Introduction
Alzheimer's disease (AD) is a neurodegenerative
disorder that occurs mainly in old age; it is characterized by
deposits of amyloid-β (Aβ) plaques and neurofibrillary tangles, and
neuronal loss (1), and its
prevalence is rapidly increasing. It is likely that AD has multiple
etiologies, although its precise cause remains unknown (2). Aβ and tau proteins constitute a prime
neurotoxic component of senile plaques in the brain of AD patients,
thus contributing to learning and memory impairment due to synaptic
dysfunction and neuronal degeneration (3). However, to date most therapeutic
interventions aimed at modifying a single pathological factor
(e.g., cholinergic dysfunction, or Aβ aberrant processing) have
failed because they target only limited pathogenic factors of AD
(4). Inflammation, mitochondrial
dysfunction, and oxidative stress are considered the most prominent
concomitant pathological events (5–7), being
potential targets of therapeutic intervention. Recently, the
ER-associated degradation (ERAD) pathway has also been drawing
widespread attention as control of protein-folding intermediaries
in AD (8,9).
Inflammation has been proposed as a main factor in
the pathogenesis of AD, including microglial activation, reactive
astrocytes and inflammatory molecules (2,10–12).
However, it has been noted that microglial activation exhibits both
beneficial and detrimental effects depending on the stage of
microglia (13). The conversion of
microglia from detrimental (M1) to beneficial (M2) phenotype may
contribute to an anti-inflammatory microenvironment in the brain
(14). Similar to microglia,
astrocytes also contribute to neuroinflammation in AD by releasing
inflammatory cytokines and other toxic molecules (15). The ubiquitin-proteasome system and
autophagy mechanisms are impaired due to the toxic effects of Aβ
and oxidative stress damage, leading to the accumulation of
oxidized/unfolded proteins that may contribute to neuronal loss
(16). In fact, non-steroidal
anti-inflammatory drugs (NSAIDS) initially garnered
enthusiasm from pre-clinical and epidemiologic studies as agents
for reducing the risk of AD (17),
but anti-inflammatory treatment failed to produce beneficial
effects in patients with severe cognitive impairment and dementia
(18).
It has been reported that Rho activity, which is
thought to contribute to AD pathogenesis (19), was elevated in the brain of AD model
mice (20). Pharmacologic inhibition
of Rho kinase (ROCK) induced protein degradation by autophagy in
mammalian cells (21), and
suppressed Aβ production in an AD mouse model (22), highlighting ROCK as a therapeutic
target to combat Aβ production in AD. Fasudil, a selective ROCK
inhibitor, increased dendrite branching and stabilized dendrite
arbors in CA1 pyramidal neurons of APP/PS1 mice (23) by preventing neurodegeneration and
stimulating neuroregeneration in various neurological disorders
(19). Our previous study also
confirmed that Fasudil treatment ameliorated memory deficits in
APP/PS1 transgenic mice, accompanied by a decrease in Aβ deposits,
p-Tau and BACE levels, an increase in PSD-95, and inhibition of the
TLRS-NF-κB-MyD88 inflammatory cytokine axis (24).
Although previous studies have demonstrated certain
beneficial effects of Fasudil intervention in the AD model
(24), several lines of evidence
suggest that there are some limitations in the clinical use of
Fasudil, including its suitability only for short-course treatment,
low oral bioavailability, a narrow safety window and blood pressure
fluctuation. Thus, novel ROCK inhibitor(s) that are more efficient,
safer, oral and suitable for long-term use for the treatment of
neurodegenerative disorders are required. In the present study, we
explored the therapeutic effect and systemic response of a novel
ROCK inhibitor, FSD-C10, and possible mechanisms of its action in
the treatment of a mouse model of AD.
Materials and methods
Animals and treatment
Experiments were performed on male APP/PS1 double
transgenic mice (APPswe/PS1dE9, 8-month-old), purchased
from Shanghai Research Center. Age-matched wild-type (WT) mice were
used as controls. All animals were housed in a room maintained at
25°C with a 12-h light/dark cycle. The mice were given free access
to food and water except during the behavioral test. The experiment
was carried out in compliance with the Guidelines for Animal Care
and Use of China, and approved by the Animal Ethics Committee of
Shanxi Datong University, Datong, China (Ethical Approval no.
1601). Every effort was made to minimize suffering of the
animals.
The experimental design was carried out in two
stages: Validation of the AD model and intervention of the AD
model. For validation of the AD model, mice were divided into two
groups: Age-matched wild-type (n=8) and APP/PS1 transgenic mice
(n=8). Mice were sacrificed at 8 months of age. Behavioral and
pathological changes were measured before and after execution. For
the intervention of FSD-C10, mice were divided into two groups:
Normal saline (NS)-control mice (n=8) and FSD-C10-treated mice
(n=8). Mice were treated with FSD-C10 (25 mg/kg/day every other
day) or NS (0.9% NaCl) for 2 months by intraperitoneal (i.p.)
injection. The concentration of FSD-C10 used in this study was
adopted based on our preliminary experiments.
Mouse spatial learning and memory
test
Spatial learning and memory of mice were assessed
using the Morris water maze (MWM) test. The MWM is a 90 cm high, 50
cm diameter circular pool, containing a submerged escape platform
(5.0×5.0 cm), 2.0 cm below the water surface. The pool was filled
with water containing an edible white pigment that made the water
opaque, and the water temperature was maintained at 23–25°C. In
each trial, the time required to escape onto the hidden platform
was recorded as escape latency. Mice were allowed a maximum of 60
sec to reach the platform, and if they failed to do so, they were
guided to the platform. After training was completed, cognitive
function was measured for 5 days, after which the platform was
removed for spatial exploration in order to determine the memory
capacity of the mouse as to the platform space position. All
behavioral parameters of mice were tracked, recorded and analyzed
using SMART 3.0 (Panlab, Barcelona, Spain).
Histopathology and
immunohistochemistry
At the end of the final behavioral test, mice were
anesthetized and transcardially perfused with NS and 4%
paraformaldehyde in phosphate-buffered saline (PBS). Brains were
sliced (10 µm) and analyzed by immunohistochemistry. Briefly,
slides were blocked with 1% BSA (Sigma) for 1 h, and permeated with
0.3% Triton X-100 in 1% BSA/PBS for 30 min at RT. Sections were
incubated with anti-Aβ1–42 (1:1,000; EMD Millipore,
Billerica, MA, USA), anti-p-Tau (1:1,000; Cell Signaling
Technology, Inc., Danvers, MA, USA), anti-BACE (1:1,00; Cell
Signaling Technology, Inc.), anti-synaptophin (1:1,000),
anti-AMPAR-1 (1:1,000), anti-AMPAR-2 (1:1,000; all from Abcam,
Cambridge, MA, USA), anti-BDNF (1:1,000; Promega Corporation,
Madison, WI, USA), and anti-GDNF (1:1,000; Promega Corporation) at
4°C overnight. After washing, the slices were incubated with
secondary antibodies at RT for 2 h. The nucleus was stained by
Hoechst 33342 (1 µg/ml, Sigma-Aldrich; Merck KGaA, Darmstadt,
Germany). Control sections were run following identical protocols,
but the primary antibodies were omitted. Results were visualized
under fluorescence microscopy by IMAGE-PRO PLUS software (Media
Cybernetics, Inc., Rockville, MD, USA). Quantification analysis of
positive cells was performed on three sections per animal, and four
mice per group were analyzed in a blinded fashion.
