Introduction
Cerebral ischemia/reperfusion (I/R) injury is a key
and common pathological process in certain diseases of the nervous
system, including stroke and traumatic brain injury (TBI), and is
also among the main causes of disability and mortality (1,2).
Although clinical trials have been conducted in an attempt to treat
I/R injury-related diseases, the only efficacious methods for the
treatment of stroke and TBI are thrombolysis and hypothermia
(1,2). Therefore, it is of great importance to
elucidate the pathophysiological mechanism of cerebral I/R and to
develop an effective neuroprotective intervention strategy such as
brain protectants. The clinical failure of many potential
neuroprotective strategies involves a lack of understanding with
regards to the choice of treatment windows for defined targets, the
importance of neurons interacting with astrocytes and, in
particular, the newly identified role of astrocytes, including the
expression of neurotransmitter receptors and the defense against
oxidative stress (1,3). Literature on the mechanism of cerebral
I/R injury is mainly focused on oxidative stress, excitatory
neurotoxicity and Ca2+ overload (2). However, the activation of astrocytes
serves a vital role in neurodegenerative changes, including
cerebral ischemia and hippocampal neuron damage by tumor necrosis
factor-α-mediated inflammatory injury (1,3,4). Astrocytes are activated and proliferate
in order to promote repair of neurons, axon growth and nerve
function restoration in the early stage of cerebral ischemia
(5).
Dexmedetomidine (Dex) is a selective α2 adrenoceptor
agonist that is used as a potent sedative for critically ill
patients in the intensive care unit and as an effective anesthetic
adjuvant for surgical patients in the operating room (6). Dex has been demonstrated to improve
neuronal survival following transient global or focal cerebral
ischemia in rats (7,8), which provides a novel neuroprotective
target for cerebral ischemia, but researchers hold different views
on the brain protective mechanism of Dex. Previous studies have
explained the neuroprotection of Dex in an α2
adrenoceptor-dependent manner. Kose et al (9) reported that intravenous drug delivery
of Dex served an important protective role in cerebral
ischemia-mediated neuron injury by inhibiting the activation of
astrocytes. Meanwhile, others have observed that reduction of
glutamic acid agonist-induced neuronal apoptosis by Dex was
associated with increased expression of brain-derived neurotrophic
factor in astrocytes (10). These
results indicate that the neuroprotective effect of Dex
pretreatment is associated with the function and α2 adrenoceptor
expression of astrocytes during cerebral I/R injury.
The neuroprotective effect of Dex has been
demonstrated to involve extracellular signal-regulated kinases 1
and 2 (ERK1/2) in an α2 adrenoceptor-independent manner. For
instance, Dex may stimulate glial cell line-derived neurotrophic
factor (GDNF) release in order to rescue neurons from neurotoxicity
induced by oxygen-glucose deprivation by upregulating hippocampal
ERK1/2 expression, and the effects may be attenuated by inhibition
of ERK (11,12). However, during cerebral I/R injury,
whether α2A adrenergic receptor (ADRA2A)-mediated phosphorylation
of ERK1/2 is involved in the neural protection of Dex remains
unknown to the best of our knowledge.
In the present study, a hypoxia/reoxygenation (H/R)
model of primary cultured astrocytes and a focal cerebral I/R model
in adult rats were used to investigate the effect of Dex
pretreatment on the expression of ADRA2A and phosphorylation of
ERK1/2 in astrocytes during hypoxic culture and cerebral ischemic
conditions, respectively. The neuroprotective mechanism of Dex
pretreatment was demonstrated to be caused by ADRA2A-mediated
phosphorylation of ERK1/2.
Materials and methods
Animals
The present study used 24 2-day-old neonatal male
Sprague-Dawley (SD) rats weighing 250–280 g, obtained from the
Experimental Animal Center of Tongji Medical College of Huazhong
University of Science and Technology (HUST), Wuhan, China. The rats
were individually fed with standard pellet chow and had access to
drinking water ad libitum under standard conditions
(22–24°C, 40–70% humidity and a regular 12-h day/night cycle). The
Ethics Committee for the Use of Experimental Animals at Tongji
Medical College of HUST approved all experimental procedures.
Chemicals
Dex was manufactured and supplied by Jiangsu Hengrui
Medicine Co., Ltd. (Jiangsu, China). Rat glial fibrillary acidic
protein (GFAP; cat. no. P90068Hu01) and recombinant rat ADRA2A
monoclonal antibodies (cat. no. CSB-YP001388MO) were purchased from
Shanghai Yanhui Biotechnology Co., Ltd. (Shanghai, China) and Wuhan
Huamei Biotechnology Co., Ltd. (Beijing, China) and used for
immunofluorescent staining and protein blotting during the
following experiments. Goat anti-rabbit (cat. no. BA1055) or goat
anti-rat (cat. no. BA1058) horseradish peroxidase-conjugated
secondary antibodies and a bicinchoninic acid (BCA) protein assay
kit were purchased from Wuhan Boster Biological Technology Ltd.
(Wuhan, China). Anti-β-actin rabbit monoclonal antibody (cat. no.
4970), rabbit ERK1/2 (cat. no. 4348) and phosphorylated (p)-ERK1/2
(Thr202/Tyr204; cat. no. 9101) antibodies, and the epidermal growth
factor receptor (EGFR) tyrosine kinase inhibitor AG1478 were
obtained from Cell Signaling Technology, Inc. (Danvers, MA, USA).
Triphenyl tetrazolium chloride (TTC) was acquired from
Sigma-Aldrich (Merck KGaA, Darmstadt, Germany).
Primary cortical astrocyte
culture
The protocol for the culture of primary cortical
astrocytes was performed according to an improved method of that
first proposed by McCarthy (13).
Briefly, the cerebral cortical tissues of the neonatal SD rats were
detached and segmented, digested for 30 min in 2.5 g/l trypsin at
37°C, and mixed with Dulbecco's modified Eagle's medium
(Sigma-Aldrich; Merck KGaA) containing 15% heat-inactivated fetal
bovine serum (Sigma-Aldrich; Merck KGaA) to end the digestion. The
digested cerebral cortices were centrifuged at 560 × g for 5 min
and the resuspended cells were filtered with a mesh bag (200 mm) in
order to achieve a single cell suspension. The prepared single cell
suspension was inoculated into a glass culture bottle without
polylysine to remove the fibroblasts for 15 min at 37°C and 5%
CO2, and then the cell suspension in the flask was
gently flipped and plated onto a 50-ml glass culture bottle coated
with L-polylysine at a density of 3×106 cells/bottle.
After 8–12 days of plating, monolayers of astrocytes were obtained.
