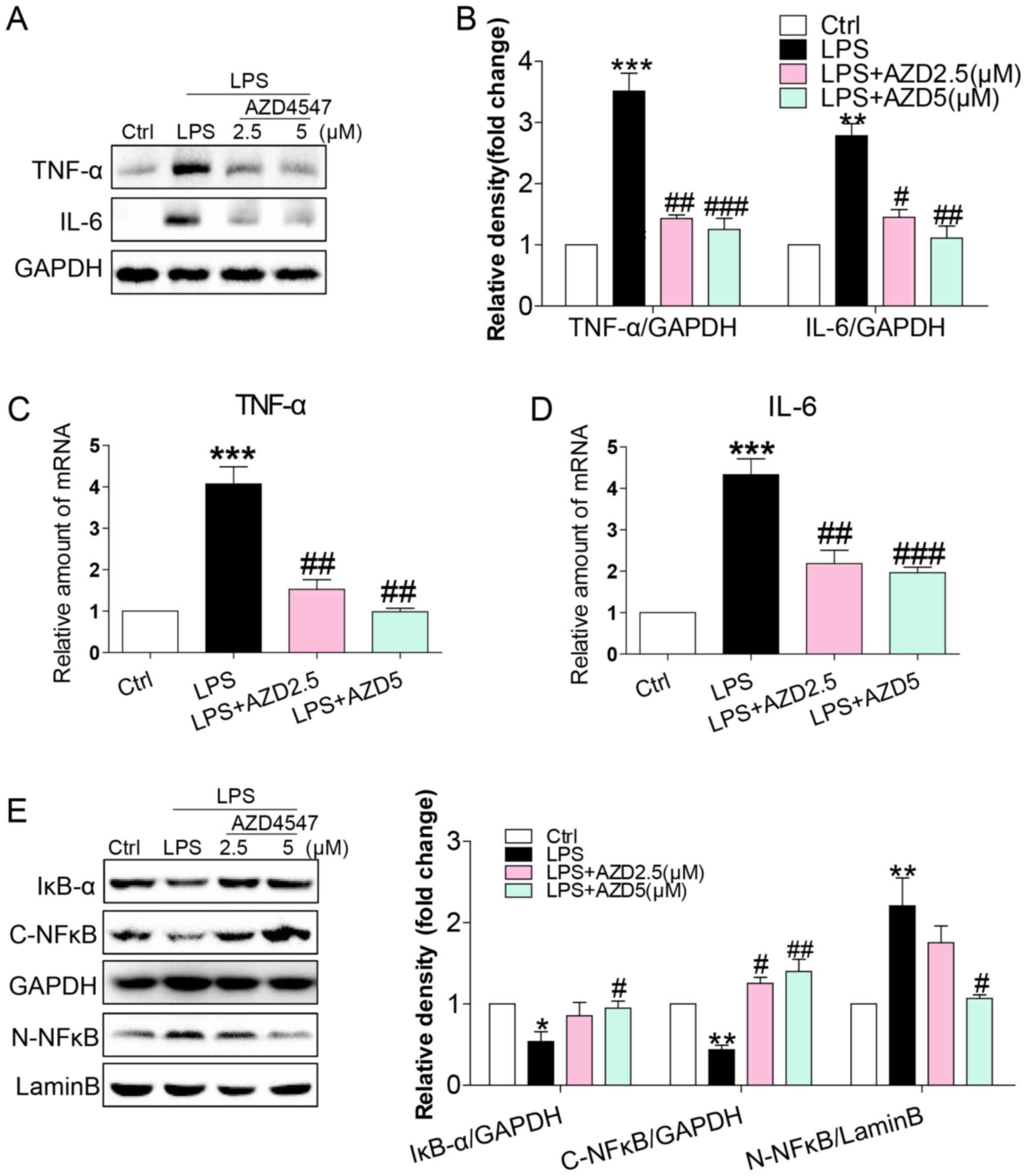
factor-α (TNF-α) and interleukin-6 (IL-6) in HSCs (Fig. 2A and B). This increase was associated
with increased mRNA levels of TNF-α and IL-6 (Fig. 2C and D). AZD4547 treatment reduced
both protein and mRNA levels of TNF-α and IL-6 (Fig. 2A-D). To uncover the signalling
mechanism underlying the anti-inflammatory activity of AZD4547, we
examined the NF-κB pathway, the signalling pathway implicated in
the expression of pro-inflammatory cytokines by LPS in HSCs
(10). We exposed HSCs to LPS and
treated the cells with AZD4547 to assess the NF-κB signalling
pathway. LPS reduced cytosolic IκB-α (Fig. 2E), cytoplasmic NF-κB p65 subunit
(Fig. 2E) and increased nuclear
NF-κB p65 subunit (Fig. 2E) levels.