Western blot analysis
Proteins of mouse brain were extracted as previously
described (17), Protein
concentrations were determined by a bicinchoninic acid (BCA) kit
(Beyotime Institute of Biotechnology, Haimen, China). Protein
samples were separated by SDS-PAGE and transferred onto PVDF
membranes. After nonspecific binding was blocked with 5% nonfat dry
milk, membranes were incubated at 4°C overnight with primary
antibodies (Aβ1–42, p-Tau, BACE, PSD-95, AMPAR-1,
AMPAR-2, BDNF, GDNF and β-actin). The next day, the membranes were
washed three to four times with 0.1% Tween-20/TBST (pH 7.6) and
incubated with horseradish peroxidase-conjugated anti-rabbit IgG or
anti-mouse IgG secondary antibodies for 2 h at 37°C. To compare
protein loading, antibodies directed against GAPDH or β-actin were
used and relative optical density was measured with Image Lab
Software (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Statistical analysis
Graph Pad Prism 5 (Cabit Information Technology Co.,
Ltd., Shanghai, China) was used for statistical analysis. Results
are presented as mean ± SEM. An unpaired, two-tailed Student's
t-test was used for comparisons of means between two groups. In all
tests, P<0.05 was considered to indicate a statistically
significant difference.
Results
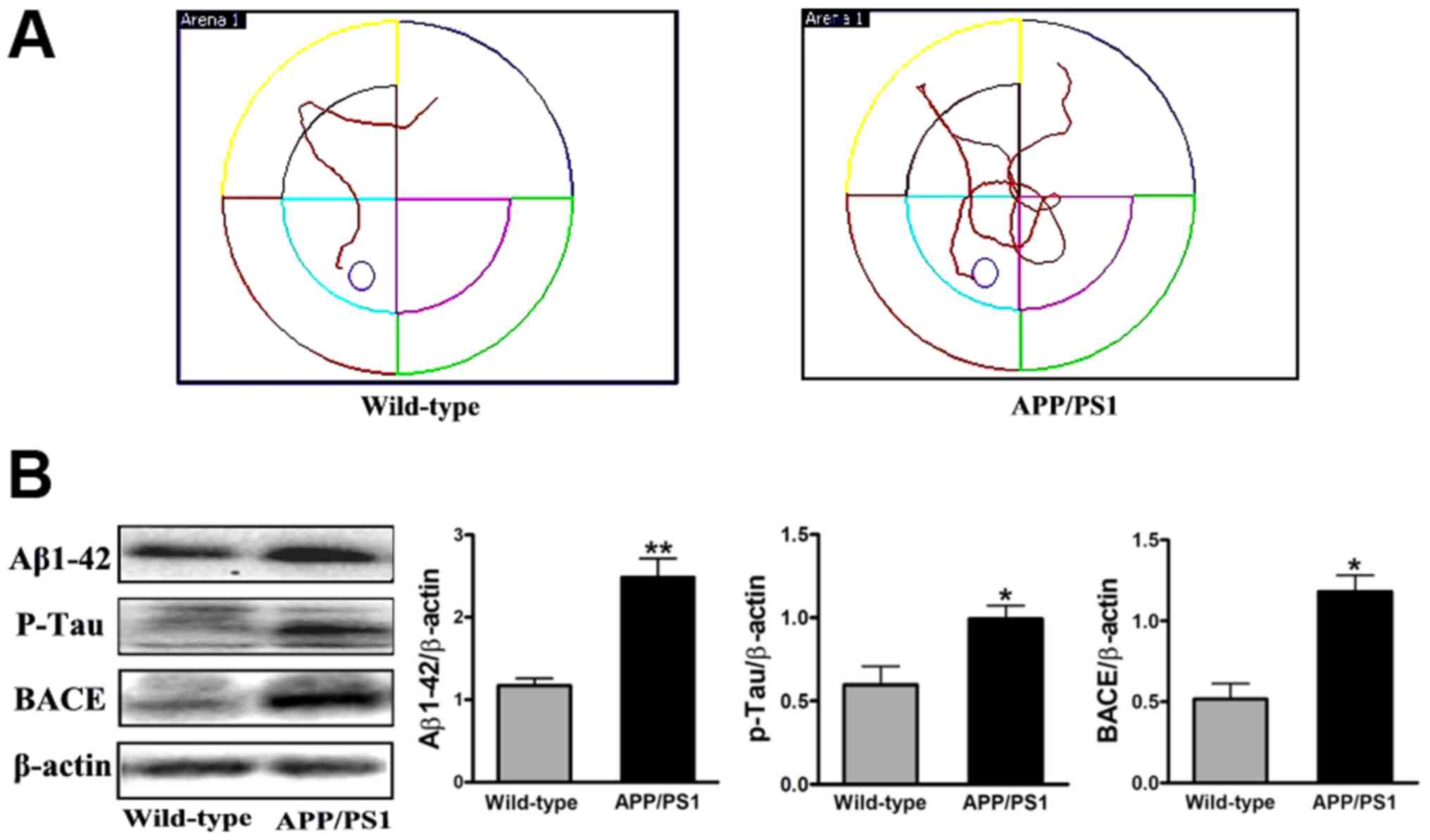
Behavioral and pathological changes in
APP/PS1 mice at 8 months of age
To observe the effect of FSD-C10 in APP/PS1
transgenic mice, we first tested the behavioral and pathological
changes at the initiation time of drug intervention as a baseline.
Memory and learning ability was measured by MWM and the expression
of Aβ1–42, p-Tau and BACE protein was determined by
western blot. As shown in Fig. 1,
APP/PS1 transgenic mice exhibited obvious space learning and memory
impairment at 8 months of age, as compared with the WT group.
Simultaneously, the levels of Aβ1–42, p-Tau and BACE
expression were more elevated in APP/PS1 transgenic mice than in
the WT group (P<0.01, P<0.05 and P<0.05, respectively;
Fig. 1B). Together, APP/PS1 mice
showed the typical behavior dysfunction and typical pathologic
abnormality of AD, thus confirming the AD model in these transgenic
mice.
