Introduction
Atherosclerotic plaque rupture or erosion is the
main cause of arterial thrombosis, which leads to a setting of
acute ischemic cardiovascular disease, i.e., acute coronary
syndrome (ACS) (1). The complex
mechanisms involved in lesion progression and instability include
the recruitment and activation of monocytes, macrophages and other
inflammatory cells, neovascularization from the vasa vasorum,
perivascular inflammation, and importantly, the synthesis of
proatherogenic cytokines, including tissue factor, which renders
the lesion more thrombogenic, and proteolytic enzymes, including
matrix metalloproteinase (MMP)-2 and −9 (2). MMPs can promote dispersion of the
fibrous lesion via the degradation of extracellular matrix (ECM)
proteins, including collagens, proteoglycans, fibronectin and
elastin. Multiple enzymes have been shown to be overexpressed in
the shoulder regions of atherosclerotic plaques and to contribute
to plaque destabilization, particularly by degrading the fibrous
cap (3). Vascular smooth muscle
cells (VSMCs) are the major cellular component of the vessel wall
and are key in vascular function. Plaque stability largely depends
on VSMC function, as these cells have a high capacity to secrete
MMPs, which regulate the balance between ECM synthesis and
degradation (4–8).
Adipose tissue, as an endocrine organ that produces
and secretes bioactive adipokines with pro- and anti-inflammatory
properties, can contribute to the inflammatory activation in acute
coronary syndrome (9). The increased
expression and secretion of resistin in epicardial adipose tissue
of patients is associated with ACS (10). Leptin is an adipocyte-derived
hormone, which was identified in 1994, and its main function is to
reflect the body's fat stores and act in maintaining energy
homeostasis. The expression of leptin is also an established
independent cardiovascular risk factor, which is important in
associated diseases, including coronary atherosclerosis (11–14).
In neurologically symptomatic patients, leptin has
been reported to be locally synthesized in carotid atherosclerotic
plaques where it can promote lesion instability by increasing the
expression of MMPs (15). In
addition, treatment with leptin promotes the progression of
abdominal aortic aneurysm in Apo-E mice by increasing the
expression of MMP-9 and −12, thereby stimulating medial
degeneration (16). It is known that
MMPs in plaques are primarily expressed by macrophages and SMCs,
and it has been demonstrated that leptin can promote SMC
proliferation and migration (17–20).
However, whether leptin can stimulate the expression of MMP in SMCs
to accelerate plaque destabilization had not been determined. The
present study hypothesized that leptin upregulates the expression
of MMP in VSMCs to promote plaque destabilization. To confirm this
hypothesis, the effect of leptin on the expression of MMP in VSMCs
was examined in vivo and in vitro, and the relevant
signaling pathways and molecular mechanisms were identified.
Materials and methods
Animal experiments
C57BL/6 and ob/ob mice (leptin deficiency,
C57BL/6 background) were purchased from Nanjing Biomedical Research
Institute of Nanjing University (Nanjing, China). The ob/ob
mouse strain is a mutant mouse, which eats excessively due to
mutations in the leptin gene (leptin deficiency). The experiments
involving animals were performed according to the guidelines and
ethical standards of the Animal Care and Use Ethics Committees of
Southern Medical University (Guangzhou, China; permit no. SCXK
2006–0116). At 8 weeks of age, the mice were anesthetized by
intraperitoneal injection of a ketamine (100 mg/kg) and xylazine
(10 mg/kg) mixture, and carotid ligation was performed. The mice
were then divided into four groups: Sham (C57BL/6, n=6), wild-type
(WT) C57BL/6 mice (n=6), ob/ob mice (n=6), and ob/ob
mice treated with leptin (n=6). Osmotic minipumps (Alzet, Durect
Corporation, Cupertino, CA, USA) filled with either recombinant
leptin (PeproTech EC, Ltd., London, UK) or phosphate-buffered
saline (PBS) were implanted into the abdominal cavity and set to
deliver a dose of 1 µg/g/d. The animals were fed a standard chow
diet and were housed at 25°C with 12-h light/dark cycles and a
humidity ≤60%. All animals were sacrificed at 4 weeks post-surgery.
The carotid artery was carefully excised, fixed in 4%
paraformaldehyde overnight at 4°C and embedded in paraffin for 30
min at 4°C. Cross-sections (5 mm) were cut and stained with
hematoxylin and eosin at room temperature for 10 min. The intima
was defined as tissue between lumen and internal elastic lamina,
and media was defined as tissue between internal elastic lamina and
external elastic lamina. The intimal and medial areas were measured
utilizing image analysis software (ImageJ 1.48; National Institutes
of Health, Bethesda, MD, USA) and the neointima/media area ratio
was calculated.
Gelatin zymography for arterial
tissue
Zymographic analysis was performed in all the
animals sacrificed at 4 weeks. The vessels were excised, washed
with Hanks' buffer (Applygen Technologies, Inc., Beijing, China)
and rapidly frozen in liquid nitrogen, prior to pulverization using
a mortar and pestle. The powders were resuspended in ice-cold lysis
buffer, containing 3M NaCl, 1M Tris-HCl (pH 7.4), 0.5M EDTA, 100 mM
PMSF and 10% Triton X-100 in ddH2O, at a ratio of 0.3
ml/10 mg wet weight. Samples were lysed on ice for 30 min and
centrifuged for 25 min (12,000 × g, 4°C). Supernatants were
retained and protein concentrations were measured using a
bicinchoninic acid assay. Protein sample loading was consistently
adjusted to protein concentration. The protein samples (80 µg) were
mixed in a SDS-PAGE 2X loading buffer [4% SDS, 100 nM Tris-Cl (pH
6.8), 20% glycerol and 0.02% bromophenol blue] and applied to an 8%
SDS-PAGE gel containing 1 mg/ml gelatin. The gels were subjected to
low current constant current electrophoresis, rinsed twice for 30
min with buffer at room temperature and then incubated for 1–5 h at
37°C. Following staining with Coomassie brilliant blue, gray-scale
analysis of gel images was performed using ImageJ 1.48 software
(National Institutes of Health).
