Introduction
Obstructive sleep apnea (OSA) is characterized by
frequent nocturnal hypoxia, microarousal, oxidative stress and
sympathetic activation, which is closely associated with
hypertension, diabetes, chronic kidney disease (CKD) and other
chronic diseases (1,2). Previously, the association between
sleep apnea and CKD has been widely discussed. The occurrence,
development and prognosis of CKD and OSA are closely associated
(3,4). OSA, as a risk factor for CKD, requires
improved control to decrease the incidence of potential injuries
that may occur. In addition to continuous positive airway pressure
(CPAP) treatment, an organ protection strategy is particularly
important in decreasing OSA-associated kidney injury. The method
for overcoming this problem is to identify the core molecular
mechanism in OSA-induced renal injury and investigate potential
drug therapies. At present, evidence suggests that the primary
mechanism of OSA-induced target organ injury is through immune
inflammation (5).
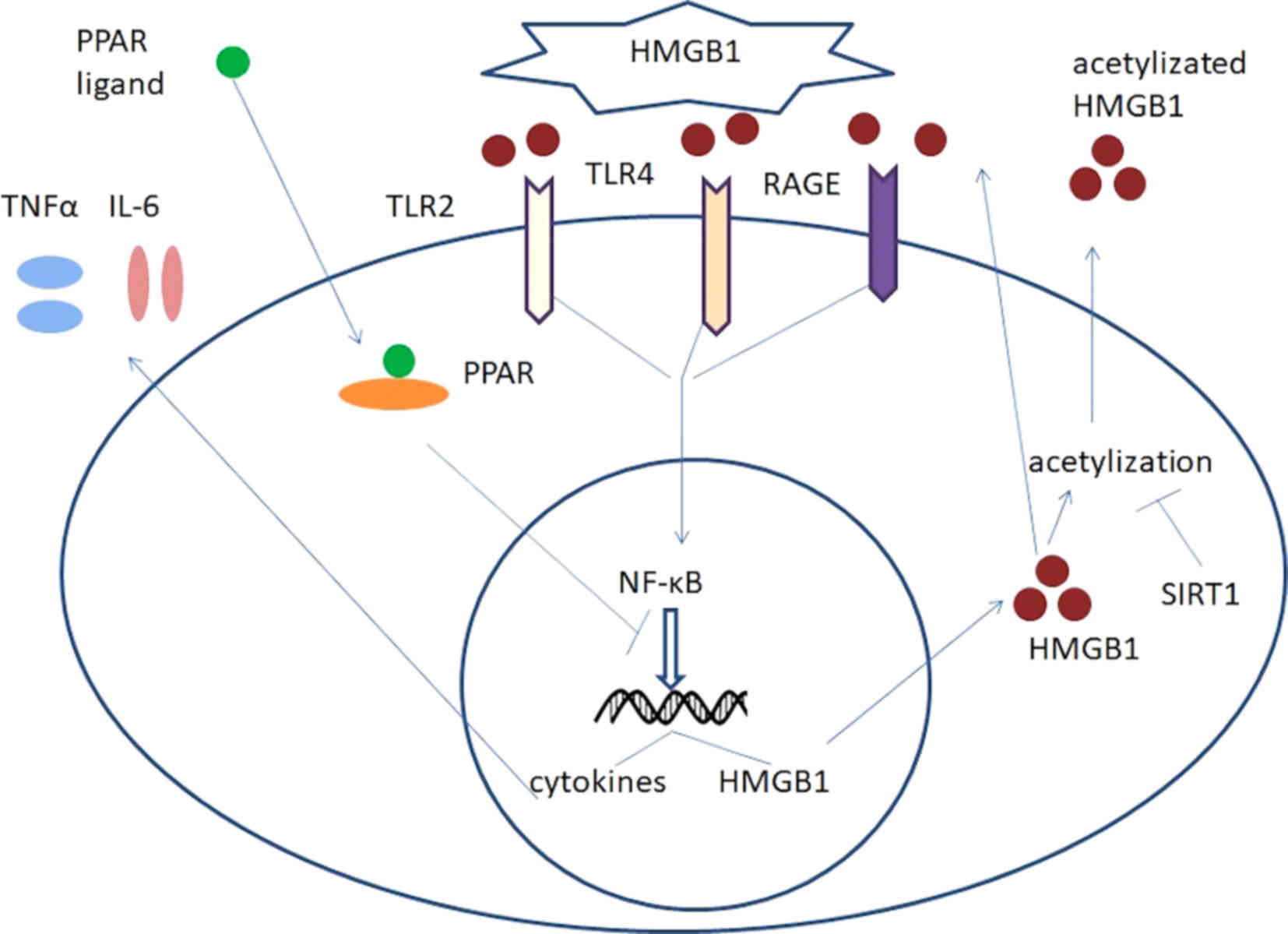
Previously, high mobility group box 1 protein
(HMGB1), as the most important representative damage-associated
molecular pattern (DAMP) (6,7), has attracted increasing attention.