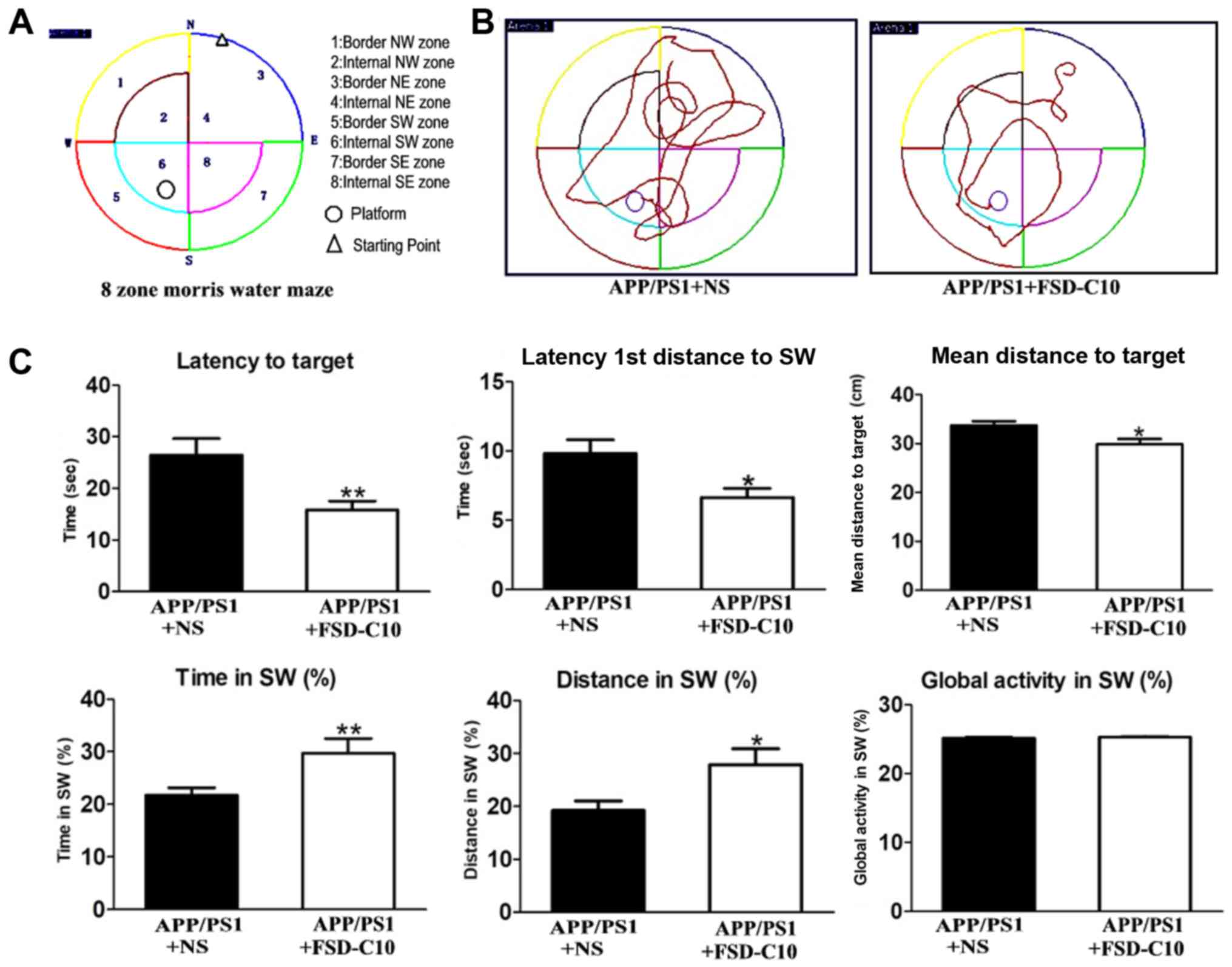
FSD-C10 improves learning and memory
abilities of the APP/PS1 Tg mice
As shown in Fig. 1,
APP/PS1 transgenic mice had obvious space learning and memory
impairment at 8 months of age compared with the WT group. To
observe whether FSD-C10 can ameliorate the deficit in learning and
memory ability, MWM measurement was used in APP/PS1 mice after 2
months of treatment (Fig. 2A and B).
As shown in Fig. 2C, FSD-C10
intervention significantly reduced the time and distance for
Latency to Target (P<0.01), Latency 1st Entrance to SW
(P<0.05), and mean distances to Target (P<0.05) of these AD
mice.
When the platform was moved, a consolidation of
spatial memory was detected. The results showed that the movement
of FSD-C10-treated mice mainly located on the position of the
target platform quadrant, while NS-controlled mice mainly moved
around the location of the target platform quadrant (Fig. 2A and B). The intervention of FSD-C10
increased Time in SW (P<0.01; Fig.
2C) and Distance in SW (P<0.05; Fig. 2C) as compared with the NS control
group. However, there were no differences in global activity
between two groups (P>0.05; Fig.
2C).
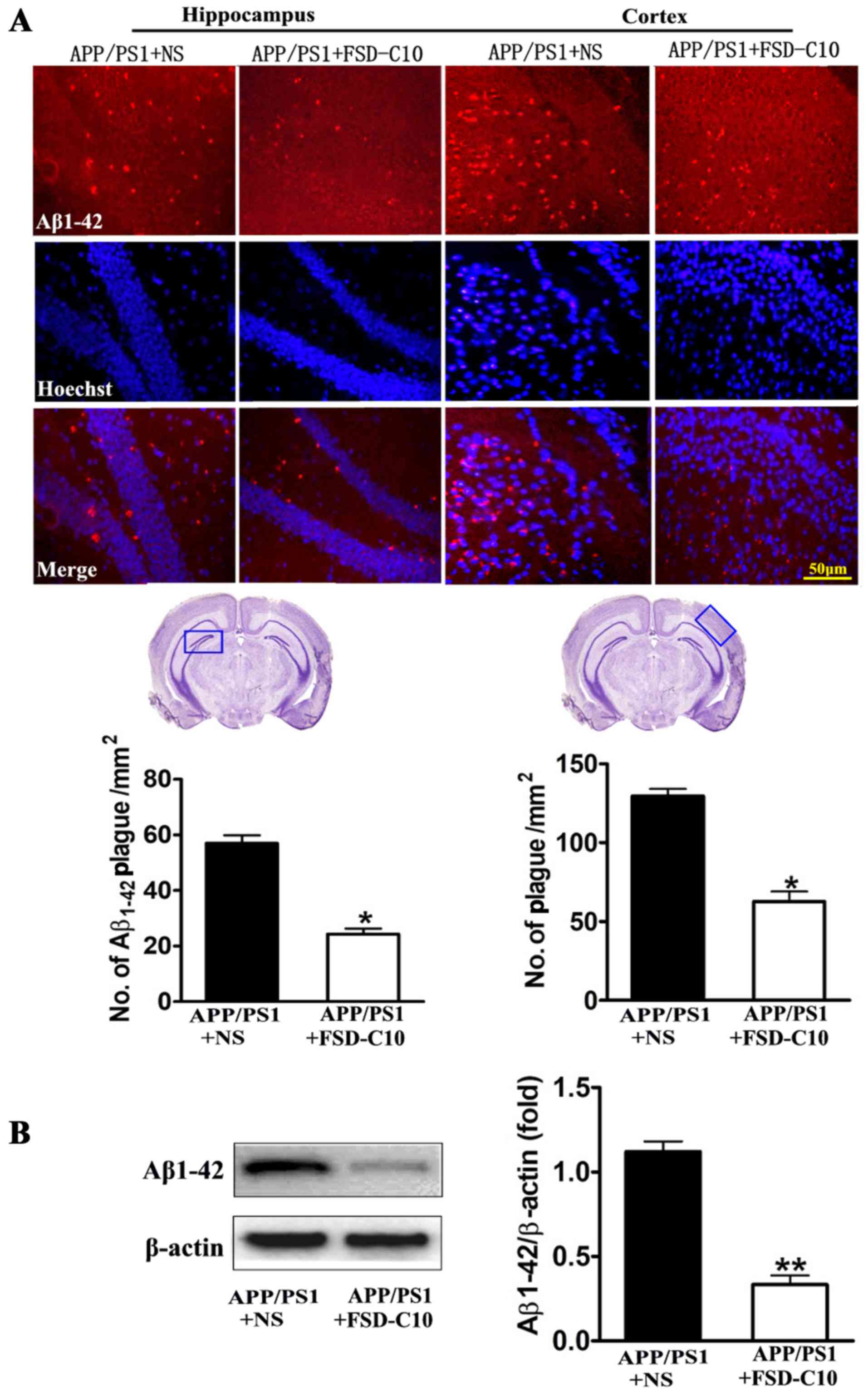
FSD-C10 attenuates Aβ burden, Tau
phosphorylation and BACE expression
We evaluated the pathological changes in APP/PS1
transgenic mice, including Aβ plaques, Tau phosphorylation and BACE
expression by immunohistochemistry and western blot. As shown in
Fig. 3A, the numbers of
Aβ1–42-positive cells in the hippocampus and cortex of
brain were lower in FSD-C10-treated mice than in NS-treated control
mice. Similarly, FSD-C10 intervention suppressed Aβ1–42
protein level in brain when compared with the NS-treated control
group (P<0.01; Fig. 3B).