The growth state of astrocytes was observed under a light
microscope (magnification, ×100). The adherent cells were digested
for 2–3 min in 300 µl of 2.5 g/l trypsin at 37°C. Immunofluorescent
staining for GFAP was used to assess the purity of the primary
cortical astrocytes. Astrocytes were cultured on cover glasses
(18×18 mm) placed in 6-well plates at 37°C until the cells attained
a confluence of 70–80%. Cells were fixed using 4% formaldehyde at
room temperature for 30 min and then incubated at 37°C for 30 min
in 0.01 mol/l PBS containing 10% goat serum (Wuhan Boster
Biological Technology Ltd.) and 0.1% Triton X-100. Following the
addition of rat GFAP (1:100; cat. no. P90068Hu01) at 4°C overnight
and washing three times with 0.01 mol/l PBS, Cy3 labeled sheep
anti-rat IgG (1:100; cat. no. A0507; Beyotime Institute of
Biotechnology, Haimen, China) was added and incubated free from
light at 37°C for 30 min. The slides with the cells were sealed
using 30 µl mounting medium containing DAPI at room temperature for
30 min. The stained sections were examined with a Leica
fluorescence microscope, and images were captured with a CCD camera
(14). Dibutyryl cyclic-adenosine
monophosphate (d-cAMP; 150 µmol, Sigma, St. Louis, MO, USA) was
added to induce cell maturation over a duration of 3 to 4 days at
37°C when >95% of the cultured cells were GFAP positive.
H/R cell model and experimental
protocol
In vitro experimental ischemia was induced by
H/R using an anoxic incubation method (15). The primary astrocytes cultured in
glucose-free medium were transferred from the normal incubator
containing 22% O2 and 5% CO2 at 37°C to an
anaerobic chamber containing 94% N2, 1% O2
and 5% CO2 at 37°C for 6 h, and then the glucose-free
medium was replaced with Dulbecco's modified Eagle medium
containing 10% heat-inactivated fetal bovine serum (Sigma-Aldrich;
Merck KGaA) and 1% penicillin/streptomycin and cells were returned
to the normal incubator for reoxygenation for 24 h. In the
preliminary experiment, cells seeded in 6-well plates prior to Dex
treatment were used to investigate the effect of Dex pretreatment
on the primary astrocytes in hypoxic conditions. The primary
in-vitro cultured astrocytes were divided into 5 groups and
were subjected to various treatments as follows: i) Control
(Con)+Dex 2, cells were treated with 500 ng/ml Dex for 3 h and then
incubated in the normal incubator for 27 h; ii) Con, cells were
incubated in the normal incubator for 30 h; iii) H/R, cells were
incubated according to the conditions of the H/R model; iv) H/R+Dex
1, cells were pretreated with 100 ng/ml Dex for 3 h and then
incubated according to the conditions of the H/R model; and v)
H/R+Dex 2, cells were pretreated with 500 ng/ml Dex for 3 h and
then incubated according to the conditions of the H/R model.
Construction of the focal cerebral I/R
model and drug administration
The cerebral I/R model was induced by transient
middle cerebral artery occlusion (tMCAO) according to a previous
study (16). Briefly, the right
carotid artery (CCA), the external carotid artery (ECA) and the
internal carotid artery (ICA) were carefully isolated under sterile
conditions following rat anesthesia with 3% isoflurane. Following
temporary closure of the ICA and CCA with arteriole clips, a
silicone line (6–0; Doccol Corporation, Sharon, MA, USA) was
inserted from the right CCA into the ICA to occlude the origin of
the right middle cerebral artery. After 30 min of occlusion, the
silicone line was withdrawn, the arteriole clip was lessened and
the ECA was ligated permanently to allow reperfusion. Following
surgery, the rats were separately fed water and food ad
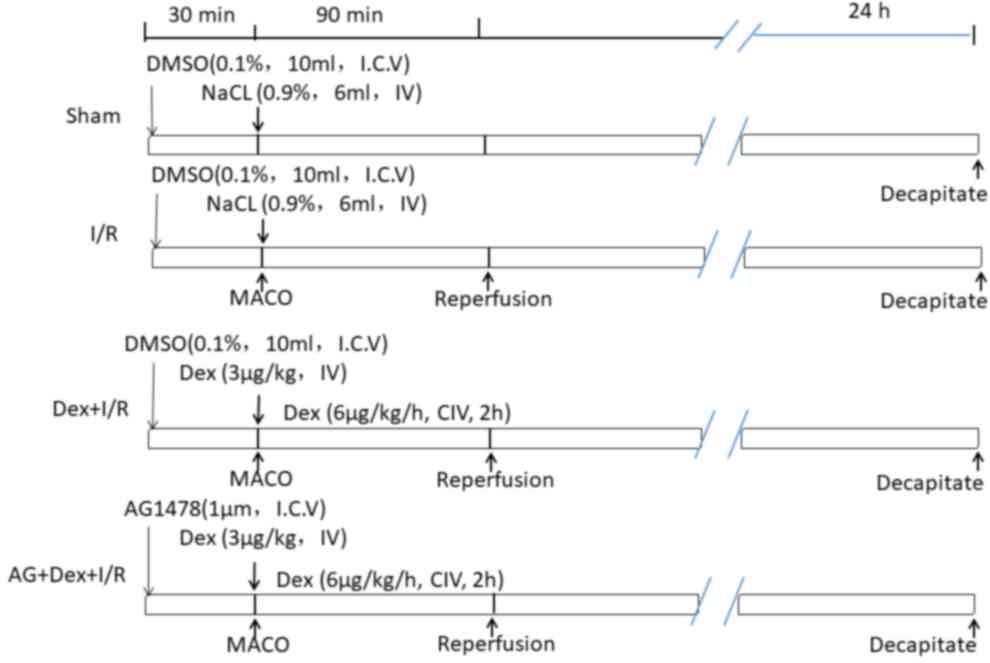
libitum at room temperature. As displayed in Fig. 1, the rats were randomly divided into
4 groups (n=6 rats/group) as follows: i) Sham, the same surgical
procedures without tMCAO were performed and the rats were
intravenously infused with DMSO and 0.9% NaCl at the same volume as
that in the Dex-pretreated rats; ii) I/R, the rats endured ischemia
for 90 min and reperfusion for 24 h; iii) Dex-pretreated (Dex+I/R),
Dex (bolus, 3 µg/kg) was infused through the left femoral vein at
the onset of ischemia and then infusion continued for a further 2 h
(6 µg/kg/h); and iv) AG+Dex+I/R, rats were injected
intracerebroventricularly with 1 µM AG1478 prior to the operation,
Dex pretreatment and reperfusion (17). The changes of physiological variables
including pCO2, pO2, MAP, pH and blood
glucose at different I/R periods, such as at the onset of ischemia,
30 min of ischemia and 24 h of reperfusion in ischemic rats were
observed and recorded.
Evaluation of neurological function
and cerebral infarct volume
A five-point scoring method was adopted to assess
the extent of neurological deficit in the rats (8,18).
Briefly, the motor findings were scored as follows: 0 points,
asymptomatic symptoms; 1 point, forelimb flexion; 2 points,
forelimb flexion and decreased resistance to lateral push; 3
points, forelimb flexion, decreased resistance to lateral push and
unilateral circling; and 4 points, forelimb flexion, and unable or
difficult to ambulate. Following decapitation, the brain tissue of
each rat was immediately collected and frozen at −20°C for 30 min.