HMGB1 serves as a proinflammatory agent and may have a central role
in inflammation (8,9). HMGB1 is a highly conserved nuclear
protein that is widely expressed in all types of mammalian cells,
and is transferred from the nucleus to the cytoplasm and then
released extracellularly upon receipt of inflammatory stimuli.
Extracellular HMGB1 may promote inflammation by
binding to receptors on effector cell membranes, including
macrophages and dendritic cells and leads to the release of
inflammatory mediators, including interleukin (IL)-6 and tumor
necrosis factor (TNF)-α. In turn, the release of IL-6 and TNF-α
leads to increased HMGB1 release, resulting a cascade amplification
of inflammation (8,9). The cell membrane receptors, in
combination with HMGB1, are primarily receptors for advanced
glycation end products (RAGE) (10)
and Toll-like receptor (TLR) 4 and TLR2 (11). RAGE is a transmembrane protein of the
immunoglobulin superfamily. It is widely expressed on a variety of
cell surfaces, but the expression levels in normal tissues are low
(12). When the ligands, including
AGEs, HMGB1, S100 proteins are aggregated, the expression of RAGE
increases (2,13). TLRs are type I transmembrane proteins
that activate the innate immune system (14). HMGB1 induces inflammation through
binding to RAGE and TLR receptors through the nuclear factor
kappa-light-chain-enhancer of active B cells (NF-κB) pathway.
The two inhibitors of this pathway are NAD-dependent
protein deacetylase sirtuin-1 (SIRT1) and peroxisome
proliferator-activated receptor (PPAR). SIRT1 is a member of the
silent mating type information regulation 2 homolog (Sirtuin)
family, and is a key anti-aging gene (15). The SIRT1 protein is an NAD+ dependent
enzyme that deacetylates proteins, which contributes to cellular
regulation (16). As an important
deacetylase, SIRT1 may decrease the extent of HMGB1 acetylation and
thereby decrease its activation and subsequent release into
extracellular regions. In addition, PPAR is a member of the nuclear
receptor transcription factor superfamily and includes three
subtypes: PPARα; PPAR β; and PPARγ, which are also types of
ligand-inducible nuclear receptors (17). A previous study indicated that when
combined with its inhibitory effect on the NF-κB signaling pathway
and therefore inhibit the transcription of HMGB1. The
HMGB1-associated signaling pathways are summarized in Fig. 1.
In the present study, various intermittent hypoxia
models were established to investigate the effects of hypoxia on
the expression of HMGB1-RAGE/TLR-TNF-α signaling pathway in kidney
tissues and peripheral blood, with the aim of identifying the
molecular markers of OSA-induced renal injury and potential targets
of drug intervention.
Materials and methods
Experimental animals
A total of 32 healthy adult male Sprague-Dawley (SD)
rats weighing 280–320 g were provided by Beijing Weitong Lihua
Experimental Animal Center (Beijing, China) and raised by
Laboratory Animal Center of Peking University First Hospital
(Beijing, China). The animals had ad libitum access to food
and water, and were maintained at a temperature of 22±2°C and
humidity of 40%, in a 12-h light/dark cycle. The experimental
animal procedures conformed with the relevant provisions of
Regulations of the People's Republic of China on the Administration
of Laboratory Animals. The present study was approved by the Animal
Ethics Committee of Peking University First Hospital.
Reagents
Reverse transcription-quantitative polymerase chain
reaction (RT-qPCR) reagents were purchased from Beijing TransGen
Biotech Co., Ltd., (Beijing, China), which included Trans Zol Up
Plus RNA kit (TransScript II All-in-One First-Strand cDNA Synthesis
SuperMix for qPCR) and qPCR kit (TransStart Top Green qPCR kit
SuperMix+Dye II).