Cell culture and treatment with
inhibitor and blocking antibody
To determine whether leptin induces the expression
of MMP in VSMCs via the mitogen-activated protein kinase
(MAPK)/extracellular signal-regulated kinase (ERK)/activator
protein-1 (AP-1) pathway, primary VSMCs were isolated by enzymatic
digestion of the thoracic aortic media from C57BL/6 male mice
(average weight 22 g, 7–8 weeks old). The isolated cells were
cultured at 37°C in a 5% CO2 humidified atmosphere. The
VSMCs were identified by immunofluorescence staining. Early passage
VSMCs at 90% confluence were exposed to serum starvation for 24 h
in high-glucose Dulbecco's modified Eagle's medium (DMEM HG; Gibco;
Thermo Fisher Scientific, Inc., Waltham, MA, USA). Cells
(1×106) were treated with leptin (1 µg/ml) or leptin (1
µg/ml) plus inhibitors (or blocking antibodies) for 24 h in 37°C,
including ERK kinase (MEK)1/2 inhibitor (U0126; 10 µM), c-Jun
N-terminal kinase (JNK) inhibitor (sp600125; #8177; 10 µM) all from
Cell Signaling Technology, Inc. (Danvers, MA, USA). SMC basal
medium served as a control treatment. The effective and safe doses
of the inhibitors or blocking antibodies were determined by
preliminary experiments. The conditioned medium was collected.
Small interfering RNA (siRNA)
The following siRNAs were purchased from Invitrogen;
Thermo Fisher Scientific, Inc. (Waltham, MA, USA): Leptin receptor
siRNA-1, 5′-ACUCCGAAACUGGUCCAUGAUCUGC-3′ and leptin receptor
siRNA-2, 5′-AUAUCCUGGUAAACGAUCUCAGUUA-3′. Negative control siRNA
(stealth RNA; negative control; cat. no. 12935-200) was also
obtained from Invitrogen; Thermo Fisher Scientific, Inc. The VSMCs
were cultured in DMEM containing 25 mM glucose and 10% (v/v)
heat-inactivated fetal calf serum (Gibco; Thermo Fisher Scientific,
Inc.) at 37°C in a 5% CO2 humidified atmosphere. To
knock down the expression of leptin receptor in VSMCs, the VMSCs
were transfected with the pcPURm6i35 shRNA expression vector
(Invitrogen; Thermo Fisher Scientific, Inc.) containing the target
sequence for mouse leptin receptor by reverse transfection using
Lipofectamine 2000 reagent (Invitrogen; Thermo Fisher Scientific,
Inc.).
Luciferase reporter assay
VSMCs were co-transfected with luciferase reporter
gene (AP-1) constructs and β-galactosidase using Lipofectamine 2000
(Invitrogen; Thermo Fisher Scientific, Inc.) for 24 h according to
the manufacturer's protocol and cells were treated with 1 µg/ml
leptin for 24 h at 37°C, with or without 1 h of pretreatment with
inhibitors. Luciferase activity was determined using a luciferase
assay kit (Promega, Madison, WI, USA).
Reverse transcription-quantitative
polymerase chain reaction analysis (RT-qPCR)
Cell RNA was extracted using TRIzol reagent
(Invitrogen; Thermo Fisher Scientific, Inc.). cDNA was synthesized
using the PrimeScript RT Reagent kit for 15 min at 42°C (Takara
Bio, Inc., Shiga, Japan). cDNA template (50 ng) was amplified using
SYBER Premix Ex Taq™ II (Takara Bio, Inc.) according to the
manufacturer's instructions. The primer sequences were as follows:
MMP-9, antisense, 5′-GCTGACTACGATAAGGACGGC-3′ and sense,
5′-AGGAAGACGAAGGGGAAGACG-3′; leptin receptor, antisense,
5′-ACCTGGCATATCCAATCTCTCC-3′ and sense,
5′-TTCAAAGCCGAGGCATTGTTT-3′; and β-actin, forward,
5′-ATGGGTCAGAAGGACTCCTACG-3′ and reverse,
5′-AGTGGTACGACCAGAGGCATAC-3′. The mRNA expression of β-actin was
used as a control. qPCR was performed using the following
conditions: 95°C for 60 sec, followed by 35 cycles of 60°C for 30
sec and 72°C for 100 sec. Each sample was replicated at least three
times. Relative quantification was determined using the
2−ΔΔCq method with β-actin as reference gene (21).
Western blot analysis
Cells were rinsed with ice-cold PBS and proteins
were extracted with lysis buffer (Thermo Fisher Scientific, Inc.)
for 30 min on ice. The extracts were centrifuged (13,400 × g; 4°C;
20 min) and the supernatants were obtained. Protein concentrations
were determined using the DC protein assay (Bio-Rad Laboratories,
Inc., Hercules, CA, USA) and proteins (40 µg) were subjected to
electrophoresis on a 10% SDS-PAGE gel (Bio-Rad Laboratories, Inc.).