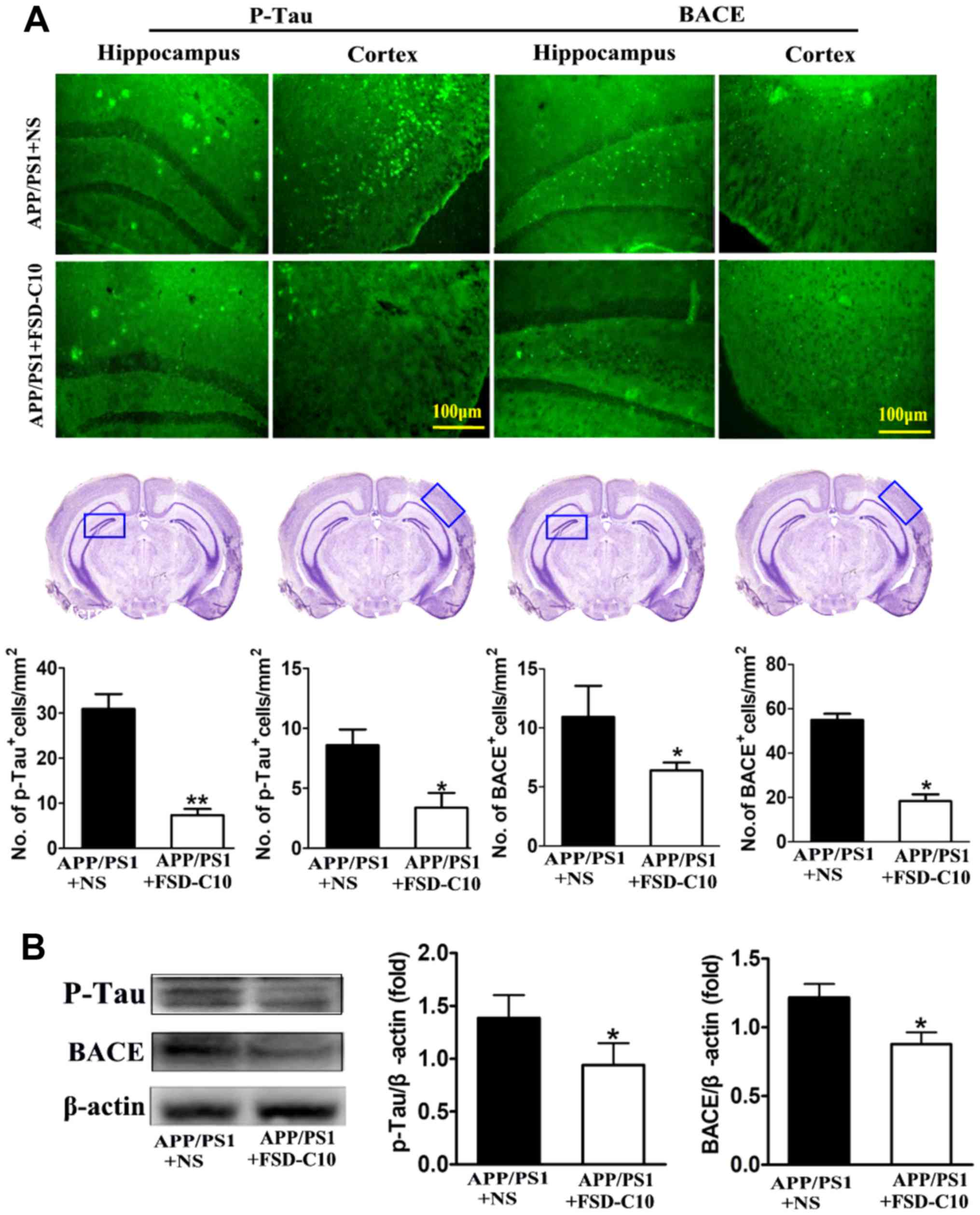
We further explored the levels of p-tau and BACE
proteins in the hippocampus and cortex with immunohistochemistry
and western blot. The results showed that the numbers of p-Tau and
BACE positive cells were significantly lower in FSD-C10-treated
group than in the NS-treated control group (P<0.05 and P<0.01
for p-Tau; P<0.05 and P<0.05 for BACE; Fig. 4A). Similarly, FSD-C10 treatment
inhibited the expression of p-Tau and BACE protein in brain when
compared with the NS-treated control mice (P<0.05; Fig. 4B). These results suggest that FSD-C10
treatment effectively alleviated pathological changes in AD
mice.