AZD4547 treatment of HSCs prevented LPS-induced reduction in
cytosolic IκB-α, p65 and increased nuclear p65 levels (Fig. 2E). These results show that LPS
induced a pro-inflammatory phenotype in HSCs and that these adverse
effects were prevented by FGFR1 inhibitor AZD4547.
Gene knockdown of FGFR1 attenuated
LPS-induced inflammatory responses and activation of HSCs
To assess the non-specificity of the small molecule
inhibitor and to provide further support for the role of FGFR1, we
silenced FGFR1 by siRNA (si-FGFR1). Transfection of si-FGFR1 led to
decreased FGFR1 protein expression in HSCs (Fig. 3A), and attenuated protein (Fig. 3B and C) and gene (Fig. 3D) expression levels of TNF-α and IL-6
in LPS-stimulated HSCs. In addition, LPS-induced NF-κB activity was
not evident following silencing of FGFR1 expression in HSCs
(Fig 3E). Subsequently, si-FGFR1
remarkably decreased LPS-induced activation of HSCs, as evidenced
by liver fibrosis markers such as TGF-β, col-1, and α-SMA at both
the protein (Fig. 3F) and mRNA
(Fig. 3G) level. These findings,
together with the results of the anti-inflammation and
anti-fibrosis effect of AZD4547, confirmed that FGFR1 had a
potential role in regulating LPS-related HSCs activation.
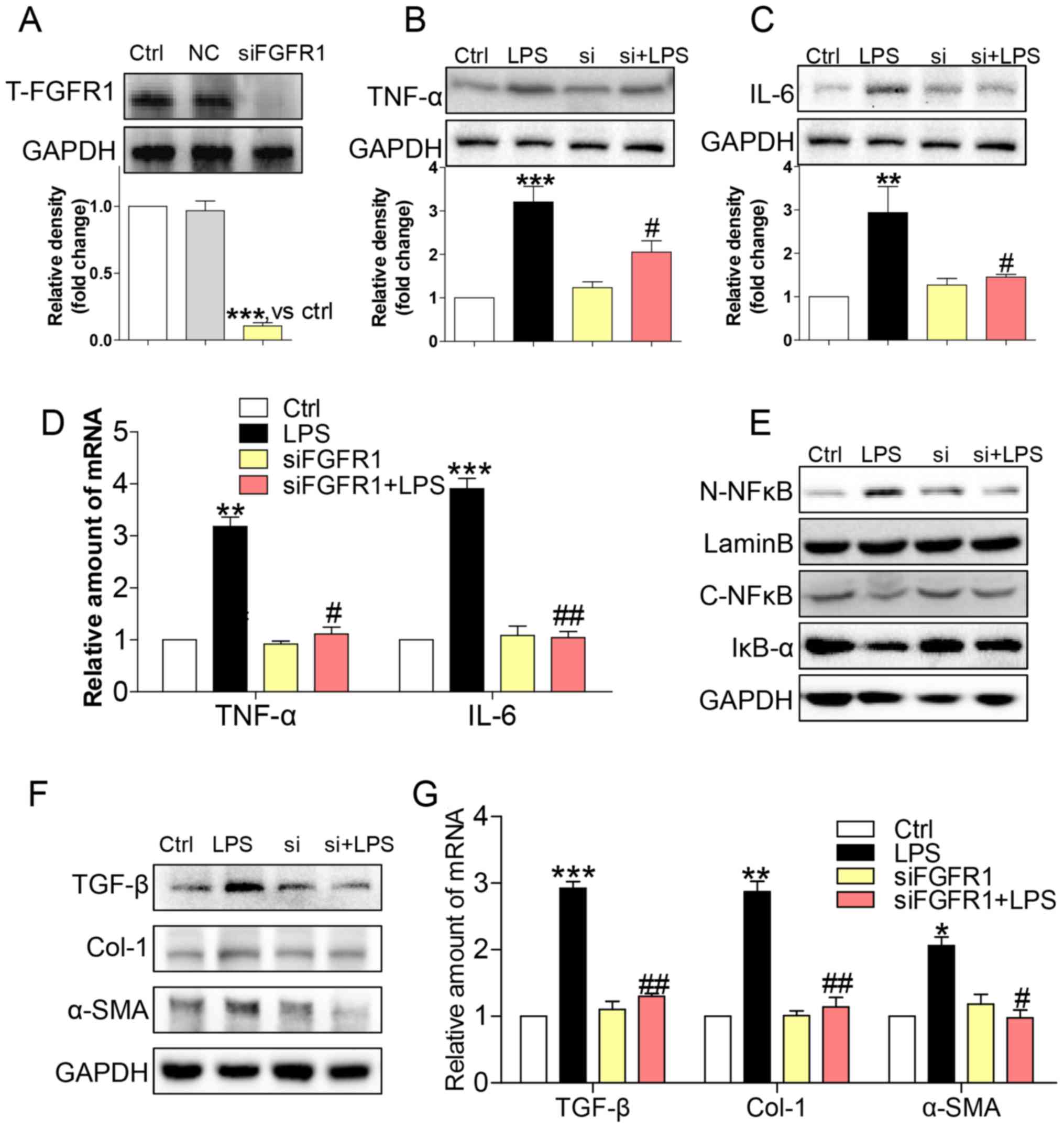 | Figure 3.siFGFR1 decreases LPS-induced HSCs
injury. (A) Western blot analysis of FGFR1 following siRNA
transfection in HSCs (NC=negative control transfection). After
incubating for 24 h, FGFR1 knockdown HSCs were stimulated with LPS
(100 ng/ml) for indicated times (Si=FGFR1 siRNA). Cells were
incubated with LPS for 24 h. (B) TNF-α and (C) IL-6 in cell lysates
were detected by western blotting (WB). (D) Incubated with LPS for
6 h. The mRNA levels of TNF-α and IL-6 were detected by RT-qPCR and
normalised by β-actin. (E) IκB-α, C-NF-κB, and N-NF-κB protein