The PCR primers were synthesized by Sangon Biotech
Co., Ltd. (Shanghai, China) and primer sequences are summarized in
Table I.
 | Table I.Quantitative polymerase chain
reaction primer sequences of HMGB1, TLR4, NF-κB, RAGE, TNF-α, IL-6,
PPAR, SIRT1 and internal reference 28S. |
Table I.
Quantitative polymerase chain
reaction primer sequences of HMGB1, TLR4, NF-κB, RAGE, TNF-α, IL-6,
PPAR, SIRT1 and internal reference 28S.
| Gene | Sequence
(5′-3′) |
|---|
| HMGB1-F |
CTAGCCCTGTCCTGGTGGTATT |
| HMGB1-R |
CCAATTTACAACCCCCAGACTGT |
| TLR4-F |
TCAACCCCCTGAAGATCTTAAGAA |
| TLR4-R |
ATGCCTTGTCTTCAATTGTCTCAA |
| RELA |
TGGCTTCTATGAGGCTGAACTCT |
| (NFκB3P65)-F |
|
| RELA |
GGATCCCCAGGTTCTGGAA |
| (NFκB3P65)-R |
|
| RAGE-F |
AGGAAAGCCCTCCTGTCAACA |
| RAGE-R |
CACAGAGCCTGCAGCTTGTC |
| TNF-α-F |
CCACCACGCTCTTCTGTCTA |
| TNF-α-R |
ACTGATGAGAGGGAGCCCATT |
| IL-6-F |
TCTGGTCTTCTGGAGTTCCGT |
| IL-6-R |
GCATTGGAAGTTGGGGTAGGA |
| PPARγ-F |
TGACTTGGCCATATTTATAGCTGTCA |
| PPARγ-R |
GATGTCCTCGATGGGCTTCA |
| Sirt1-F |
GCTGGCCTAATAGACTTGCAAAG |
| Sirt1-R |
ATGTAACGATTTGGTGGTACAAACA |
| 28S ribosomal |
TGCCATGGTAATCCTGCTCA |
| RNA-F |
|
| 28S ribosomal |
CCTCAGCCAAGCACATACACC |
| RNA-R |
|
For the ELISA protocols: Rat TLR2 ELISA kit (cat.
no., H299) and Rat PPAR ELISA kit (cat. no., H271; Nanjing
Jiancheng Bioengineering Institute, Nanjing, China), TLR4 (cat.
no., SEA753Ra; Cloud-Clone Corp., Katy, TX, USA), IL-6 (cat. no.,
R6000B) and TNF-α (cat. no., RTA00; both R&D Systems, Inc.,
Minneapolis, MN, USA) were purchased.
For the immunohistochemistry protocols, the primary
antibody was HMGB1 rabbit mAb (cat. no., 6893S; Cell Signaling
Technology, Inc., Danvers, MA, USA) at a dilution of 1:400. The
secondary antibody used was a horseradish peroxide (HRP)-conjugated
goat anti-rabbit IgG (cat. no., Ab6721; Abcam, Cambridge, UK) at a
dilution of 1:200.
Establishment of hypoxia models and
specimen collection
Hypoxia models
A hypoxia chamber was jointly developed with the
Academy for Advanced Interdisciplinary Studies, Peking University
(Beijing, China).
Continuous hypoxia (CH)
The rats were placed in the hypoxia chamber for 8
h/day for 2 weeks. The oxygen concentration in the hypoxia chamber
was maintained at 10.0±0.5% (n=8).
Intermittent hypoxia (IH)
The rats were placed in the hypoxia chamber for 8
h/day for 2 weeks. The oxygen concentration in the hypoxia chamber
was maintained at between 10–21% with a 2 min cycle time (10%
O2 for 1 min-21% O2 for 1 min; n=8).
Intermittent hypoxia with hypercapnia
(IHH)
The rats were placed in the hypoxia chamber for 8
h/day for 2 weeks. The oxygen and CO2 concentration in
the hypoxia chamber was maintained with a 2 min circulation time
(10% O2+5% CO2 for 1 min then 21%
O2+0–0.5% CO2 for 1 min; n=8).