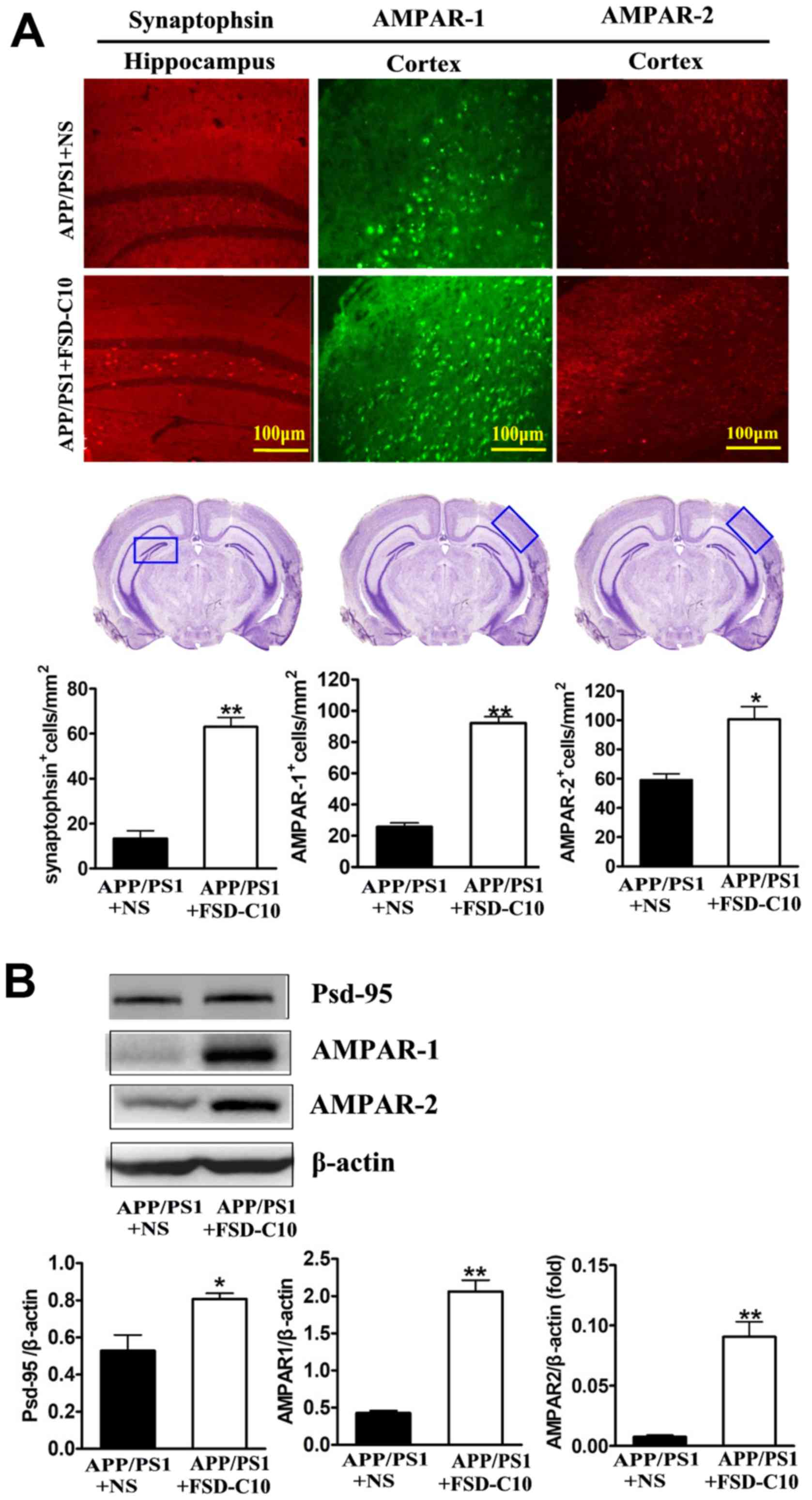
FSD-C10 induces the expression of
synapse-associated proteins
It has become clear that cognitive dysfunction more
strongly correlates with synapse loss in AD than with counts of
plaques, tangles, and neuronal loss (25,26). Our
results showed that FSD-C10 treatment up-regulated synaptophsin
expression in the hippocampus (P<0.01; Fig. 5A) as well as AMPAR-1 and AMPAR-2
expression in the cortex (P<0.01 and P<0.05, respectively;
Fig. 5A) as compared with NS-treated
control mice. Data from western blot also confirmed that FSD-C10
treatment up-regulated the expression of AMPAR-1 and AMPAR-2
protein in brain (both P<0.01; Fig.
5B).
PSD-95 is a synaptic protein that regulates synaptic
distribution, synaptic stability and certain types of memory
(27). As shown in Fig. 5B, FSD-C10 treatment elevated the
level of PSD-95 expression in brain as compared with the NS-treated
control group (P<0.05; Fig. 5B).
These results indicate that the improvement in learning and memory
impairment mediated by FSD-C10 may be related to the up-regulation
of these synapse-associated proteins in both hippocampus and
cortex.
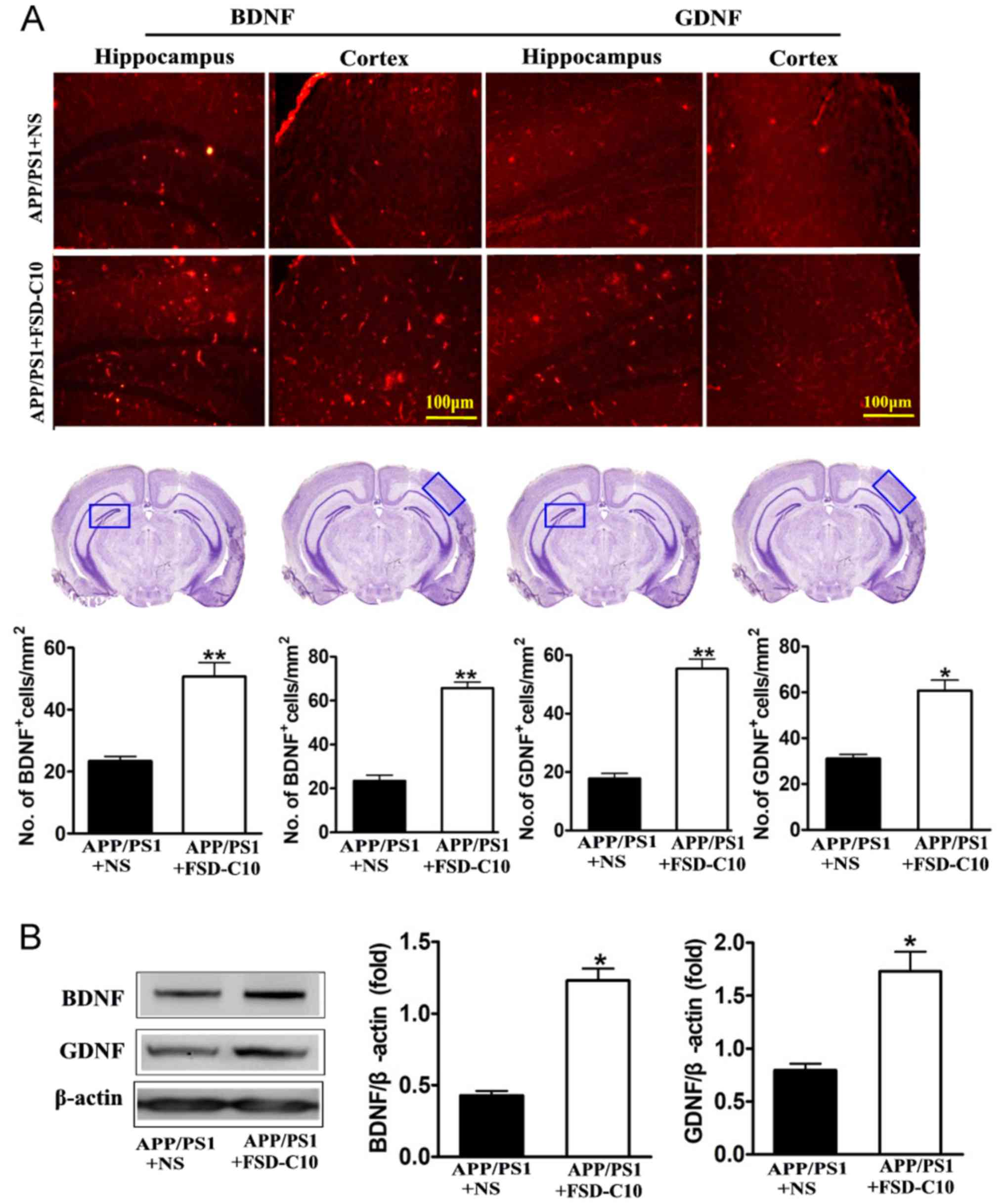
FSD-C10 increases the expression of
neurotrophic factors
Neurotrophic factors are well known to be important
for the survival, differentiation, growth and regeneration of
neurons (28), as well as for
synaptic transmission and plasticity (29). In this research, we explored the role
of FSD-C10 in production of neurotrophic factors.