levels were determined in cells with LPS treatment for 1 h. (F)
Incubated with LPS for 24 h. The levels of TGF-β, col-1, and α-SMA
were detected by WB. (G) Exposing to LPS for 6 h, the mRNA levels
of TGF-β, col-1, and α-SMA were detected by RT-qPCR and normalised
by β-actin (*P<0.05, **P<0.01, ***P<0.001, vs. Ctrl group;
#P<0.05, ##P<0.01, vs. LPS group). |
LPS triggered FGFR1 phosphorylation in
HSCs through the TLR4/c-Src signalling cascade
How LPS activates FGFR1 remained unaddressed. LPS
directly binds to TLR4 (9),
subsequently activating downstream NF-κB signalling and
inflammatory responses (21,22). TLR4 signalling activation promoted
c-Src phosphorylation (23). It was
also reported that c-Src via FGFR1 transactivation and subsequent
downstream activation of multiple pathways mediated lymphoma and
myeloproliferative disorders (24).
Therefore, we tested whether LPS activated FGFR1 in HSCs through
the TLR4/c-Src signalling cascade. AZD4547 did not block expression
of TLR4 (Fig. 4A) or phosphorylation
of c-Src (Fig. 4B) induced by LPS,
suggesting that FGFR1 may be not be the upstream regulator of
TLR4/c-Src. We then evaluated the role of TLR4 blocker TAK-242 and
c-Src blocker PP2 in LPS induced-FGFR1 phosphorylation. As shown in
Fig. 4C, both TAK-242 and PP2
reduced FGFR1 activation in HSCs. In addition, gene knockdown of
FGFR1 did not suppress LPS-induced increases in TLR4 (Fig. 4D) or activation of c-Src (Fig. 4E). We also assessed c-Src/FGFR1
complex formation in the context of LPS. Co-immunoprecipitation
showed that LPS challenge of HSCs for 5 to 30 min remarkably
increased the recruitment of FGFR1 to c-Src (Fig. 4F). These findings suggested a novel
mechanism of LPS-induced FGFR1 activation, one that involves
TLR4/c-Src signalling cascade.