The frozen brain was cut into 4-mm thick slices and the slices were
stained in 2% TTC solution (pH 7.4) at 37°C for 30 min and fixed in
10% formalin at room temperature for 20 min. Normal brain tissue
stained red and the infarcted area white. The volume of cerebral
infarction was measured and calculated using Image-Pro Plus
software, version 6.0 (Media Cybernetics, Inc., Rockville, MD,
USA). Infarct volume (%)=(left cerebral hemispheric volume-right
cerebral non-infarct volume)/(left cerebral hemisphere volume ×2)
×100 (19).
Hematoxylin and eosin (H&E)
staining
Following immersion of rat brain tissues in 4%
paraformaldehyde solution for 24 h at 4°C, the water in the brain
tissue was gradually removed with 70, 80, 90 and 95% ethanol
solutions, which was followed by xylene clearing and paraffin
embedding. The embedded wax blocks were sectioned into 5-µm
continuous coronal brain tissue slices, dried and dewaxed, and then
were stained with H&E at room temperature for 30 min (Solarbio
Co., Ltd, Beijing, China). Morphological characteristics of each
brain tissue section were observed with an optical microscope
(Leica DM1000; Leica Microsystems GmbH, Wetzlar, Germany).
Western blotting
Total protein from the rat right ischemic hemisphere
or primary cultured astrocytes were extracted on ice with cell
lysis buffer [1% Triton X-100, 50 mM Tris (pH 7.4), 150 mM NaCl,
0.1% sodium dodecyl sulfate (SDS) and 1 mM EDTA] containing 1
mmol/l complete proteinase inhibitor cocktail. The BCA method was
used to determined protein concentration. Total protein (40–50 mg)
was separated by 10% SDS-polyacrylamide gel electrophoresis and
then electrotransferred onto nitrocellulose membranes. The
membranes were blocked with 5% non-fat powdered milk and then
incubated overnight at 4°C with the antibodies against rabbit
ERK1/2 (1:1,000) and p-ERK1/2 (1:1,000), rat ADRA2A (1:1,000) and
rabbit β-actin (1:1,000). Subsequently, the membranes were washed
in tris-buffered saline with Tween-20, goat anti-rabbit or goat
anti-rat secondary antibodies were added and the color was
developed using an enhanced chemiluminescence kit (cat. no.
orb90504; Wuhan Boster Biological Technology Ltd.). The values were
assayed using Quantity One software (version 4.62, Bio-Rad
Laboratories, Inc., Hercules, CA, USA). The relative optical
density of p-ERK1/2/ERK1/2 was calculated as the phosphorylation
level of ERK1/2. The relative optical density of ADRA2A/β-actin was
calculated as the expression level of ADRA2A.
Statistical analysis
SPSS statistical software version 19.0 (IBM Corp.,
Armonk, NY, USA) was used for data analysis. All quantitative data
are expressed as the mean ± standard deviation. One-way analysis of
variance followed by the Newman-Keuls test was used to make
statistical comparisons. P<0.05 was considered to indicate a
statistically significant difference.
Results
Morphological changes and GFAP
expression in the primary cultured astrocytes
To investigate the role of astrocytes in cerebral
ischemic injury, primary cultured astrocytes from the cerebral
cortex of newborn SD rats were separated, cultivated and purified
through an improved McCarthy method combined with trypsin
digestion, differential adhesion and vibration on a thermostatic
table. The astrocytes with a purity of 98% and which exhibited
typical layers of growth were collected and cultured. The adherent
cells covered the bottom of the bottle and exhibited clear growth
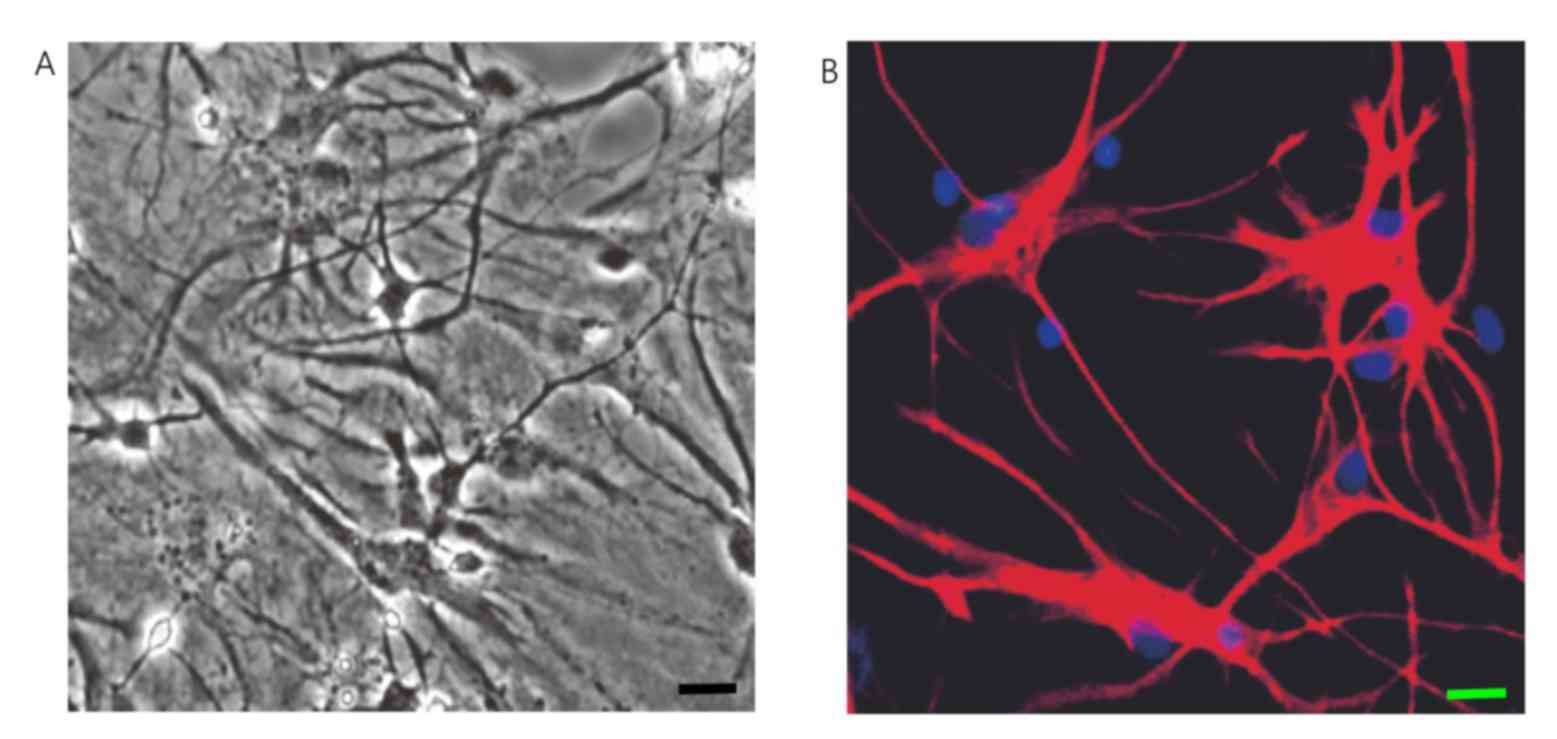
in a stratified manner under a light microscope after 9 to 12 days
culture (Fig. 2A). The
immunofluorescence results indicated that there were numerous
astrocytes expressing GFAP to a high level in the primary culture
after 12 days. When cells grew to fuse, the cell protrusions were
not clear. They were diverse in shape and size and the cell
protrusions were elongated at the edges of cells (Fig. 2B).