Control (C)
SD rats were placed in the chamber with ad
libitum access to food and water, breathing normal air for 8
h/day for 2 weeks (n=8).
Specimen collection
On the day following the completion of the
intervention, the rats were intraperitoneally anesthetized with 4
ml/kg 10% chloral hydrate (0.4 g/kg). Then, blood samples were
collected for a blood routine test and for collecting plasma
following centrifugation (716 × g; 10 min; room temperature). Rats
were sacrificed and the kidney tissues of the rats were collected
and preserved in a refrigerator at −80°C, fixed in 4%
paraformaldehyde at room temperature for 24–48 h or 2.5%
glutaraldehyde at 4°C for 2–12 h for subsequent analysis.
Specimen detection
Hematoxylin and eosin (HE) and
immunohistochemistry (IHC) staining sample preparation
Kidney tissues were immediately fixed in 4%
paraformaldehyde at room temperature for 24–48 h and then
dehydrated with an ethanol concentration gradient (50, 70, 85, 95
and 100%) then cleared in xylene of room temperature and finally
embedded in paraffin. The tissue blocks were sectioned at 5 µm
thickness, and then used for HE and IHC staining.
HE staining
All steps were performed at room temperature. Prior
to staining, the sections were dewaxed in xylene (100%; 20 min),
rehydrated through decreasing concentrations of ethanol (100% for
10 min; and 95, 90, 80 and 70% for 5 min each) and washed in PBS.
Samples were incubated in hematoxylin solution for 8–15 min and
wash with water 1–2 min. Samples were then placed in 1%
hydrochloric acid in alcohol for differentiation for a 30 sec,
followed by washing with water (30–60 min). Samples were immersed
in 1% eosin solution for 2–5 min followed by washing with water.
Following staining, sections were dehydrated through increasing
concentrations of ethanol and xylene (ethanol, 95 and 100% for 5
min; xylene, 100% 5 min).
IHC
Hydrogen peroxide (3%) was used for 10 min at room
temperature for blocking of endogenous peroxides. Sections were
blocked with 10% fetal bovine serum (FBS; ExCell Biology, Inc.,
Shanghai, China) at room temperature for 1 h. Following the removal
of FBS, sections were incubated with the HMGB1 primary antibody
(rabbit; cat. no., 6893S; Cell Signaling Technology, Inc.) at 1:400
dilution at 4°C overnight. Incubation with the secondary antibody
(goat anti-Rabbit IgG; 1:200; cat. no., Ab6721; Abcam) was
performed at room temperature for 1 h. Staining of HE and IHC were
observed by light microscope (magnifications, ×100, ×200 and
×400).
Transmission electron microscope
observation
A small section (1–2 mm3) of kidney
tissue was fixed in 2.5% glutaraldehyde (4°C; 2–12 h), post-fixed
in 1% osmium tetroxide, dehydrated in serial acetone (50, 70, 90
and 100%), and embedded in Epon 812 resin (37°C for 6 h; 60°C for
48 h). The ultrathin sections (70–90 nm) were then stained using
saturated uranyl acetate (15 min; room temperature) and standard
lead citrate solution (A, 1.33 g Pb(NO3)2 and
1.76 g
Na3C6H5O7x2H2O
in 30 ml water; B, 1M NaOH; 30 ml A plus 8 ml B to 50 ml with
water; 15 min; room temperature), then examined by a transmission
electron microscope (magnifications, ×8,000 and ×15,000; JEM-1230;
JEOL Ltd., Tokyo, Japan).
RT-qPCR
Total RNA was extracted from the kidney tissues
using TRIZOL method. Total RNA quantification and purity were
determined by ultraviolet spectrophotometry. cDNA was reverse
transcribed by random primers using the Trans Script II All-in-One
First-Strand cDNA Synthesis SuperMix for qPCR kit according to the
protocol of the manufacturer. The reverse transcription cDNA
products were stored at −80°C for PCR amplification.