The proteins were then transferred onto polyvinylidene difluoride
membranes (Immobilon-P; EMD Millipore, Billerica, MA, USA). The
membranes were blocked in PBS with Tween 20 (PBST, pH 7.4)
containing 5% bovine serum albumin and were probed with antibodies
targeting phosphorylated (p-)ERK (AF1018; 0.1 µg/ml); ERK
(MAB15761; 1 µg/ml; F1018); p-JNK (AF1205; 0.5 µg/ml); JNK
(MAB1387; 0.2 µg/ml); leptin receptor (AF497; 0.1 µg/ml); MMP-2
(AF1488; 0.1 µg/ml); and MMP-9 (AF909; 0.25 µg/ml; all R&D
Systems, Inc., Minneapolis, MN, USA) for 18 h at 4°C. The membranes
were washed and incubated for 1 h at room temperature with
horseradish peroxidase-conjugated secondary antibody (1:5,000;
BM2002; Wuhan Boster Biological Technology, Ltd., Wuhan, China).
Immunoreactive bands were visualized using enhanced
chemiluminescence reagent (EMD Millipore) and the optical density
of bands was measured using ImageJ 1.42 (National Institutes of
Health).
Statistical analysis
All in vitro experiments were performed in
triplicate. One-way analysis of variance and Student's t-tests were
used with SPSS software 16.0 (SPSS, Inc., Chicago, IL, USA). The
statistical significance of the differences among multiple groups
was tested using one-way analysis of variance and pairwise
comparisons were performed using Tukey's post hoc analysis. Error
bars indicate the standard error of the mean. P<0.05 was
considered to indicate a statistically significant difference.
Results
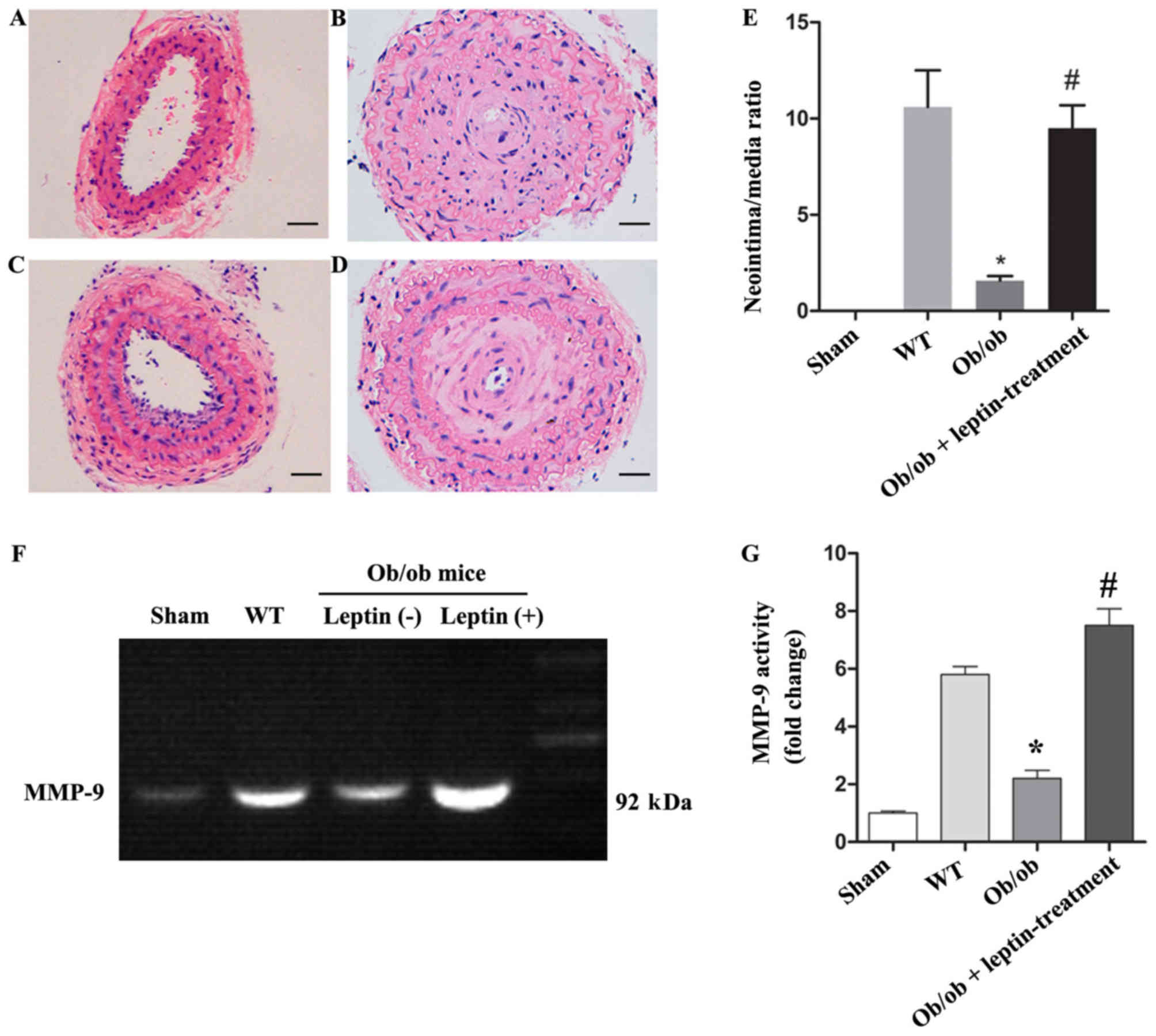
Leptin treatment significantly
increases the neointima/media ratio and expression of MMP-9 in the
carotid artery of ob/ob mice following carotid ligation
The mean body weight of the ob/ob mice at the
beginning of the study was 56.5±4.5 g and after 4 weeks, the mean
body weights of leptin-treated mice were lower compared with the
untreated animals (50.5±5.2 vs. 57.1±4.8 g; P<0.05). When the
ob/ob mice reached a certain weight (55–60 g), the body
weight gain slowed in control group and leptin-treated group
(56.5±4.5 vs. 57.1±4.8 g; data not shown) (22). The body weight of the mice was also
affected by invasive carotid ligation surgery. The neointima/media
ratio in the carotid artery following carotid ligation of the
ob/ob mice was lower compared with the WT mice (1.67 vs.