Effect of DEX pretreatment on ADRA2A
and ERK1/2 expression in primary cultured astrocytes under H/R
conditions
Astrocytes have important roles in normal and
pathological functioning of the central nervous system (CNS). A
previous study reported that ADRA2A colocalizes with GFAP in
primary astrocytes as identified by a double immunofluorescence
assay (14). To observe the
neuroprotective role and corresponding mechanism of DEX
pretreatment in cerebral I/R injury involving ADRA2A in
vitro in the present study, an astrocyte H/R model was
constructed.
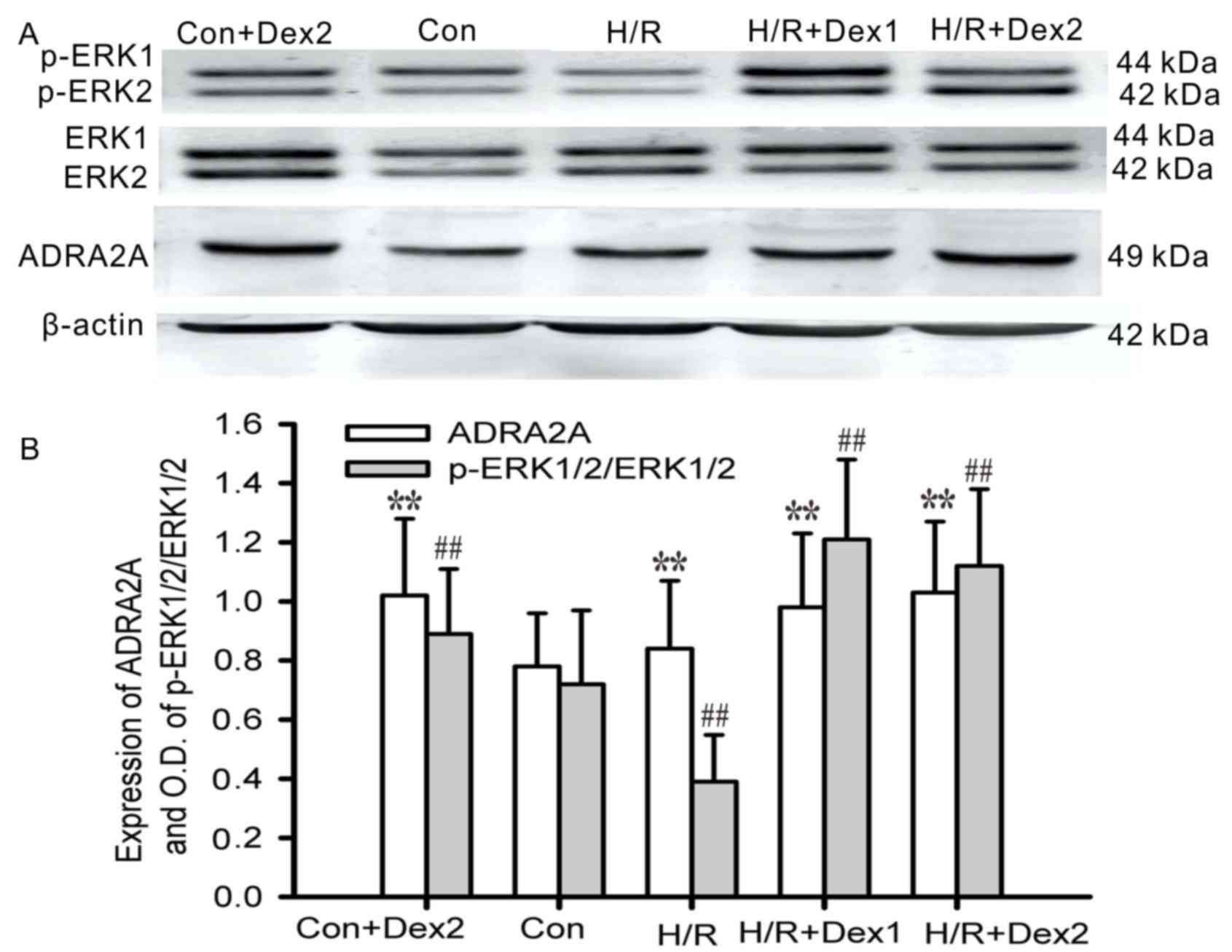
The levels of ERK1/2 expression in the astrocytes
were not notably different among the groups except for those in the
control group. Under the normal oxygen conditions, 500 ng/ml Dex
pretreatment increased the expression levels of ADRA2A and p-ERK1/2
in the astrocytes, compared with those in the control group
(P<0.01). Hypoxic culture for 6 h and then reoxygenation for 24
h decreased the levels of p-ERK1/2 expression in the astrocytes
compared with those in the control group (P<0.01), and this was
prevented by Dex pretreatment (100 or 500 ng/ml) for 3 h in the H/R
+ Dex groups, in which p-ERK1/2 expression was increased
(P<0.01). The hypoxic culture and then reoxygenation increased
the expression levels of ADRA2A (P<0.01; Fig. 3). These results suggested that Dex
promotes phosphorylation of ERK1/2 in astrocytes under H/R
conditions. As a specific agonist of ADRA2A, Dex may activate
phosphorylation of ERK1/2 through ADRA2A in astrocytes.
Successful construction of the focal
cerebral ischemia model
A focal cerebral I/R model was constructed to
investigate the neuroprotective role of Dex pretreatment in
cerebral ischemic injury. The regular variables were detected
during the surgical procedure. There were no notable differences in
the baseline physiological values such as pCO2,
pO2 and pH between the I/R and I/R Dex groups (Table I). Compared to those following 30 min
of ischemia and 2 h of reperfusion in the I/R group, the levels of
MAP were significantly reduced and then returned to normal, while
blood glucose levels were markedly increased and then returned to
normal in the Dex+I/R group (P<0.01). These results indicated
that the focal cerebral ischemia model was successful and suitable
for Dex pretreatment protection of brain function when subject to
cerebral ischemic injury.
 | Table I.Change of physiological variables at
different I/R periods in ischemic rats. |
Table I.