According to the instruction of the SYBR Green PCR
kit, the following reagents were added to the PCR tube: 1 µl cDNA
template, 0.4 µl forward primer (10 µM), 0.4 µl reverse primer (10
µM), 10 µl 2X TransStar Tip Green qPCR SuperMix, 0.4 µl Passive
Reference Dye (50X) and 7.8 µl ddH2O to make a 20 µl
total reaction system. The thermocycling conditions for the PCR
were as follows: 40 cycles of 94°C for 30 sec; 94°C for 5 sec and
60°C for 30 sec. The thermocycling conditions for the melting curve
analysis were: 95°C for 15 sec, 60°C for 60 sec, 95°C for 30 sec
and 60°C for 15 sec. The real time qPCR was completed using an ABI
7500 Realtime PCR system. The 28S gene was used as the reference
gene. The 2−ΔΔCq method (18) was used to analyze the data.
ELISA
These experiments were performed according to the
protocols of the kit manufacturers. All reagents and working fluids
were prepared and maintained at room temperature. The 96-well
enzyme-labeled plates were prepared. The standard solution and
peripheral blood samples were added, with two repeated wells for
each sample. HRP was added, mixed and incubated. The plates were
washed and dried. The color reagent was added to each well. Then,
the termination fluid was added. The optical density value of each
well was measured at 450 nm. A standard curve was calculated and
the concentration of each specimen was calculated.
Statistical analysis
SPSS 17 statistical software (SPSS, Inc., Chicago,
IL, USA) was used for data processing. Continuous variables are
presented as the mean ± standard deviation. Comparisons between the
groups were evaluated by one-way analysis of variance followed by
Least Significant Difference multiple comparison, or by the
nonparametric Kruskal-Wallis test, according to the results of the
homogeneity of variance test. P<0.05 was considered to indicate
a statistically significant difference.
Results
Routine blood testing
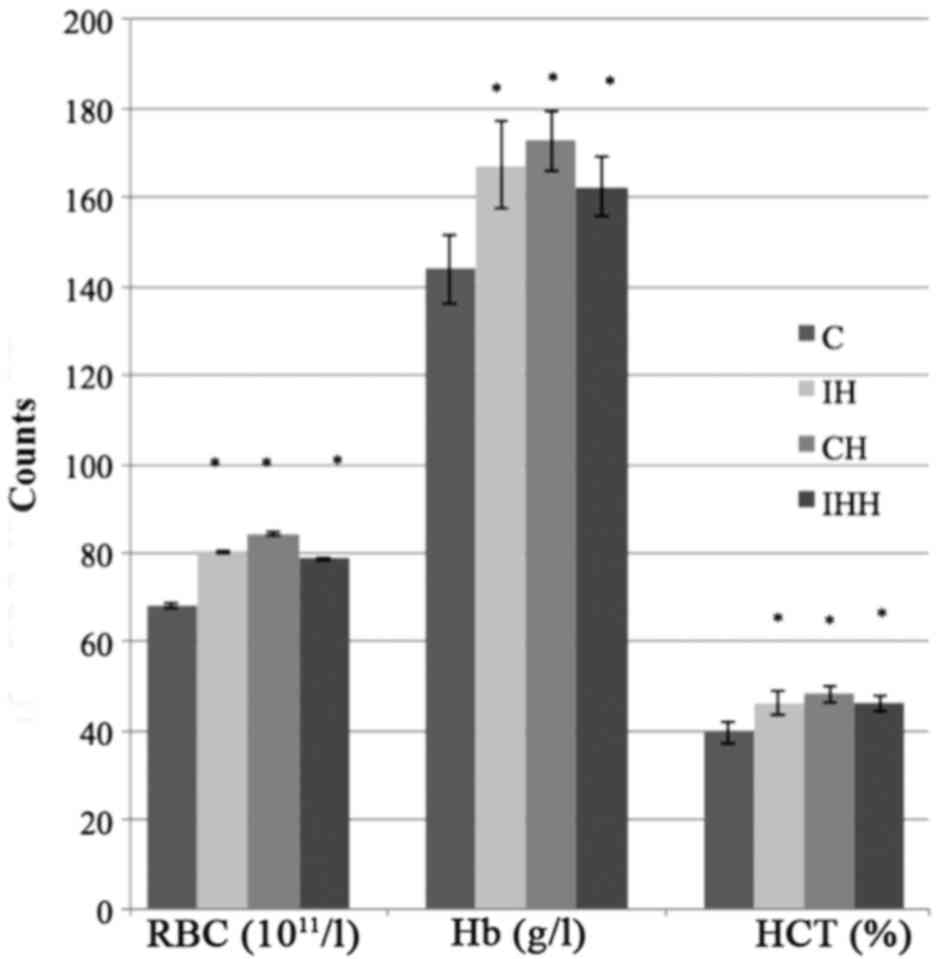
The red-cell count, hemoglobin and hematocrit levels
of the three different models of hypoxia were significantly
increased compared with those of control group (P<0.05; Fig. 2), suggesting that successful hypoxia
models were established.