Immunohistochemical analysis revealed that FSD-C10 intervention
increased the numbers of both BDNF and GDNF positive cells in the
hippocampus and cortex of brain compared with the NS-treated
control group (P<0.01 and P<0.05 respectively; Fig. 6A). Expression of BDNF and GDNF
protein was also higher in FSD-C10-treated mice than in the
NS-treated control group measured by western blot (both P<0.05;
Fig. 6B). FSD-C10 thus has a
potentially neuroprotective effect through the production of
neurotrophic factors.
Discussion
AD is an age-related and chronic neurodegenerative
disorder presenting as progressive cognitive decline. Although the
exact pathogenesis is not yet clear, multiple etiologic pathways
have been considered (2). The major
histopathological hallmarks of AD include Aβ plaques and
neurofibrillary tangles (NFTs) formed by hyperphosphorylated Tau
protein (30), and the neurite
atrophy and synaptic loss induced by Aβ are considered to be the
major cause of gradual cognitive detetrioration in AD (31). Neurotoxicity of Aβ becomes apparent
via induction of oxidative stress, neuronal excitotoxicity,
neuroinflammation and apoptosis (32,33). One
of the most promising strategies for treatment of AD is to directly
target Aβ by decreasing the production and clearing aggregation of
Aβ (34). In addition to Aβ
accumulation, tau hyperphosphorylation is also an important
pathological hallmark of AD (35).
Abnormal phosphorylation of tau protein promotes the loss of
microtubule stabilizing ability and may contribute to neurite
degeneration as well as NFT formation (36). Current drugs for AD treatment, such
as acetylcholinesterase inhibitor and NMDA antagonist, show limited
benefits in most AD patients (37).
There is thus an urgent need for novel therapeutic strategies that
can halt the disease process and are suitable for long-term
clinical management. In our previous studies, a comparative study
of Fasudil and FSD-C10 has been reported, including cell viability,
cell death, neurite outgrowth and dendritic formation, vasodilation
insensitivity and animal mortality (38). In this study, we demonstrate that
treatment with FSD-C10, as a new ROCK inhibitor, was able to
reverse cognitive impairment, possibly through decreasing Aβ
accumulation and Tau phosphorylation in the cortex and hippocampus
of APP/PS1 transgenic mice.
A series of studies have demonstrated that ROCK was
elevated in the brain of AD patients compared to controls (39,40), and
inhibition of ROCK activity decreased Aβ levels by enhancing APP
protein degradation (41). Abnormal
activation of ROCK has also been found in AD experimental models,
and may be involved in the occurrence and development of diseases
(41). ROCK activation increased Aβ
production, CRMP-2 phosphorylation and hindered tubulin assembly,
leading to the inhibition of synapses (20,42).
Inhibition of ROCK reduced the production of Aβ (41) and increased the synaptic density and
length of hippocampal pyramidal neurons (43). Fasudil has been proved to protect
against nerve degeneration induced by Aβ, and to improve spatial
memory and learning ability in AD rats (44). Our present study provides further
evidence that inhibiting ROCK activity can improve cognitive
deficits in APP/PS1 transgenic mice.
In clinical practice, Fasudil has several
limitations, including a relatively narrow safety window, poor oral
bioavailability, and unsuitability for long-term use. Researchers
are looking for novel ROCK inhibitors that are more efficient and
safer, can be taken orally and over a long period of time for the
treatment of neurodegenerative disorders. We have in previous
studies found that FSD-C10, as a novel ROCK inhibitor, has the same
therapeutic effect, but is safer than Fasudil (38). In EAE, FSD-C10 ameliorated the
clinical severity of disease, accompanied by improvement in
demyelination and the inhibition of inflammatory cells in the CNS
of EAE mice, clearly showing a therapeutic effect (45).
Glutamate, the major excitatory neurotransmitter in
the CNS, is involved in synaptic transmission, neuronal growth and
differentiation, synaptic plasticity and learning and memory
(46). When a neuron is depolarized,
glutamate is released into the synaptic cleft where it binds
glutamate receptors (47). Glutamate
receptors, such as NMDA receptor and AMPA receptor, are involved in
rapid excitatory synaptic transmission and the release of
neurotransmitters, which is closely related with learning and
memory (48). In the postsynaptic
membrane, an AMPAR insert can induce and promote learning and
memory behavior. Aβ-induced decline in AMPAR number and synaptic
function through endocytosis is a plausible mechanism for the
cognitive impairment that occurs in the very early stages of AD
(49). Further study found that lack
of AMPAR can cause dendritic spine reduction and loss of NMPAR
(50), both of which are related to
cognitive impairment. In our study, increased expression of AMPAR,
synaptophsin and PSD-95 protein after FSD-C10 administration could
be an important mechanism for the improvement in cognitive function
in transgenic APP/PS1 mice. PSD-95 organizes synaptic proteins to
mediate the functional and structural plasticity of the excitatory
synapse and to maintain synaptic homeostasis (51). The stabilization of a new synaptic
protrusion is associated with activity-driven PSD proteinaceous
network formation. In this proteinaceous network, PSD-95 is
believed to play a role in synapse maturation, given that it is
particularly vulnerable to the toxic effects of Aβ (28). Synaptophysin is the major integral
membrane protein of presynaptic vesicles required for vesicle
formation and exocytosis (52).
Taken together, FSD-C10 may maintain the normal function of the
synapse, possibly through promoting the expression of these
synaptic related proteins.
Neurotrophic factors play an essential role in the
survival of neurons affected by degenerative processes (53,54).
Increased levels of BDNF are associated with improved learning and
memory, and a reduction in BDNF leads to age-related memory
deficits (55). The potential of
GDNF against age-related cognitive deterioration has not been fully
explored. It was reported that serum GDNF levels were significantly
reduced in AD patients (56,57), and expression of GDNF transgene in
astrocytes improved cognitive deficits in aged rats (58). Thus, increased BDNF, GDNF, AMPAR,
PSD-95, or synaptophysin levels may reflect increased synaptic
density, activity, and vesicles, revealing improved functioning of
synapses. Inducing expression of these molecules could therefore be
a mechanism underlying improved learning and memory abilities of
the APP/PS1 transgenic mice after FSD-C10 treatment. The
limitations of this study may include that further mechanisms
underlying the inhibitory effect on Aβ after blocking ROCK activity
in AD remain unknown, and the oral effect of this reagent in AD
mice needs be tested. These questions will be further explored in
the near future.
Acknowledgements
The authors would like to thank Ms. Katherine Regan
(Department of Neurology, Thomas Jefferson University) for
editorial assistance.
Funding
The present study was supported by grants from The
National Natural Science Foundation of China (grant nos. 81272163,
81471412 and 81371414), Shanxi University of Traditional Chinese
Medicine (grant no. 2011PY-1), research projects supported by
Datong Municipal Science and Technology Bureau (grant no. 2017136)
and Shanxi Datong University (grant no. 2016K10), and an open
project of The State Key Laboratory of Molecular Developmental
Biology of China (grant no. 2018-MDB-KF-07 to YQY).
Availability of data and materials
The analysed data sets generated during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
QFG and JZY designed the study, performed the mouse
behavioral tests, participated in the statistical analysis and
revised the manuscripts. CGM conceived the study, participated in
its design and coordination, and helped to draft the manuscript.