Change of physiological variables at
different I/R periods in ischemic rats.
|
| Baseline | 30 min of
ischemia | 2 h of
reperfusion |
|---|
|
|
|
|
|
|---|
| Variable | I/R | Dex+I/R | I/R | Dex+I/R | I/R | Dex+I/R |
|---|
| pH | 7.23±0.01 | 7.25±0.02 | 7.24±0.02 | 7.23±0.03 | 7.22±0.03 | 7.23±0.01 |
| MAP (mmHg) | 94±6 | 93±7 | 108±10 | 87±6a | 96±9 | 95±8 |
| pCO2
(mmHg) | 41±1 | 40±2 | 42±1 | 43±2 | 40±2 | 43±2 |
| pO2
(mmHg) | 93±6 | 94±5 | 90±2 | 89±3 | 92±6 | 92±7 |
| Blood glucose
(mM/l) | 5.9±0.7 | 6.6±0.6 | 5.0±0.7 |
9.5±0.7a | 5.3±0.5 | 6.2±0.5 |
Dex pretreatment increases neural
protection in focal cerebral I/R rat models
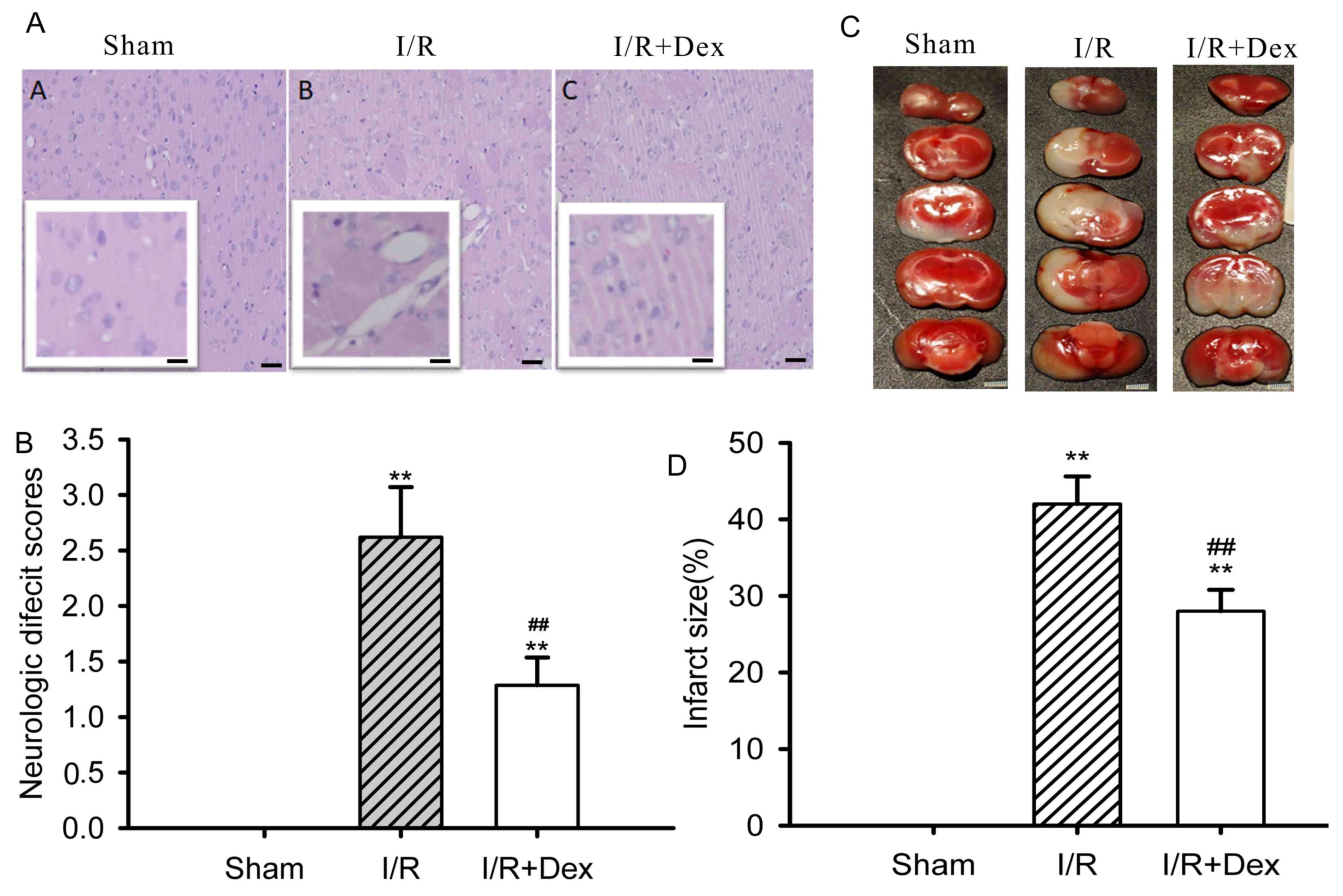
The neurological deficit score and cerebral infarct
volume in the I/R group were higher than those in the sham-operated
group (P<0.01; Fig. 4A). Dex
pretreatment notably reduced the damage to neurological function
(P<0.01; Fig. 4B) and brain
infarct volume induced by I/R (P<0.01; Fig. 4C and D).
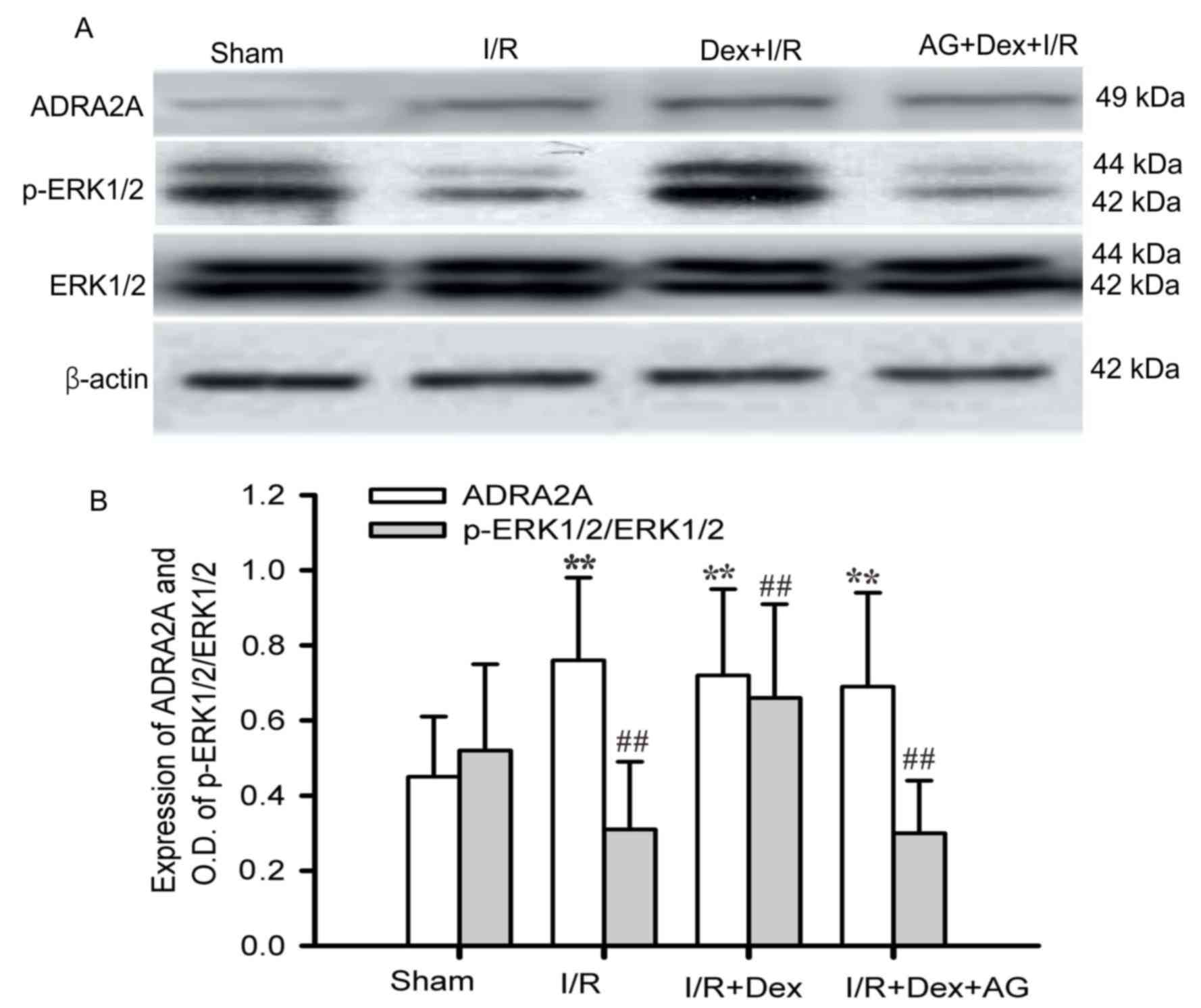
Dex pretreatment upregulates ADRA2A
and ERK1/2 expression in focal cerebral I/R brain tissues
The expression levels of ADRA2A in the ischemic
brain tissues of the I/R group were higher than those of the sham
group, while p-ERK1/2/ERK expression was higher in the Sham group
than in the I/R group. These effects were prevented by Dex
pretreatment. The expression levels of ADRA2A and p-ERK1/2 were
0.72±0.23 and 0.66±0.25 following Dex treatment, respectively,
compared with 0.76±0.22 and 0.31±0.18, respectively, prior to Dex
treatment. The effect of Dex pretreatment on increasing p-ERK1/2
expression was attenuated by AG1478 pretreatment in the ischemic
brain tissues (Fig. 5A and B). These
results indicated that the neuroprotective role of Dex pretreatment
in cerebral ischemic injury functions may occur via ADRA2A-mediated
phosphorylation of ERK1/2.
Discussion
Recent in-vitro experimental studies have
indicated that Dex pretreatment against cerebral ischemic damage in
cultured brain slices from the hippocampus induced neuroprotection
via ERK1/2 phosphorylation (20,21). In
the present study, Dex promoted phosphorylation of ERK1/2 in
astrocytes under H/R conditions. As a specific agonist of ADRA2A,
Dex may activate phosphorylation of ERK1/2 through ADRA2A in
astrocytes. Thus, the neuroprotective role of Dex pretreatment
against cerebral ischemia injury may function via ADRA2A-mediated
phosphorylation of ERK1/2.
As astrocytes are the most abundant subtype of cells
in the CNS, they are structurally and functionally involved in
normal brain function and responses to an ischemic lesion (22,23). A
number of studies have indicated that astrocytes are better
preserved than neurons in animal models of stroke outside of the
core in which all cells die, and that astrocytes are important
therapeutic targets for improving functional outcome, including
after stroke (1–4,22). The
many essential functions of astrocytes include K+
homeostasis, neurotransmitter synthesis and uptake, synapse
formation and regulation of the blood brain barrier (3,23). GFAP
expression occurs during reperfusion periods and increases in the
peri-infarct area (24). In the
present study, GFAP expression was preserved following MCAO-induced
focal cerebral I/R, which indicates that astrocytes serve key roles
in ischemic brain diseases.
An increasing number of studies have indicated that