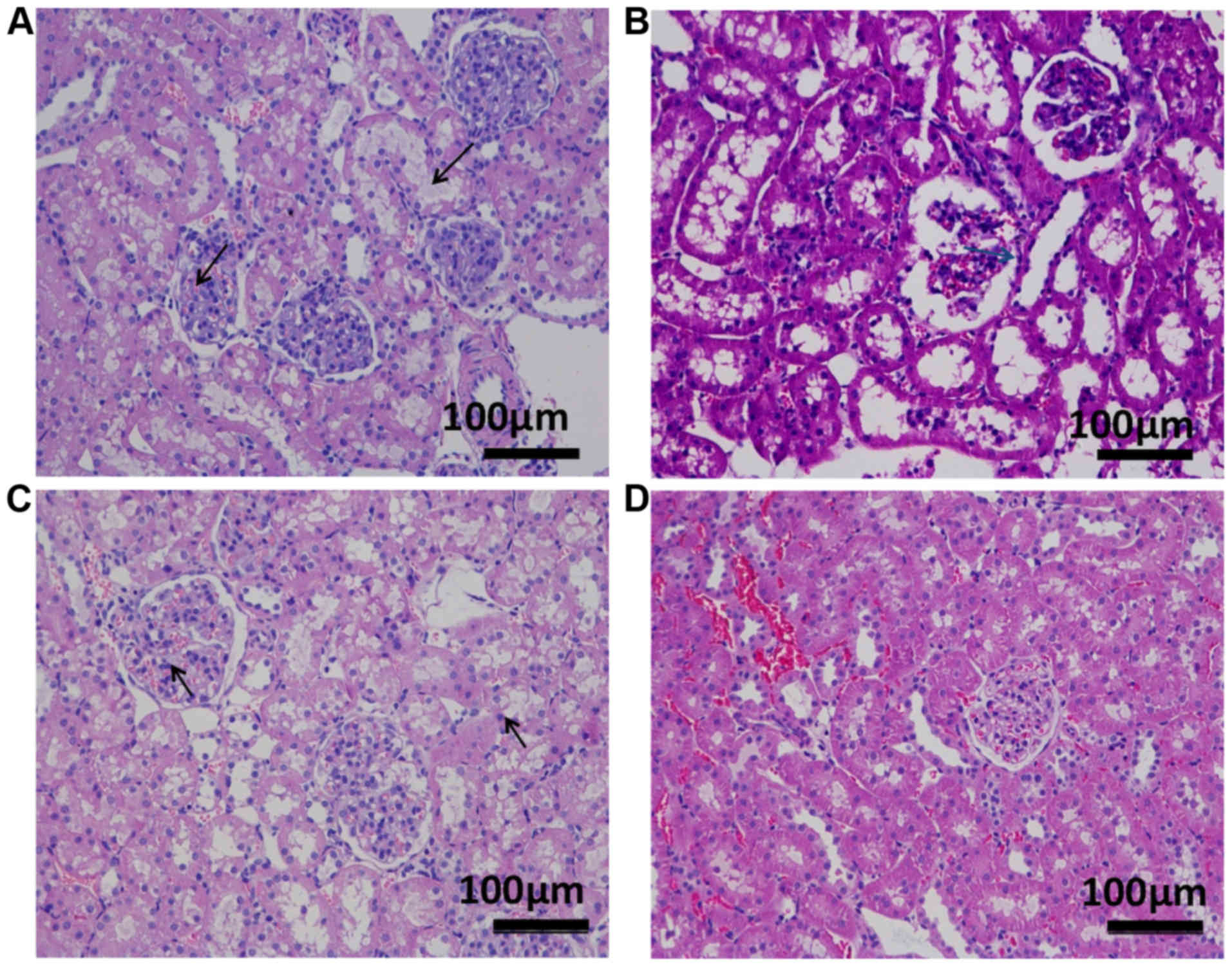
Renal HE staining
Overall, kidney injury was not serious in any of the
different hypoxia groups, as indicated by the HE staining. The HE
staining demonstrated a certain degree of renal tubular epithelial
cell vacuolation in the different types of hypoxia groups, as
presented in Fig. 3.