10.78, P<0.05), but was significantly higher in the ob/ob
mice treated with leptin (8.76 vs. 1.67, P<0.05; Fig. 1A-E). Gelatin zymography also showed
that the activity of MMP-9 in the injured carotid artery was
significantly higher in the leptin-treated group compared with that
in the other groups (P<0.05; Fig. 1F
and G).
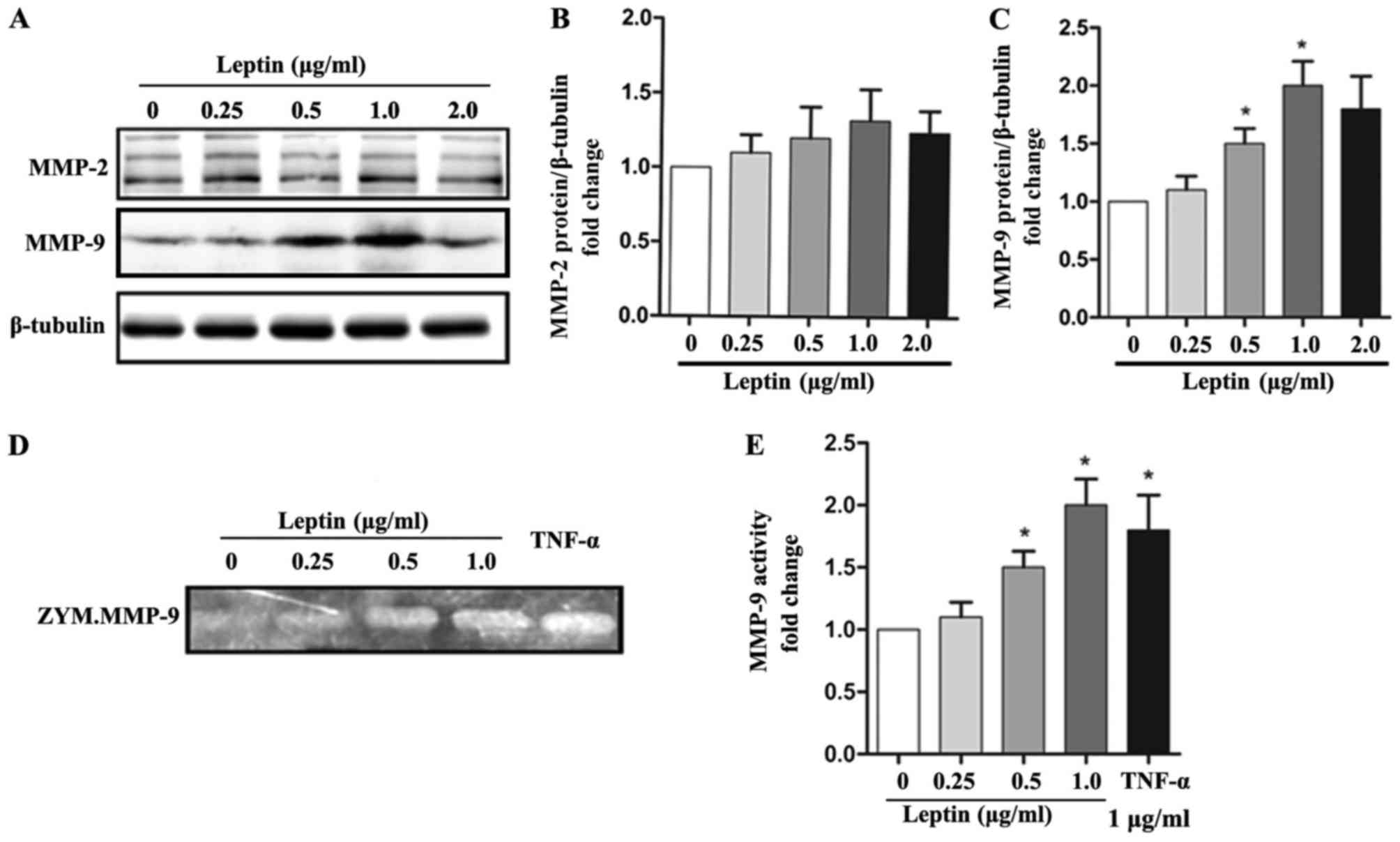
Leptin upregulates the expression and
activity of MMP-9 in SMCs
The present study examined the levels of MMPs in
SMCs following leptin treatment. Leptin treatment significantly
increased the protein levels of MMP-9 in a dose-dependent manner;
however, it did not affect the protein expression of MMP-2
(Fig. 2A-C). Furthermore, using
gelatin zymography, it was found that leptin treated induced MMP-9
proteolytic activity in the supernatant of SMCs in a dose-dependent
manner (Fig. 2D and E). Together,
these findings suggested that leptin upregulated the expression and
activity of MMP-9 in SMCs.