astrocytes have important supporting and regulatory roles in brain
function (1–3). The essential metabolic needs of neurons
include K+ buffering, glutamate clearance, brain
antioxidant defense and the modulation of neuronal excitability
(23). The astrocytic contributions
to brain injury are extremely complex and not completely
understood, but it is established that astrocytes cause brain
injury-associated neuronal death (22). A previous study reported that failure
of astrocytes to support these needs of neurons leads to this
secondary injury (22).
Which of the three subtypes of α2 adrenoceptor,
namely α2A, α2B and α2C, mediates the neuroprotective effect of Dex
was examined in cell culture and in an in vivo model of
neonatal asphyxia (25). Although
the α2A and α2C subtypes are dominant in the CNS, all three
subtypes are widely distributed in the nervous system (25). The different functions of the
receptor subtypes result from their specific distributions in brain
tissues. In the mammalian CNS, the α2B receptor subtype is mainly
located in the thalamus, the α2A subtype is highly expressed in the
locus coeruleus (in the brain stem), and the α2A and α2C subtypes
are widely distributed in the brain (25). ADRA2A also exists in the presynaptic
and postsynaptic terminals, where it is mainly involved with the
inhibition of norepinephrine release and the excitement of neurons
(26,27). Dex mediates its main pharmacological
and cranial nerve protective effects via ADRA2A (28). In vivo activation of the locus
coeruleus may produce α2A adrenergic effects in astrocytes, while
in the CNS, the target cell type of α2A adrenergic agonists is the
astrocytes (28). Notably,
astrocytes treated with d-cAMP mainly express ADRA2A (29). Compared with neurons, endothelial
cells and microglia, adult mouse astrocytes have been demonstrated
to express ADRA2A more highly in the cerebral cortex (30); thus, the key site of the effect of
Dex in the whole brain appears to be astrocytes. It was previously
reported that ADRA2A was expressed and colocalized with GFAP in
astrocytes (14). In the present
study, there were numerous astrocytes highly expressing GFAP in
primary culture; however, a similar experiment to confirm that GFAP
was expressed together with ADRA2A was not performed and should be
verified in future study. In vitro studies have further
demonstrated that in cultured astrocytes and brain slices, Dex
activation of ADRA2A is dependent on the concentration of Dex
(8,29). Although doses of 1–100 µg/kg Dex may
have a neuroprotective effect during ischemia (31), it is not clear if Dex pretreatment
prior to focal cerebral I/R injury regulates ADRA2A expression in
astrocytes in ischemic brain diseases. In the present study, 500
ng/ml Dex pretreatment increased the expression of ADRA2A in the
astrocytes.
Dex has been demonstrated to evoke no ERK1/2
phosphorylation in cultured neurons but to activate phosphorylation
of ERK1/2 in primary cultures of mouse astrocytes, and neurons
could respond to astrocyte-conditioned medium with ERK1/2
phosphorylation (32). Dex may
activate astrocytes through ADRA2A, and promote the release of
different factors including GDNF or heparin-binding epidermal
growth factor (EGF)-like growth factor (HB-EGF) to protect neurons
following brain ischemia, potentially via different signaling
pathways (10–12,32).