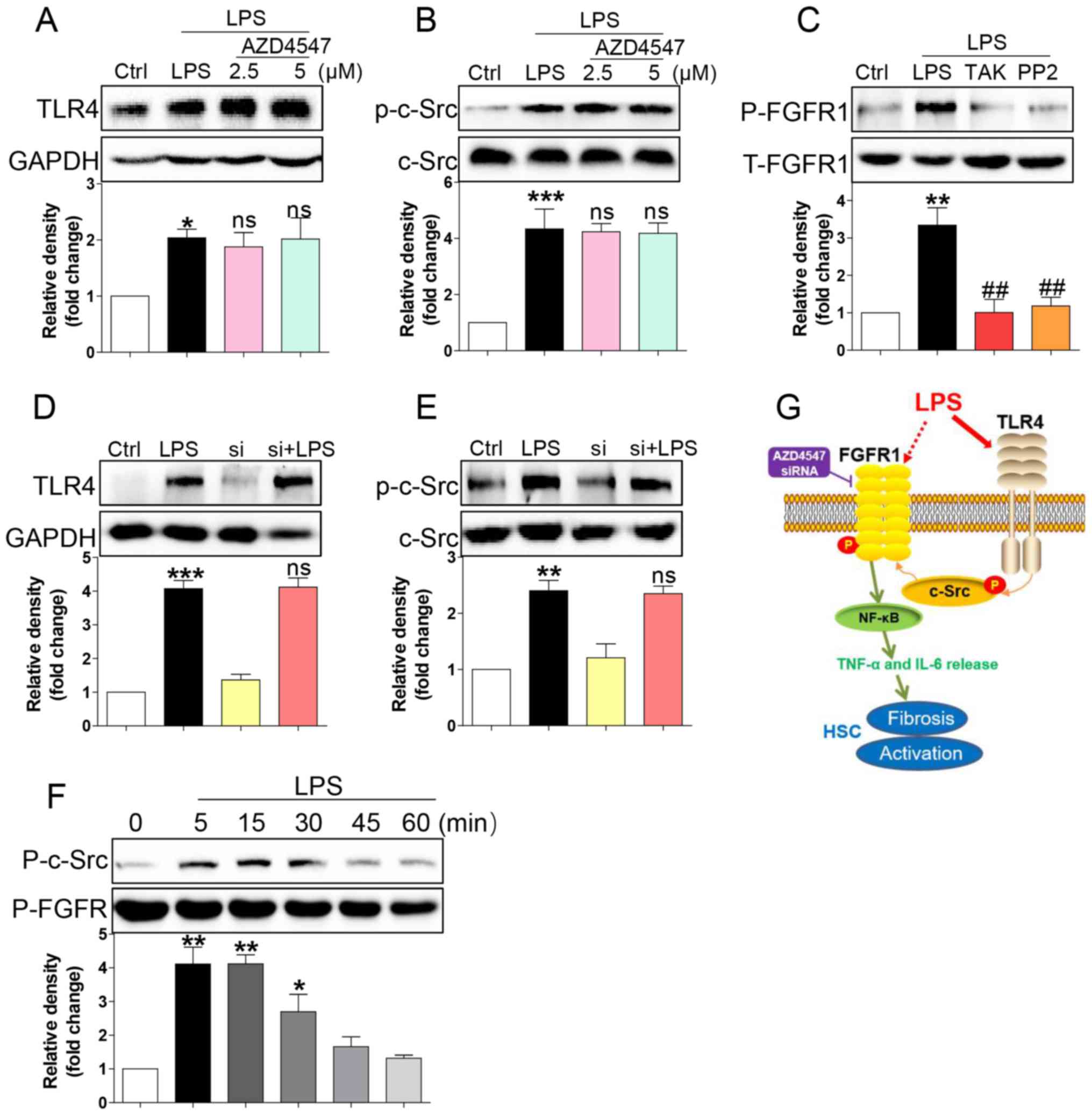 | Figure 4.LPS triggers FGFR1 phosphorylation in
HSCs through the TLR4/c-Src signalling cascade. HSCs were
pre-treated with AZD4547 (AZD, 2.5, 5 µM) for 1 h. After LPS
treatment for 15 min, the expression of (A) TLR4 and (B)
phosphorylation levels of c-Src were detected by western blotting
(WB). (C) HSCs were pre-treated with TAK-242 (TLR4 inhibitor), or
PP2 (c-Src inhibitor) for 1 h, followed by LPS treatment for 15
min. Phosphorylation of FGFR1 was determined via WB. (D and E)
siFGFR1 did not reduce LPS (15 min)-induced increase expression of
TLR4 and c-Src activation. (F) LPS-induced interaction between
c-Src and FGFR1. HSCs were exposed to LPS for the indicated times.