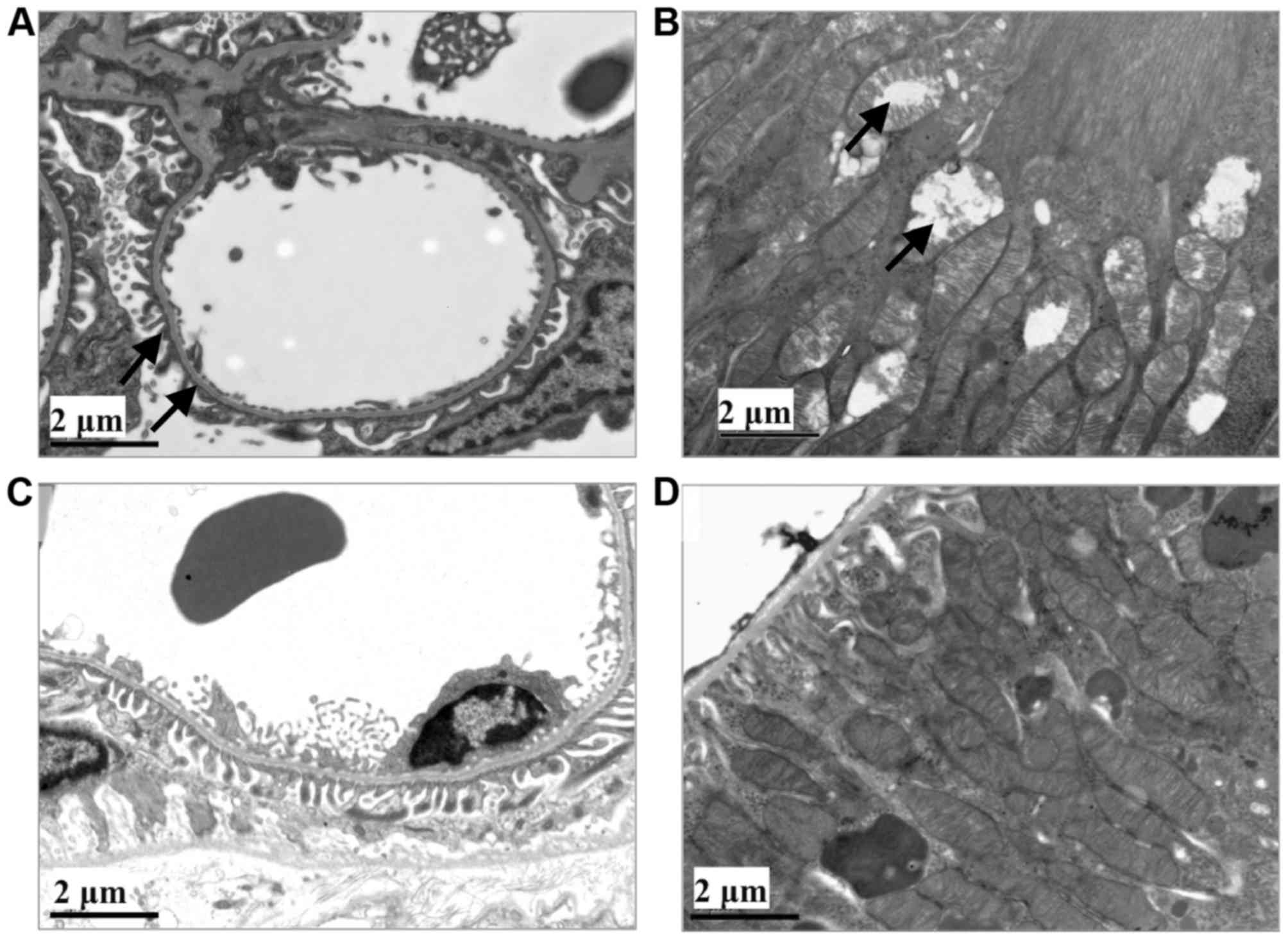
Ultrastructural observation
Transmission electron micrographs of rat kidney
tissues were captured. Fusion of the foot process was visualized in
the glomerulus. In the proximal convoluted tubules, additional
secondary lysosomes and vacuoles were observed. Swelling and
degeneration of mitochondria was identified in the tubules
(Fig. 4).