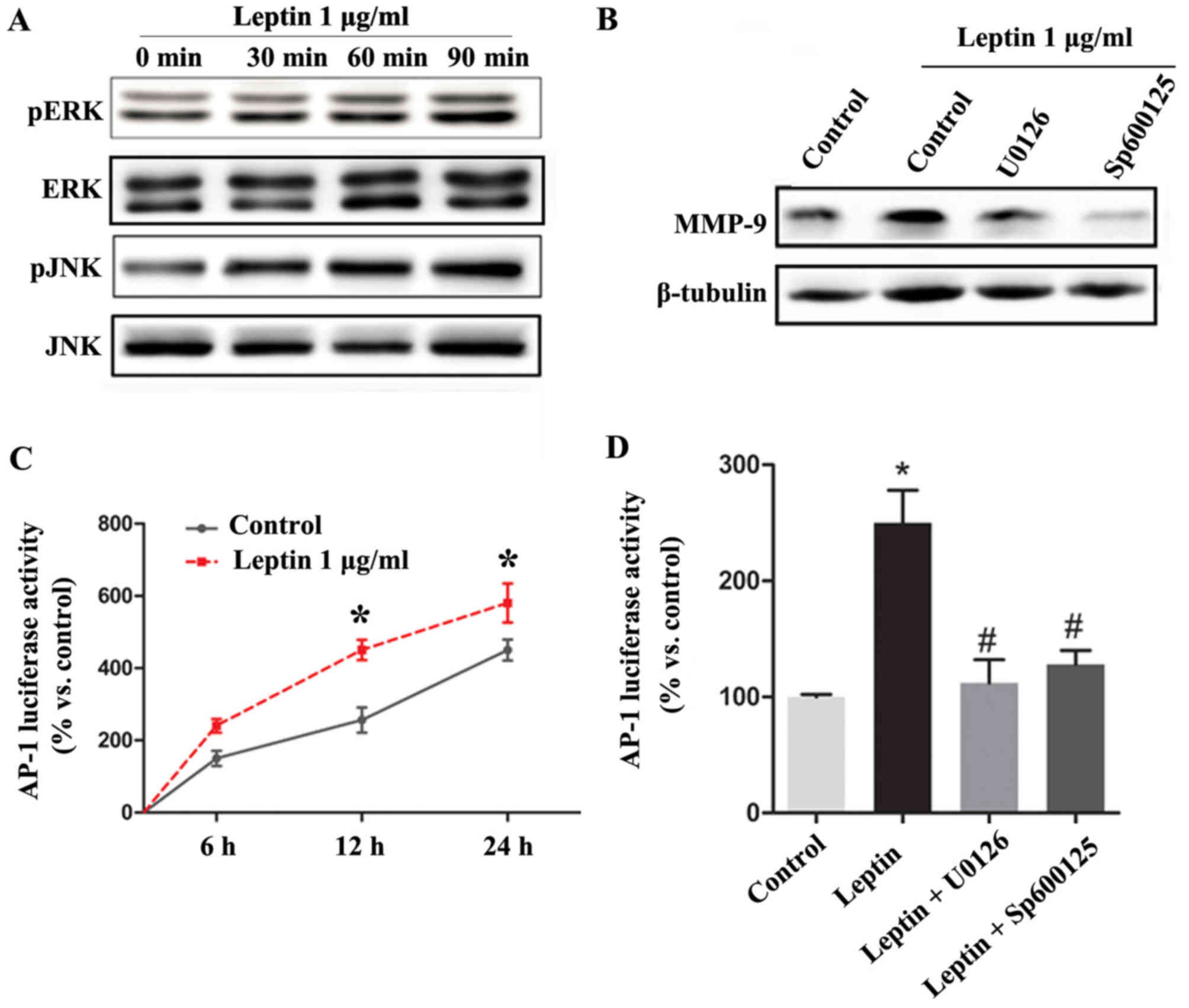
Leptin upregulates the expression of
MMP-9 by activating AP-1 via the MAPK/ERK/JNK signaling pathways in
SMCs
It was observed that leptin treatment markedly
induced cytoplasmic ERK and JNK phosphorylation in the SMCs in a
time-dependent manner, and it was found that treatment of the cells
with leptin in addition to pharmacological inhibitors of the
MAPK/ERK pathway (U0126) or JNK (SP600125) abrogated the
leptin-mediated effects on the expression of MMP-9 (Fig. 3A and B).
AP-1 is known to be a major transcription factor
that regulates the expression of MMP-9. Therefore, to understand
the possible mechanisms involved in the leptin-mediated
upregulation of MMP-9, the present study determined whether AP-1
was activated by leptin using a Luciferase reporter assay. Leptin
significantly increased the transcriptional activity of AP-1 in a
time-dependent manner, whereas pharmacological inhibitors of the
MAPK/ERK pathway (U0126) or JNK (SP600125) inhibited the
transcriptional activity of AP-1 by ~90% in the absence of leptin
stimulation (Fig. 3C and D).