Lysates were then subjected to c-Src IP and FGFR1 was measured. (G)
Schematic illustration of the major findings of this study: LPS
activated TLR4 and increased signalling through the c-Src pathway
to cause FGFR1 activation and production of inflammatory cytokines.
This proinflammatory response produced HSCs activation and liver
fibrosis (*P<0.05, **P<0.01, ***P<0.001, vs. Ctrl group;
ns, not significant vs. LPS group; ##P<0.01, vs. LPS group). |
Discussion
In this study, we revealed that LPS mediated FGFR1
activation in HSCs, which then contributed to NF-κB activation,
IL-6 and TNF-α release, fibrosis and proliferation in HSCs.
Application of an FGFR1 inhibitor or genetic knockdown of FGFR1 in
LPS-challenged cells produced a great reduction in HSC viability,
fibrosis and inflammatory responses, suggesting that inhibition of
FGFR1 may be a therapeutic approach for LPS-induced HSC activation
(Fig. 4G).
Evidence implicated FGFR1 in a host of liver
fibrosis diseases (13–15). Böhm et al (13) generated mice with hepatocytes that
lacked FGFR1 and subjected them to acute and chronic CCl4-induced
liver injury and partial hepatectomy. In hepatocytes, loss of FGFR1
eliminated responsiveness to FGF7 but did not affect toxin-induced
liver injury and fibrosis. However, mortality after partial
hepatectomy increased because of severe hepatocyte necrosis
(13). Using a tissue microarray of
89 primary liver tumours, including a subset of 10 fibrolamellar
carcinomas, Riehle et al (15) provided evidence of FGFR1
overexpression in human fibrolamellar carcinoma and supported the
use of FGFR1 inhibitors in the treatment of patients with
unresectable fibrolamellar carcinoma. Our results indicated that
FGFR1 inhibitor or genetic silencing by siRNA significantly
decreased the expression of ECMs, including TGF-β, α-SMA, collagen
I, and reduced cell viability in HSCs related to LPS.
The NF-κB signalling pathway, a conserved mediator
of inflammatory responses, plays a central role in regulating the
progression of liver fibrogenesis (25). Inhibition of IκB kinases stimulated
HSC apoptosis, indicating that NF-κB signalling played a central
role in the activation of HSC (10).
Liver fibrosis is often associated with LPS-induced
pro-inflammatory cytokines IL-6, and TNF-α release (26). We showed that the same FGFR1/NF-κB
activation pathway enhanced inflammatory responses in HSCs that
were markedly reversed by the FGFR1 inhibitor AZD4547 or
siRNA-silencing FGFR1.
There is a pressing need to understand how LPS
activates FGFR1 signalling. Yao et al (27) found that dioscin exhibited protective
effects against LPS-induced liver injury via altering TLR4
signalling. We identified TLR4, the classic receptor for LPS, as a
potential activator of FGFR1 in HSCs. In HFD-fed mice, TLR4 and
FGFR1 appeared to be implicated in the expression of
proinflammatory cytokines and hepatic steatosis (28). It has been reported that Src
activation played an important key role in FGFR1 kinase activation
(24,29). A.E. Medvedev and colleagues found
that c-Src kinase played a key role in LPS-dependent NF-κB
activation (30). Here, we
identified for the first time that TLR4 interacted with LPS to
facilitate c-Src/FGFR1 interactions, thereby activating downstream
NF-κB signalling and inflammatory responses (Fig. 4F).
In summary, we demonstrated that FGFR1 mediated
LPS-induced NF-κB activation, inflammatory responses and activation
of HSCs. LPS-induced FGFR1 activation appeared to require upstream
TLR4-Src-related mechanisms. Our data suggested that FGFR1
inhibition may be a feasible strategy for treating LPS-related
liver fibrosis.
Acknowledgements
Not applicable.