Furthermore, Dex may induce GDNF release and increase the
expression of pERK1/2 via mechanisms independent of ADRA2A
activation. The I1-imidazoline receptors likely contribute to these
effects (11). However, EGF receptor
(EGFR) transactivation has been reported to lead to phosphorylation
of ERK1/2 in astrocytes themselves and in adjacent neurons, because
the astrocytes from the in vivo mature brain respond to
a2-adrenoceptor stimulation (33).
In the present study, Dex may have activated astrocytes through
ADRA2A, dependent on p-ERK1/2 activation, indicating that other
mechanisms enabling Dex to promote HB-EGF release through ADRA2A to
protect neurons cannot be ruled out, and such need to be validated
in further studies.
Dex concentrations of 25–500 nM can cause ERK1/2
phosphorylation in in-vitro cultured mouse astrocytes
(32). The EGFR tyrosine kinase
inhibitor AG1478, Zn2 +-dependent metal protease
inhibitor GM6001 and HB-EGF antagonist heparin can inhibit
phosphorylation of ERK1/2 and EGFR (32). However, whether Dex pretreatment
induces ERK1/2 phosphorylation remains unclear in in-vivo
ischemic brain tissues with MCAO-induced focal cerebral I/R. In the
present study, the results indicated that Dex increased ADRA2A
expression in focal cerebral I/R brain tissues and mediated
phosphorylation of ERK1/2.
The protective effects of Dex in cerebral ischemia
were achieved presently with pre- and post-conditioning (drug
administration prior to and following the ischemic episode), which
is important in a clinical context, since its ability to protect
brain tissue even after the onset of ischemia may increase its
therapeutic potential as a neuroprotective agent (34,35). The
present results indicated that Dex decreased the inflammatory
response of brain cells to ischemia, even when administrated 90 min
after the H/R period, which indicated that it appeared to be a
suitable and reasonable choice that Dex was administrated within 90
min after stoke.
In conclusion, a H/R cell model and focal cerebral
ischemia model were successfully constructed in the present study
in order to investigate the neuroprotective role and corresponding
mechanism of Dex pretreatment in cerebral ischemia injury. Dex
pretreatment appeared to increase ADRA2A and p-ERK1/2 expression in
primary cultured astrocytes and rat ischemic brain tissues.
Furthermore, the neuroprotective role of Dex pretreatment against
cerebral ischemia injury seemed to occur via ADRA2A-mediated
phosphorylation of ERK1/2. Since in vitro experiments cannot
recapitulate all aspects of an in vivo model, the
neuroprotective mechanism of Dex in astrocytes from cerebral
ischemia tissues should further be identified through in
vivo studies. Although many previous studies have used a sham
group as a control to investigate the neuroprotective role and
mechanism of Dex in cerebral ischemia injury in adult rats
(8,34,36,37), it
may be more effective to have a sham group that also receives Dex.
Elucidation of the neuroprotective mechanism of Dex in cerebral
ischemia will better guide the treatment of ischemic disease, and
targeting of astrocytes may be a novel treatment strategy for
ischemic cerebral disease.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Foundation of China (grant no. 81172467) and the Foundation
of Health and family Planning Commission of Hubei Province (grant
no. WJ2015MB153).
Availability of data and materials
All data generated or analyzed during the present
study are included in this published article.
Authors' contributions
YS contributed to drafting the manuscript and the
statistical analysis. XL constructed the in vivo model and
experiments that followed. GL contributed to in vitro
experiments. XP collected the data. MW designed the study and
drafted and revised the manuscript. All authors have critically
reviewed this manuscript and approved the final version to be
published.
Ethics approval and consent to
participate
The Ethics Committee for the Use of Experimental
Animals at Tongji Medical College of Huazhong University of Science
and Technology approved all animal experimental procedures.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
Dex
|
dexmedetomidine
|
|
I/R
|
ischemia/reperfusion
|
|
GFAP
|
glial fibrillary acidic protein
|
|
ADRA2A
|
α2A adrenergic receptor
|
|
ERK1/2
|
extracellular-signal regulated kinases
1 and 2
|
|
H/R
|
hypoxia/reoxygenation
|
|
(t)MCAO
|
(transient) middle cerebral artery
occlusion
|
|
GDNF
|
glial cell line-derived neurotrophic
factor
|
|
SD
|
Sprague-Dawley
|
|
BCA
|
bicinchoninic acid
|
|
p-
|
phosphorylated
|
|
EGFR
|
epidermal growth factor receptor
|
|
TTC
|
triphenyl tetrazolium chloride
|
|
d-cAMP
|
dibutyryl cyclic-adenosine
monophosphate
|
|
CCA
|
carotid artery
|
|
ECA
|
external carotid artery
|
|
ICA
|
internal carotid artery
|
|
CNS
|
central nervous system
|
|
EGF
|
epidermal growth factor
|
|
EGFR
|
epidermal growth factor receptor
|
|
HB-EGF
|
heparin-binding epidermal growth
factor-like growth factor
|
References
|
1
|
Ouyang YB, Xu L, Yue S, Liu S and Giffard
RG: Neuroprotection by astrocytes in brain ischemia: Importance of
microRNAs. Neurosci Lett. 565:53–58. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Tuttolomondo A, Di Sciacca R, Di Raimondo
D, Arnao V, Renda C, Pinto A and Licata G: Neuron protection as a
therapeutic target in cute ischemic stroke. Curr Top Med Chem.
9:1317–1334. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Barreto GE, Gonzalez J, Torres Y and
Morales L: Astrocytic-neuronal crosstalk: Implications for
neuroprotection from brain injury. Neurosci Res. 71:107–113. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Takano T, Oberheim N, Cotrina ML and
Nedergaard M: Astrocytes and ischemic injury. Stroke. 40 Suppl
3:S8–S12. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lee TH, Kato H, Chen ST, Kogure K and
Itoyama Y: Expression of nerve growth factor and trkA after
transient focal cerebral ischemia in rats. Stroke. 29:1687–1697.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Afonso J and Reis F: Dexmedetomidine:
Current role in anesthesia and intensive care. Rev Bras Anestesiol.
62:118–133. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kuhmonen J, Pokorný J, Miettinen R,
Haapalinna A, Jolkkonen J, Riekkinen P Sr and Sivenius J:
Neuroprotective effects of dexmedetomidine in the gerbil
hippocampus after transient global ischemia. Anesthesiology.
87:371–377. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhu YM, Wang CC, Chen L, Qian LB, Ma LL,
Yu J, Zhu MH, Wen CY, Yu LN and Yan M: Both PI3K/Akt and ERK1/2
pathways participate in the protection by dexmedetomidine against
transient focal cerebral ischemia/reperfusion injury in rats. Brain
Res. 1494:1–8. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kose EA, Bakar B, Kasimcan O, Atilla P,
Kilinc K, Muftuoglu S and Apan A: Effects of intracisternal and
intravenous dexmedetomidine on ischemia-induced brain injury in
rat: A comparative study. Turk Neurosurg. 23:208–217.