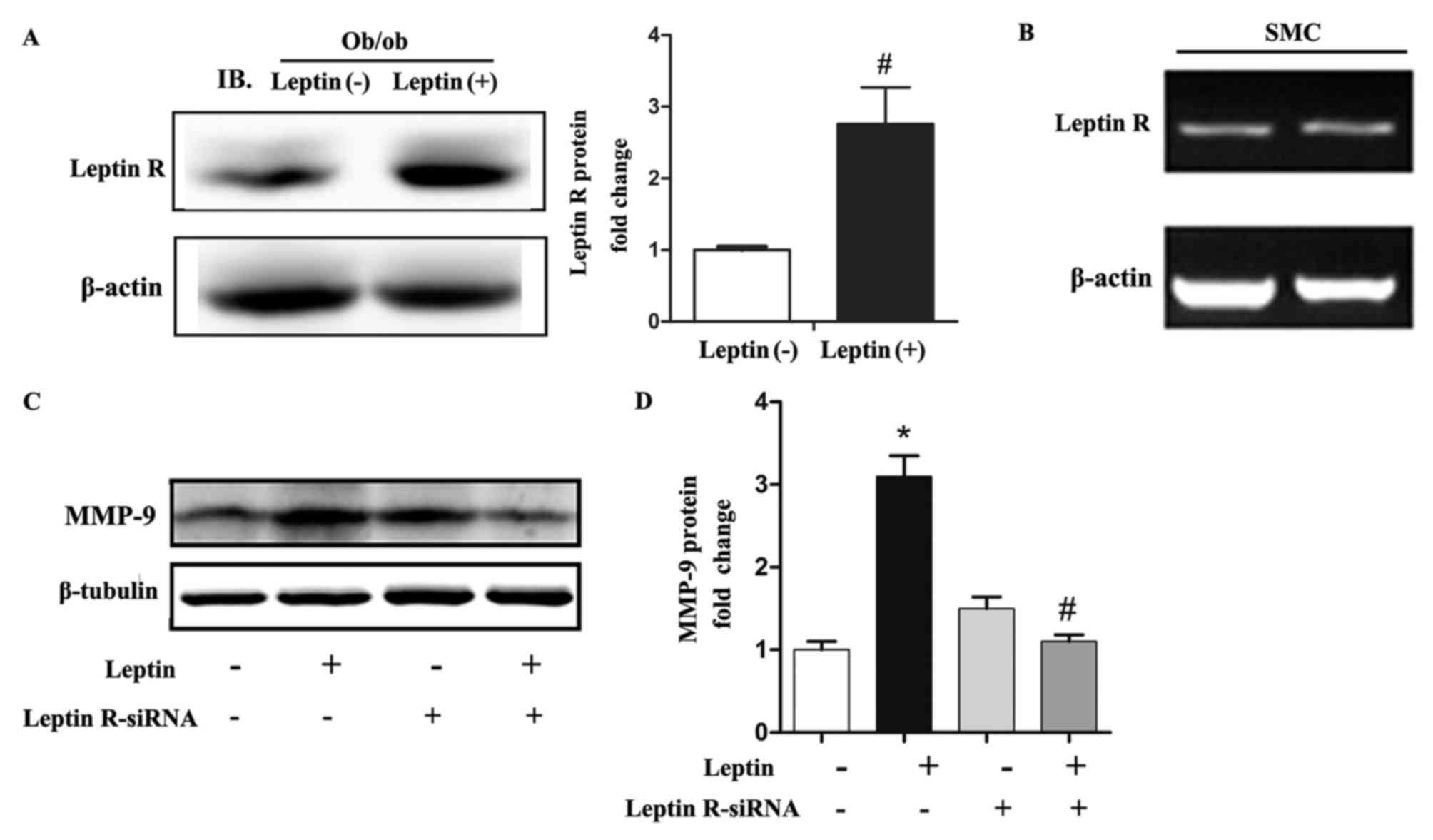
Leptin receptor mediated
leptin-induced expression of MMP-9 in SMCs
To elucidate how leptin acts on SMCs, further
experiments were performed to identify the receptor that leptin
binds to on SMCs. Following systemic leptin treatment, the carotid
artery of ob/ob mice showed significantly increased leptin
receptor expression via western blot analysis (Fig. 4A). In addition, the results of
RT-qPCR analysis and agarose gel electrophoresis indicated that the
leptin receptor was expressed by SMCs (Fig. 4B). Finally, the siRNA-mediated
knockdown of leptin receptor abrogated the effect of leptin
treatment (Fig. 4C and D).
Discussion
The secretory functions of SMCs and macrophages are
crucial in intimal hyperplasia and plaque lesion instability
(23). The MMP family of proteases,
also known as the metzincin superfamily, are important in tissue
remodeling through degrading denatured collagens, gelatins and
various ECM components in different tissues. MMPs are mainly
produced by macrophages and SMCs in atherosclerotic plaques
(3–5). MMP-2 and −9 belong to a sub-group of
gelatinases, which share similar proteolytic activity and are
involved in atherosclerotic plaque rupture (24,25).
Soluble cytokines and cell-cell interactions have been shown to
upregulate the level and bioactivity of MMPs in cells present in
the normal and diseased blood vessel wall. The activation of MMP in
response to inflammatory molecules, including interleukin (IL)-1,
IL-4 and tumor necrosis factor-α (TNF-α), may contribute to
pathological matrix destruction and plaque rupture (6).
Leptin is an important endocrine factor, which
affects cholesterol synthesis (26),
but it may also act as an inflammatory factor, and plasma leptin
concentrations have been found to be increased in patients with
ACS, and the expression of leptin is high in vulnerable plaques
(27). However, whether the
expression of leptin is simply an accompanying phenomenon or a
pathogenic factor in atherosclerotic plaque rupture remains to be
elucidated. A previous study suggested that leptin is involved in
matrix remodeling by promoting the expression of MMPs and tissue
inhibitor of MMPs in vascular endothelial cells (28). In the present study, in vivo
leptin treatment significantly induced the expression and activity
of MMP-9 in the carotid artery following ligation in ob/ob
mice. In vitro, leptin upregulated the expression and
activity of MMP-9, but not MMP-2, in SMCs. This indicated that
leptin may have a substantial effect on plaque rupture by inducing
the SMC production of MMP-9, which promotes degradation of the
ECM.