Funding
The project supported by research grants from the
Zhejiang Provincial Program of Chinese Medical and Health Science
Funding (grant no. 20172A141), Zhejiang Provincial Program of
Medical and Health Science Funding (grant no. 2017KY679), Zhuji
City Natural Science Funding and Zhejiang Pharmaceutical
Association Science Funding (grant no. 2016ZYY30).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
DL, JH, and LZ performed the experiments. YQ, JH, DL
and LJ designed the research study. YQ and LJ contributed essential
reagents or tools. HM, JX, JS, LJ and ZX analysed the data. LJ and
JH wrote the paper.
Ethics approval and consent to
participate
Animal care and experimental protocols were approved
by the Committee on Animal Care of Zhuji People's Hospital of
Zhejiang Province (Zhuji, China; approval no. zjdw2017-008).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Koyama Y and Brenner DA: Liver
inflammation and fibrosis. J Clin Invest. 127:55–64. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Schuppan D and Kim YO: Evolving therapies
for liver fibrosis. J Clin Invest. 123:1887–1901. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gu L, Tao X, Xu Y, Han X, Qi Y, Xu L, Yin
L and Peng J: Dioscin alleviates BDL- and DMN-induced hepatic
fibrosis via Sirt1/Nrf2-mediated inhibition of p38 MAPK pathway.
Toxicol Appl Pharmacol. 292:19–29. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Xu L, Yin L, Tao X, Qi Y, Han X, Xu Y,
Song S, Li L, Sun P and Peng J: Dioscin, a potent ITGA5 inhibitor,
reduces the synthesis of collagen against liver fibrosis: Insights
from SILAC-based proteomics analysis. Food Chem Toxicol.
107:318–328. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhang X, Han X, Yin L, Xu L, Qi Y, Xu Y,
Sun H, Lin Y, Liu K and Peng J: Potent effects of dioscin against
liver fibrosis. Sci Rep. 5:97132015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yin C, Evason KJ, Asahina K and Stainier
DY: Hepatic stellate cells in liver development, regeneration, and
cancer. J Clin Invest. 123:1902–1910. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yin L, Qi Y, Xu Y, Xu L, Han X, Tao X,
Song S and Peng J: Dioscin inhibits HSC-T6 cell migration via
adjusting SDC-4 expression: Insights from iTRAQ-based quantitative
proteomics. Front Pharmacol. 8:6652017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fouts DE, Torralba M, Nelson KE, Brenner
DA and Schnabl B: Bacterial translocation and changes in the
intestinal microbiome in mouse models of liver disease. J Hepatol.
56:1283–1292. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hedayat M, Netea MG and Rezaei N:
Targeting of Toll-like receptors: A decade of progress in combating
infectious diseases. Lancet Infect Dis. 11:702–712. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Oakley F, Meso M, Iredale JP, Green K,
Marek CJ, Zhou X, May MJ, Millward-Sadler H, Wright MC and Mann DA:
Inhibition of inhibitor of kappaB kinases stimulates hepatic
stellate cell apoptosis and accelerated recovery from rat liver
fibrosis. Gastroenterology. 128:108–120. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mohammadi M, Olsen SK and Ibrahimi OA:
Structural basis for fibroblast growth factor receptor activation.
Cytokine Growth Factor Rev. 16:107–137. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Fischer H, Taylor N, Allerstorfer S,
Grusch M, Sonvilla G, Holzmann K, Setinek U, Elbling L, Cantonati
H, Grasl-Kraupp B, et al: Fibroblast growth factor
receptor-mediated signals contribute to the malignant phenotype of
non-small cell lung cancer cells: Therapeutic implications and
synergism with epidermal growth factor receptor inhibition. Mol
Cancer Ther. 7:3408–3419. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Böhm F, Speicher T, Hellerbrand C, Dickson
C, Partanen JM, Ornitz DM and Werner S: FGF receptors 1 and 2
control chemically induced injury and compound detoxification in
regenerating livers of mice. Gastroenterology. 139:1385–1396. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lin N, Chen S, Pan W, Xu L, Hu K and Xu R:
NP603, a novel and potent inhibitor of FGFR1 tyrosine kinase,
inhibits hepatic stellate cell proliferation and ameliorates
hepatic fibrosis in rats. Am J Physiol Cell Physiol. 301:C469–C477.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Riehle KJ, Yeh MM, Yu JJ, Kenerson HL,
Harris WP, Park JO and Yeung RS: mTORC1 and FGFR1 signaling in
fibrolamellar hepatocellular carcinoma. Mod Pathol. 28:103–110.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mederacke I, Dapito DH, Affò S, Uchinami H
and Schwabe RF: High-yield and high-purity isolation of hepatic
stellate cells from normal and fibrotic mouse livers. Nat Protoc.