BGX designed and performed all analyses, and drafted and revised
the manuscript. GXZ participated in the study design and revised
the manuscript. YHL performed the immunoassays. CYL performed the
western blot analysis. HW and LF performed the statistical analysis
and the immunohistochemistry. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
The experiment was carried out in compliance with
the Guidelines for Animal Care and Use of China, and approved by
the Animal Ethics Committee of Shanxi Datong University, Datong,
China (Ethical Approval no. 1601).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Goedert M and Spillantini MG: A century of
Alzheimer's disease. Science. 314:777–781. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Khalil Bou R, Khoury E and Koussa S:
Linking multiple pathogenic pathways in Alzheimer's disease. World
J Psychiatry. 6:208–214. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ferrer I: Defining Alzheimer as a common
age-related neurodegenerative process not inevitably leading to
dementia. Prog Neurobiol. 97:38–51. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Scheltens P, Blennow K, Breteler MM, de
Strooper B, Frisoni GB, Salloway S and Van der Flier WM:
Alzheimer'sdisease. Lancet. 388:505–517. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Jiang T, Sun Q and Chen S: Oxidative
stress: A major pathogenesis and potential therapeutic target of
antioxidative agents in Parkinson's diseaseand Alzheimer's disease.
Prog Neurobiol. 147:1–19. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Onyango IG, Dennis J and Khan SM:
Mitochondrial dysfunction in alzheimer's disease and the rationale
for bioenergetics based therapies. Aging Dis. 7:201–214. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ransohoff RM: How neuroinflammation
contributes to neurodegeneration. Science. 353:777–783. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cai Y, Arikkath J, Yang L, Guo ML,
Periyasamy P and Buch S: Interplay of endoplasmic reticulum stress
and autophagy in neurodegenerative disorders. Autophagy.
12:225–244. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Duran-Aniotz C, Cornejo VH and Hetz C:
Targeting endoplasmic reticulum acetylation to restore proteostasis
in Alzheimer's disease. Brain. 139:650–652. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hoeijimakers L, Heinen Y, van Dam AM,
Lucassen PJ and Korasi A: Microglial priming and Alzheimer's
disease: A possible Role for (Early) immune challenges and
epigenetics? Front Hum Neurosci. 10:3982016.PubMed/NCBI
|
|
11
|
Olsen I and Singhrao SK: Inflammation
involvement in alzheimer's disease. J Alzheimer's Dis. 54:45–53.
2016. View Article : Google Scholar
|
|
12
|
Torika N, Asraf K, Roasso E, Danon A and
Fleisher-Berkovich S: Angiotensin converting enzyme inhibitors
ameliorate braininflammation associated with microglial activation:
Possible implication for Alzheimer's disease. J Neueoimmune
Pharmacol. 11:774–785. 2016. View Article : Google Scholar
|
|
13
|
Wallker DG and Lue LF: Immune phenotypes
of microglial in humane neurodegenerativedisease: Challenges to
detecting microglial polarization in human brains. Alzheimers Res
Ther. 7:562015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Heneka MT, Kummer MP, Stutz A, Delekate A,
Schwartz S, Vieira-Saecker A, Gripe A, Axt D, Remus A, Remus A, et
al: NLRP3 is actived in Alzheimer's disease and contributes to
pathology in APP/PS1mice. Nature. 493:674–678. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Glass CK, Saijo K, Winner B, Marchetto MC
and Gage FH: Mechanisms underlying inflammation in
neurodegeneration. Cell. 140:918–934. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Trammtola A, Di Domenico F, Barone E,
Perluigi M and Butterfield DA: It is all about (U)biquitin: role of
altered ubiquitin-proteasome system and UCHLI in Alzheimer's
Disease. Oxid Med Cell Longev 2756068. 2016. View Article : Google Scholar
|
|
17
|
Deardorff WJ and Grossberg GT: Targeting
neuroinflammation in Alzheimer's disease: Evidence for NASIDs and
novel therapeutics. Expert Rev Neurother. 17:17–32. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Miguel-Álvarez M, Santos-Lozano A,
Sanchis-Gomar F, Fiuza-Luces C, Pareja-Galeano H, Garatachea N and
Lucia A: Non-steroidal anti-inflammmatory drugs as a treatment for
Alzheimer's disease: A systematic review and meta-analysis of
treatment effect. Drugs Aging. 32:139–147. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Mueller BK, Mack H and Teusch N: Rho
kinase, a promising drug target for neurogical disorders. Nat Rev
Drug Discov. 4:387–398. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Petraos S, Li QX, George AJ, Hou X, Kerr
ML, Unabia SE, Hatzinisiriou I, Maksel D, Aguilar MI and Small DH:
The beta-amyloid protein of Alzheimer's disease increase neuronal
CRMP-2 phosphorylation by a Rho-GTP mechanism. Brain. 131:90–108.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bauer PO, Wong HK, Oyama F, Goswami A,
Okuno M, Kino Y, Miyazaki H and Nukina N: Inhbition of Rho kinases
enhances the degradation of mutant huntingtin. J Boiol Chem.
284:13153–13164. 2009. View Article : Google Scholar
|
|
22
|
Herskowitz JH, Feng Y, Mattheyses AL,
Hales CM, Higginbotham LA, Duong DM, Montine TJ, Troncoso JC,
Thambisetty M, Seyfried NT, et al: Pharmacologic inhibition of
ROCK2 suppresses amyloid-β production in an Alzheimer's disease
mouse model. J Neurosci. 33:19086–19098. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Couch BA, DeMarco GJ, Gourley SL and
Koleske AJ: Increased dendrite branching in AbetaPP/PS1 mice and
elongation of dendrite arbors by fasudil administration. J
Alzheimers Dis. 20:1003–1008. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yu JZ, Li YH, Liu CY, Wang Q, Gu QF, Wang
HQ, Zhang GX, Xiao BG and Ma CG: Multitarget therapeutic effect of
fasudil in APP/PS1transgenic mice. CNS Neurol Disord Drug Targets.
16:199–209. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hong S, Beja-Glasser VF, Nfonoyim BM,
Frouin A, Li S, Ramakrishnan S, Merry KM, Shi Q, Rosenthal A,
Barres BA, et al: Complement and microglia mediate early synapse
loss in Alzheimer mouse models. Science. 352:712–716. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Terry RD, Masliah E, Salmon DP, Butters N,
DeTeresa R, Hill R, Hansen LA and Katzman R: Physical basis of
cognitive alterations in Alzheimer's disease: Synapse loss is the
major correlate of cognitive impairment. Ann Neurol. 30:572–580.
1991. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Almeida CG, Tampellini D, Takahashi RH,
Greengard P, Lin MT, Snyder EM and Gouras GK: Beta-amyloid
accumulation in APP mutant neurons reduces PSD-95 and GluR1 in
synapses. Neurobiol Dis. 20:187–198. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Huang EJ and Reichardt LF: Neurotrophins:
Roles in neuronal development and function. Annu Rev Neurosci.