Studies have demonstrated that the transcription
factor AP-1 is a key transcriptional regulator responsible for the
induction of MMP-9 in various types of cells (29). In the present study, it was found
that leptin significantly induced the phosphorylation of ERK1/2 and
JNK in SMCs. Furthermore, the results demonstrated that ERK1/2 and
JNK were indispensable for AP-1-mediated expression of MMP-9 in
leptin-treated SMCs. Therefore, in the present study, it was found
that leptin induced the activation of AP-1 through the activity of
ERK1/2 and JNK, which then facilitated AP-1 binding to promoter
regions of the MMP-9 gene to promote the transcription of
MMP-9.
Leptin and its receptor are expressed in SMCs
(17,18). Leptin receptor expression is elevated
in carotid plaques relative to unstable plaques, and thus, may be
involved in intimal neovascularization (30,31). In
the present study, leptin receptor was expressed at a high level in
the mice aorta and then, following leptin treatment, a
statistically significant change in the mRNA expression of aortic
leptin receptor was observed in comparison with that in the control
group. These results suggested that the leptin receptor served as
the major receptor of leptin on SMCs and may be a novel therapeutic
target for preventing leptin-induced plaque rupture.
In terms of limitations, whether the leptin receptor
mediates the leptin-induced expression of MMP-9 in db/db
mice in vivo requires verification; however, similar
published studies have shown that leptin is involved via combining
leptin receptors in db/db mice (32,33). The
present study focused on leptin stimulation of the secretion of
MMP-9 in SMCs to promote plaque instability, and further
examination is to be continued in future investigations.
In conclusion, the results of the present study
showed that leptin significantly increased neointima hyperplasia
and the expression of MMP-9 in the mouse carotid artery following
carotid ligation. Leptin also significantly stimulated the
expression of MMP-9, mainly by activating AP-1 via the leptin
receptor/MAPK/ERK signal transduction pathways. These findings
provide novel evidence that leptin may have a substantial effect on
plaque rupture by promoting ECM degradation via the upregulation of
MMP-9.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
RL performed experiments. BC was involved in data
collection, analysis and interpretation, and in manuscript writing.
JC performed cellular experiments and immunohistochemistry, and
helped drafting the manuscript. JL conceived and designed the
study, and analyzed and interpreted the data. All authors read and
approved the final version of the manuscript.
Ethics approval and consent to
participate
The experimental animal protocol was approved by the
Animal Ethics Committee of Southern Medical University (permit no.
SCXK2006-0116).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
MMPs
|
matrix metalloproteinases
|
|
SMC
|
smooth muscle cell
|
|
ACS
|
acute coronary syndrome
|
|
AP-1
|
luciferase reporter gene
|
References
|
1
|
Falk E, Nakano M, Bentzon JF, Finn AV and
Virmani R: Update on acute coronary syndromes: The pathologists'
view. Eur Heart J. 34:719–728. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Beaudeux JL, Giral P, Bruckert E,
Foglietti MJ and Chapman MJ: Matrix metalloproteinases,
inflammation and atherosclerosis: Therapeutic perspectives. Clin
Chem Lab Med. 42:121–131. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ketelhuth DF and Bäck M: The role of
matrix metalloproteinases in atherothrombosis. Curr Atheroscler
Rep. 13:162–169. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Plutzky J: Atherosclerotic plaque rupture:
Emerging insights and opportunities. Am J Cardiol. 84:J15–J20.
1999. View Article : Google Scholar
|
|
5
|
Mason DP, Kenagy RD, Hasenstab D,
Bowen-Pope DF, Seifert RA, Coats S, Hawkins SM and Clowes AW:
Matrix metalloproteinase-9 overexpression enhances vascular smooth
muscle cell migration and alters remodeling in the injured rat
carotid artery. Circ Res. 85:1179–1185. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Newby AC: Dual role of matrix
metalloproteinases (matrixins) in intimal thickening and
atherosclerotic plaque rupture. Physiol Rev. 85:1–31. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ha KT, Lee TK, Kwak KH, Kim JK, Kim DI,
Choi DY and Kim CH: Inhibitory effect of Cho-Deung-San on human
aortic smooth muscle cell migration induced by TNF-alpha through
inhibition of matrix metalloproteinase-2 and −9 activity. Vasc
Pharmacol. 41:83–90. 2004. View Article : Google Scholar
|
|
8
|
Johnson JL, van Eys GJ, Angelini GD and
George SJ: Injury induces dedifferentiation of smooth muscle cells
and increased matrix-degrading metalloproteinase activity in human
saphenous vein. Arterioscler Thromb Vasc Biol. 21:1146–1151. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mattu HS and Randeva HS: Role of
adipokines in cardiovascular disease. J Endocrinol. 216:T17–T36.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li R, Chen LZ, Zhao SP and Huang XS:
Inflammation activation contributes to adipokine imbalance in
patients with acute coronary syndrome. PloS One. 11:e01519162016.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Couillard C, Lamarche B, Mauriège P,
Cantin B, Dagenais GR, Moorjani S, Lupien PJ and Després JP:
Leptinemia is not a risk factor for ischemic heart disease in men.
Prospective results from the Quebec Cardiovascular Study. Diabetes
Care. 21:782–786. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Piemonti L, Calori G, Mercalli A, Lattuada
G, Monti P, Garancini MP, Costantino F, Ruotolo G, Luzi L and
Perseghin G: Fasting plasma leptin, tumor necrosis factor-alpha
receptor 2, and monocyte chemoattracting protein 1 concentration in
a population of glucose-tolerant and glucose-intolerant women:
Impact on cardiovascular mortality. Diabetes Care. 26:2883–2889.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liu J, Butler KR, Buxbaum SG, Sung JH,
Campbell BW and Taylor HA: Leptinemia and its association with
stroke and coronary heart disease in the Jackson Heart Study. Clin
Endocrinol (Oxf). 72:32–37. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tahergorabi Z and Khazaei M: Leptin and
its cardiovascular effects: Focus on angiogenesis. Adv Biomed Res.