10:305–315. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tucker JA, Klein T, Breed J, Breeze AL,
Overman R, Phillips C and Norman RA: Structural insights into FGFR
kinase isoform selectivity: Diverse binding modes of AZD4547 and
ponatinib in complex with FGFR1 and FGFR4. Structure. 22:1764–1774.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liu M, Xu Y, Han X, Yin L, Xu L, Qi Y,
Zhao Y, Liu K and Peng J: Dioscin alleviates alcoholic liver
fibrosis by attenuating hepatic stellate cell activation via the
TLR4/MyD88/NF-κB signaling pathway. Sci Rep. 5:180382015.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Pinzani M and Marra F: Cytokine receptors
and signaling in hepatic stellate cells. Semin Liver Dis.
21:397–416. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ceccarelli S, Panera N, Mina M, Gnani D,
De Stefanis C, Crudele A, Rychlicki C, Petrini S, Bruscalupi G,
Agostinelli L, et al: LPS-induced TNF-α factor mediates
pro-inflammatory and pro-fibrogenic pattern in non-alcoholic fatty
liver disease. Oncotarget. 6:41434–41452. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Qi M, Yin L, Xu L, Tao X, Qi Y, Han X,
Wang C, Xu Y, Sun H, Liu K and Peng J: Dioscin alleviates
lipopolysaccharide-induced inflammatory kidney injury via the
microRNA let-7i/TLR4/MyD88 signaling pathway. Pharmacol Res.
111:509–522. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Qi M, Zheng L, Qi Y, Han X, Xu Y, Xu L,
Yin L, Wang C, Zhao Y, Sun H, et al: Dioscin attenuates renal
ischemia/reperfusion injury by inhibiting the TLR4/MyD88 signaling
pathway via up-regulation of HSP70. Pharmacol Res. 100:341–352.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shan X, Zhang Y, Chen H, Dong L, Wu B, Xu
T, Hu J, Liu Z, Wang W, Wu L, et al: Inhibition of epidermal growth
factor receptor attenuates LPS-induced inflammation and acute lung
injury in rats. Oncotarget. 8:26648–26661. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ren M, Qin H, Ren R, Tidwell J and Cowell
JK: Src activation plays an important key role in lymphomagenesis
induced by FGFR1 fusion kinases. Cancer Res. 71:7312–7322. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Xiao C and Ghosh S: NF-kappaB, an
evolutionarily conserved mediator of immune and inflammatory
responses. Adv Exp Med Biol. 560:41–45. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Paik YH, Schwabe RF, Bataller R, Russo MP,
Jobin C and Brenner DA: Toll-like receptor 4 mediates inflammatory
signaling by bacterial lipopolysaccharide in human hepatic stellate
cells. Hepatology. 37:1043–1055. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yao H, Hu C, Yin L, Tao X, Xu L, Qi Y, Han
X, Xu Y, Zhao Y, Wang C and Peng J: Dioscin reduces
lipopolysaccharide-induced inflammatory liver injury via regulating
TLR4/MyD88 signal pathway. Int Immunopharmacol. 36:132–141. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Park S, Choi Y, Um SJ, Yoon SK and Park T:
Oleuropein attenuates hepatic steatosis induced by high-fat diet in
mice. J Hepatol. 54:984–993. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zou L, Cao S, Kang N, Huebert RC and Shah
VH: Fibronectin induces endothelial cell migration through β1
integrin and Src-dependent phosphorylation of fibroblast growth
factor receptor-1 at tyrosines 653/654 and 766. J Biol Chem.
287:7190–7202. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Medvedev AE, Piao W, Shoenfelt J, Rhee SH,
Chen H, Basu S, Wahl LM, Fenton MJ and Vogel SN: Role of TLR4
tyrosine phosphorylation in signal transduction and endotoxin
tolerance. J Biol Chem. 282:16042–16053. 2007. View Article : Google Scholar : PubMed/NCBI
|