24:677–736. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Waterhouse EG and Xu B: New insights into
the role of brain-derived neurotrophic factor in synaptic
plasticity. Mol Cell Neurosci. 42:81–89. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Prvulovic D and Hampel H: Amyloid β (Aβ)
and phospho-tau (p-tau) as diagnostic biomarkers in Alzheimer's
disease. Clin Chem Lab Med. 49:367–374. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tohda C, Ichimura M, Bai Y, Tanaka K, Zhu
S and Komatsu K: Inhibitory effects of Eleutherococcus senticosus
extracts on amyloid beta(25–35)-induced neuritic atrophy and
synaptic loss. J Pharmacol Sci. 107:329–339. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhang ZH, Yu LJ, Hui XC, Wu ZZ, Yin KL,
Yang H and Xu Y: Hydroxy-safflor yellow A attenuates
Aβ1–42-induced inflammation by modulating the
JAK2/STAT3/NF-κB pathway. Brain Res. 1563:72–80. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Morroni F, Sita G, Tarozzi A, Rimondini R
and Hrelia P: Early effects of Aβ1–42 oligomers injection in mice:
Involvement of PI3K/Akt/GSK3 and MAPK/ERK1/2 pathways. Behav Brain
Res. 314:106–115. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Sevigny J, Chiao P, Bussière T, Weinreb
PH, Williams L, Maier M, Dunstan R, Salloway S, Chen T, Ling Y, et
al: The antibody aducanumab reduces Aβ plaques in Alzheimer's
disease. Nature. 537:50–56. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Mateo I, Vázquez-Higuera JL, Sánchez-Juan
P, Rodríguez-Rodríguez E, Infante J, García-Gorostiaga I, Berciano
J and Combarros O: Epistasis between tau phosphorylation regulating
genes (CDK5R1 and GSK-3beta) and Alzheimer's disease risk. Acta
Neurol Scand. 120:130–133. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Goedert M: Neurofibrillary pathology of
Alzheimer's disease and other tauopathies. Prog Brain Res.
117:287–306. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Cummings J, Aisen PS, DuBois B, Frölich L,
Jack CR Jr, Jones RW, Morris JC, Raskin J, Dowsett SA and Scheltens
P: Drug development in Alzheimer's disease: The path to 2025.
Alzheimers Res Ther. 8:392016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Xin YL, Yu JZ, Yang XW, Liu CY, Li YH,
Feng L, Chai Z, Yang WF, Wang Q, Jiang WJ, et al: FSD-C10: A more
promising novel ROCK inhibitor than Fasudil for treatment of CNS
autoimmunity. Biosci Rep. 35:pii: e00247. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Salminen A, Suuronen T and Kaarniranta K:
ROCK, PAK, and Toll of synapses in Alzheimer's disease. Biochem
Biophys Res Commun. 371:587–590. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Bobo-Jiménez V, Delgado-Esteban M,
Angibaud J, Sánchez-Morán I, de la Fuente A, Yajeya J, Nägerl UV,
Castillo J, Bolaños JP and Almeida A: APC/CCdh1-Rock2
pathway controls dendritic integrity and memory. Proc Natl Acad Sci
USA. 114:4513–4518. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Henderson BW, Gentry EG, Rush T, Troncoso
JC, Thambisetty M, Montine TJ and Herskowitz JH: Rho-associated
protein kinase 1 (ROCK1) is increased in Alzheimer's disease and
ROCK1 depletion reduces amyloid-β levels in brain. J Neurochem.
138:525–531. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Arimura N, Ménager C, Kawano Y, Yoshimura
T, Kawabata S, Hattori A, Fukata Y, Amano M, Goshima Y, Inagaki M,
et al: Phosphorylation by Rho kinase regulates CRMP-2 activity in
growth cones. Mol Cell Biol. 25:9973–9984. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Swanger SA, Mattheyses AL, Gentry EG and
Herskowitz JH: ROCK1 and ROCK2 inhibition alters dendritic spine
morphology in hippocampal neurons. Cell Logist. 5:e11332662016.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Song Y, Chen X, Wang LY, Gao W and Zhu MJ:
Rho kinase inhibitor fasudil protects against β-amyloid-induced
hippocampal neurodegeneration in rats. CNS Neurosci Ther.
19:603–610. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Li YH, Yu JZ, Liu CY, Zhang H, Zhang HF,
Yang WF, Li JL, Feng QJ, Feng L, Zhang GX, et al: Intranasal
delivery of FSD-C10, a novel Rho kinase inhibitor, exhibits
therapeutic potential in experimental autoimmune encephalomyelitis.
Immunology. 143:219–229. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Revett TJ, Baker GB, Jhamandas J and Kar
S: Glutamate system, amyloid ß peptides and tau protein: Functional
interrelationships and relevance to Alzheimer disease pathology. J
Psychiatry Neurosci. 38:6–23. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Masoudian N, Riazi GH, Afrasiabi A,
Modaresi SM, Dadras A, Rafiei S, Yazdankhah M, Lyaghi A, Jarah M,
Ahmadian S and Seidkhani H: Variations of glutamate concentration
within synaptic cleft in the presence of electromagnetic fields: An
artificial neural networks study. Neurochem Res. 40:629–642. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Anggono V, Tsai LH and Götz J: Glutamate
receptors in Alzheimer's disease: Mechanisms and therapies. Neural
Plast. 2016:82561962016. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Dong Z, Han H, Li H, Bai Y, Wang W, Tu M,
Peng Y, Zhou L, He W, Wu X, et al: Long-term potentiation decay and
memory loss are mediated by AMPAR endocytosis. J Clin Invest.
125:234–247. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Hsieh H, Boehm J, Sato C, Iwatsubo T,
Tomita T, Sisodia S and Malinow R: AMPAR removal underlies
Abeta-induced synaptic depression and dendritic spine loss. Neuron.
52:831–843. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Keith D and El-Husseini A: Excitation
control: Balancing PSD-95 function at the synapse. Front Mol
Neurosci. 1:42008. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Valtorta F, Pennuto M, Bonanomi D and
Benfenati F: Synaptophysin: Leading actor or walk-on role in
synaptic vesicle exocytosis? Bioessays. 26:445–453. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Poo MM: Neurotrophins as synaptic
modulators. Nat Rev Neurosci. 2:24–32. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Lessmann V, Gottmann K and Malcangio M:
Neurotrophin secretion: Current facts and future prospects. Prog
Neurobiol. 69:341–374. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Bimonte HA, Nelson ME and Granholm AC:
Age-related deficits as working memory load increases:
Relationships with growth factors. Neurobiol Aging. 24:37–48. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Straten G, Eschweiler GW, Maetzler W,
Laske C and Leyhe T: Glial cell-line derived neurotrophic factor
(GDNF) concentrations in cerebrospinal fluid and serum of patients
with early Alzheimer's disease and normal controls. J Alzheimers
Dis. 18:331–337. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Forlenza OV, Miranda AS, Guimar I, Talib
LL, Diniz BS, Gattaz WF and Teixeira AL: Decreased neurotrophic
support is associated with cognitive decline in non-demented
subjects. J Alzheimers Dis. 46:423–429. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Pertusa M, García-Matas S, Mammeri H,
Adell A, Rodrigo T, Mallet J, Cristòfol R, Sarkis C and Sanfeliu C:
Expression of GDNF transgene in astrocytes improves cognitive
deficits in aged rats. Neurobiol Aging. 29:1366–1379. 2008.
View Article : Google Scholar : PubMed/NCBI
|