2013.PubMed/NCBI
|
|
10
|
Degos V, Charpentier TL, Chhor V, Brissaud
O, Lebon S, Schwendimann L, Bednareck N, Passemard S, Mantz J and
Gressens P: Neuroprotective effects of dexmedetomidine against
glutamate agonist-induced neuronal cell death are related to
increased astrocyte brain-derived neurotrophic factor expression.
Anesthesiology. 118:1123–1132. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Dahmani S, Paris A, Jannier V, Hein L,
Rouelle D, Scholz J, Gressens P and Mantz J: Dexmedetomidine
increases hippocampal phosphorylated extracellular signal-regulated
protein kinase 1 and 2 content by an alpha
2-adrenoceptor-independent mechanism: Evidence for the involvement
of imidazoline I1 receptors. Anesthesiology. 108:457–466. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yan M, Dai H, Ding T, Dai A, Zhang F, Yu
L, Chen G and Chen Z: Effects of dexmedetomidine on the release of
glial cell line-derived neurotrophic factor from rat astrocyte
cells. Neurochem Int. 58:549–557. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jana M, Jana A, Pal U and Pahan K: A
simplified method for isolating highly purified neurons,
oligodendrocytes, astrocytes, and microglia from the same human
fetal brain tissue. Neurochem Res. 32:2015–2022. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liu H, Davis JR, Wu ZL and Faez Abdelgawad
A: Dexmedetomidine attenuates lipopolysaccharide induced MCP-1
expression in primary astrocyte. Biomed Res Int.
2017:63521592017.PubMed/NCBI
|
|
15
|
Chai L, Guo H, Li H, Wang S, Wang YL, Shi
F, Hu LM, Liu Y and Adah D: Scutellarin and caffeic acid ester
fraction, active components of Dengzhanxixin injection, upregulate
neurotrophins synthesis and release in hypoxia/reoxygenation rat
astrocytes. J Ethnopharmacol. 150:100–107. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Longa EZ, Weinstein PR, Carlson S and
Cummins R: Reversible middle cerebral artery occlusion without
craniectomy in rats. Stroke. 20:84–91. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Levitzki A and Gazit A: Tyrosine kinase
inhibition: An approach to drug development. Science.
267:1782–1788. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Alessandrini A, Namura S, Moskowitz MA and
Bonventre JV: MEK1 protein kinase inhibition protects against
damage resulting from focal cerebral ischemia. Proc Natl Acad Sci
USA. 96:12866–12869. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang L, Lu YY, Zhou Q and Shui XL:
Establishment and evaluation of a murine cerebral
ischemia-reperfusion model. Hainan Med J. 25:2965–2969. 2014.(In
Chinese).
|
|
20
|
Justicia C and Planas AM: Transforming
growth factor-alpha acting at the epidermal growth factor receptor
reduces infarct volume after permanent middle cerebral artery
occlusion in rats. J Cereb Blood Flow Metab. 19:128–132. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gu L, Li B, Yang X, Hu X, Huang X, Hertz L
and Peng L: Depolarization-induced, glutamate receptor-mediated,
and transactivation-dependent extracellular-signal regulated kinase
phosphorylation in cultured cerebellar granule neurons.
Neuroscience. 147:342–353. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liu Z and Chopp M: Astrocytes, therapeutic
targets for neuroprotection and neurorestoration in ischemic
stroke. Prog Neurobiol. 144:103–120. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Barreto G, White RE, Ouyang Y, Xu L and
Giffard RG: Astrocytes: Targets for neuroprotection in stroke. Cent
Nerv Syst Agents Med Chem. 11:164–173. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhang S, Wu M, Peng C, Zhao G and Gu R:
GFAP expression in injured astrocytes in rats. Exp Ther Med.
14:1905–1908. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ma D, Hossain M, Rajakumaraswamy N, Arshad
M, Sanders RD, Franks NP and Maze M: Dexmedetomidine produces its
neuroprotective effect via the alpha 2A-adrenoceptor subtype. Eur J
Pharmacol. 502:87–97. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Milner TA, Lee A, Aicher SA and Rosin DL:
Hippocampal alpha2a-adrenergic receptors are located predominantly
presynaptically but are also found postsynaptically and in
selective astrocytes. J Comp Neurol. 395:310–327. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hein L, Altman JD and Kobilka BK: Two
functionally distinct alpha2-adrenergic receptors regulate
sympathetic neurotransmission. Nature. 402:181–184. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Enkvist MO, Hämäläinen H, Jansson CC,
Kukkonen JP, Hautala R, Courtney MJ and Akerman KE: Coupling of
astroglial alpha 2-adrenoreceptors to second messenger pathways. J
Neurochem. 66:2394–2401. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hertz L, Lovatt D, Goldman SA and
Nedergaard M: Adrenoceptors in brain: Cellular gene expression and
effects on astrocytic metabolism and [Ca(2+)]i. Neurochem Int.
57:411–420. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Huang R and Hertz L: Receptor subtype and
dose dependence of dexmedetomidine-induced accumulation of
[14C]glutamine in astrocytes suggests glial involvement in its
hypnotic-sedative and anesthetic-sparing effects. Brain Res.
873:297–301. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Jolkkonen J, Puurunen K, Koistinaho J,
Kauppinen R, Haapalinna A, Nieminen L and Sivenius J:
Neuroprotection by the alpha2-adrenoceptor agonist,
dexmedetomidine, in rat focal cerebral ischemia. Eur J Pharmacol.
372:31–36. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Du T, Li B, Liu S, Zang P, Prevot V, Hertz
L and Peng L: ERK phosphorylation in intact, adult brain by
alpha(2)-adrenergic transactivation of EGF receptors. Neurochem
Int. 55:593–600. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Peng L, Li B, Du T, Kong EK, Hu X, Zhang
S, Shan X and Zhang M: Astrocytic transactivation by
alpha2A-adrenergic and 5-HT2B serotonergic signaling. Neurochem
Int. 57:421–431. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wang SL, Duan L, Xia B, Liu Z, Wang Y and
Wang GM: Dexmedetomidine preconditioning plays a neuroprotective
role and suppresses TLR4/NF-κB pathways model of cerebral ischemia
reperfusion. Biomed Pharmacother. 93:1337–1342. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Rodríguez-González R, Sobrino T, Veiga S,
López P, Rodríguez-García J, del Río SV, Baluja A, Castillo J and
Álvarez J; Neuroprotective effects of dexmedetomidine conditioning
strategies, . Evidences from an in vitro model of cerebral
ischemia. Life Sci. 144:162–169. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Cheng J, Zhu P, Qin H, Li X, Yu H, Yu H
and Peng X: Dexmedetomidine attenuates cerebral
ischemia/reperfusion injury in neonatal rats by inhibiting TLR4
signaling. J Int Med Res. 46:2925–2932. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yuan F, Fu H, Sun K, Wu S and Dong T:
Effect of dexmedetomidine on cerebral ischemia-reperfusion rats by
activating mitochondrial ATP-sensitive potassium channel. Metab
Brain Dis. 32:539–546. 2017. View Article : Google Scholar : PubMed/NCBI
|