4:792015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Schneiderman J, Schaefer K, Kolodgie FD,
Savion N, Kotev-Emeth S, Dardik R, Simon AJ, Halak M, Pariente C,
Engelberg I, et al: Leptin locally synthesized in carotid
atherosclerotic plaques could be associated with lesion instability
and cerebral emboli. J Am Heart Assoc. 1:e0017272012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tao M, Yu P, Nguyen BT, Mizrahi B, Savion
N, Kolodgie FD, Virmani R, Hao S, Ozaki CK and Schneiderman J:
Schneiderman, locally applied leptin induces regional aortic wall
degeneration preceding aneurysm formation in apolipoprotein
E-deficient mice. Arterioscler Thromb Vasc Biol. 33:311–320. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Schroeter MR, Eschholz N, Herzberg S,
Jerchel I, Leifheit-Nestler M, Czepluch FS, Chalikias G,
Konstantinides S and Schäfer K: Leptin-dependent and
leptin-independent paracrine effects of perivascular adipose tissue
on neointima formation. Arterioscler Thromb Vasc Biol. 33:980–987.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li H, Wang YP, Zhang LN and Tian G:
Perivascular adipose tissue-derived leptin promotes vascular smooth
muscle cell phenotypic switching via p38 mitogen-activated protein
kinase in metabolic syndrome rats. Exp Biol Med (Maywood).
239:954–965. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Schäfer K, Halle M, Goeschen C, Dellas C,
Pynn M, Loskutoff DJ and Konstantinides S: Leptin promotes vascular
remodeling and neointimal growth in mice. Arterioscler Thromb Vasc
Biol. 24:112–117. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Oda A, Taniguchi T and Yokoyama M: Leptin
stimulates rat aortic smooth muscle cell proliferation and
migration. Kobe J Med Sci. 47:141–150. 2001.PubMed/NCBI
|
|
21
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Pelleymounter MA, Cullen MJ, Baker MB,
Hecht R, Winters D, Boone T and Collins F: Effects of the obese
gene product on body weight regulation in ob/ob mice. Science.
269:540–543. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ross R: Rous-Whipple award lecture.
Atherosclerosis: A defense mechanism gone awry. Am J Pathol.
143:987–1002. 1993.PubMed/NCBI
|
|
24
|
Galis ZS, Sukhova GK, Lark MW and Libby P:
Increased expression of matrix metalloproteinases and matrix
degrading activity in vulnerable regions of human atherosclerotic
plaques. J Clin Invest. 94:2493–2503. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kampoli AM, Tousoulis D, Papageorgiou N,
Antoniades C, Androulakis E, Tsiamis E, Latsios G and Stefanadis C:
Matrix metalloproteinases in acute coronary syndromes: Current
perspectives. Curr Top Med Chem. 12:1192–1205. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kosztáczky B, Fóris G, Paragh G Jr, Seres
I, Zsiros E, Koncsos P, Balogh Z and Paragh G: Leptin stimulates
endogenous cholesterol synthesis in human monocytes: New role of an
old player in atherosclerotic plaque formation. Leptin-induced
increase in cholesterol synthesis. Int J Biochem Cell Biol.
39:1637–1645. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lee K, Santibanez-Koref M, Polvikoski T,
Birchall D, Mendelow AD and Keavney B: Increased expression of
fatty acid binding protein 4 and leptin in resident macrophages
characterises atherosclerotic plaque rupture. Atherosclerosis.
226:74–81. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Park HY, Kwon HM, Lim HJ, Hong BK, Lee JY,
Park BE, Jang Y, Cho SY and Kim HS: Potential role of leptin in
angiogenesis: Leptin induces endothelial cell proliferation and
expression of matrix metalloproteinases in vivo and in vitro. Exp
Mol Med. 33:95–102. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Moon SK, Kim HM and Kim CH: PTEN induces
G1 cell cycle arrest and inhibits MMP-9 expression via the
regulation of NF-kappaB and AP-1 in vascular smooth muscle cells.
Arch Biochem Biophys. 421:267–276. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Schneiderman J, Simon AJ, Schroeter MR,
Flugelman MY, Konstantinides S and Schaefer K: Leptin receptor is
elevated in carotid plaques from neurologically symptomatic
patients and positively correlated with augmented macrophage
density. J Vasc Surg. 48:1146–1155. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kang SM, Kwon HM, Hong BK, Kim D, Kim IJ,
Choi EY, Jang Y, Kim HS, Kim MS and Kwon HC: Expression of leptin
receptor (Ob-R) in human atherosclerotic lesions: Potential role in
intimal neovascularization. Yonsei Med J. 41:68–75. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Taleb S, Herbin O, Ait-Oufella H, Taleb S,
Herbin O, Ait-Oufella H, Verreth W, Gourdy P, Barateau V, Merval R,
et al: Defective leptin/leptin receptor signaling improves
regulatory T cell immune response and protects mice from
atherosclerosis. Arterioscler Thromb Vasc Biol. 27:2691–2698. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Luo W, Bodary PF, Shen Y, Wickenheiser KJ,
Ohman MK, Guo C, Bahrou KL, Myers MG Jr and Eitzman DT: Leptin
receptor-induced STAT3-independent signaling pathways are
protective against atherosclerosis in a murine model of obesity and
hyperlipidemia. Atherosclerosis. 214:81–85. 2011. View Article : Google Scholar : PubMed/NCBI
|