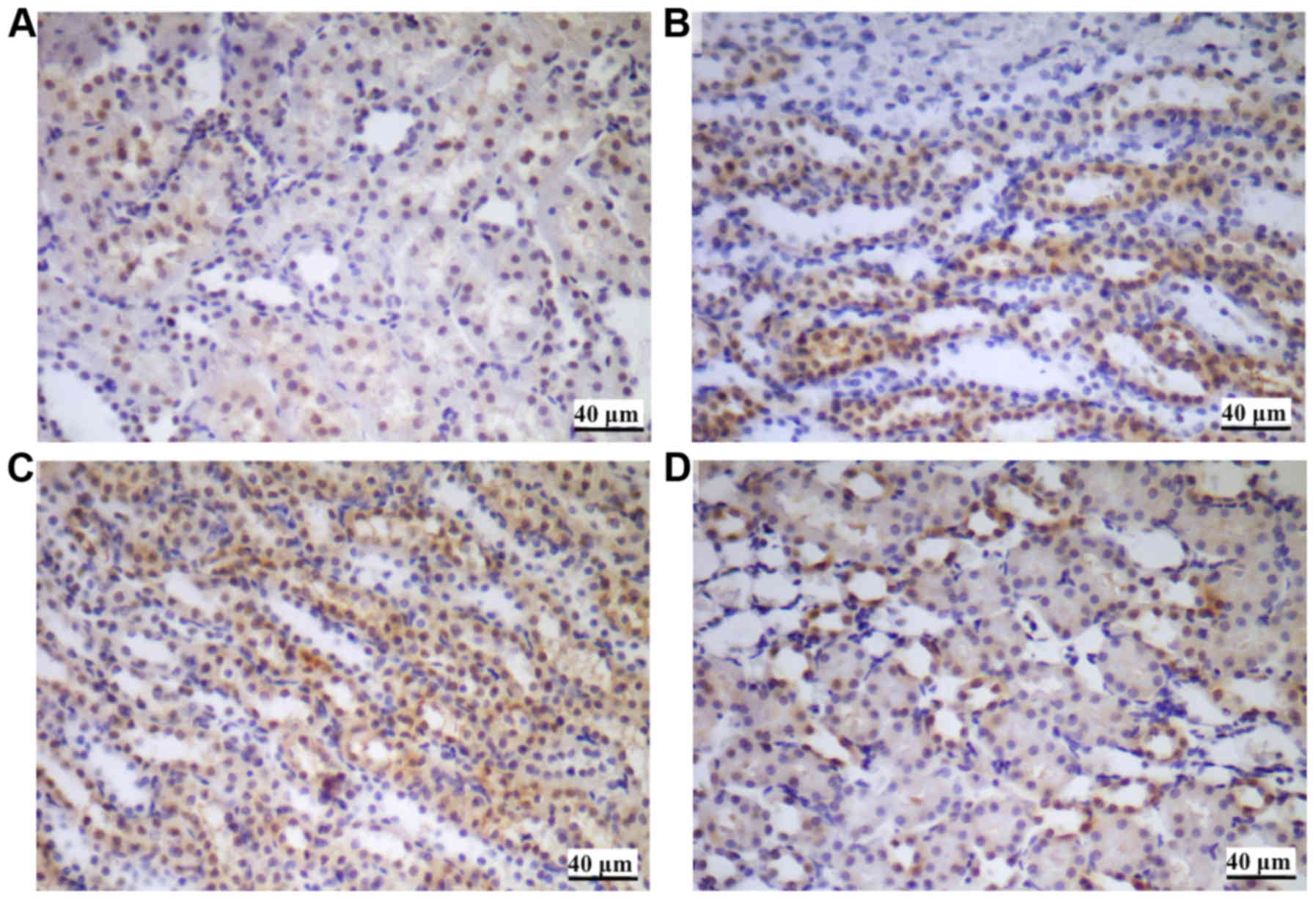
HMGB1 immunohistochemical staining of
renal tissues
HMGB1 immunohistochemical staining of renal tissues
suggested that the HMGB1 protein was primarily expressed in the
nucleus of the renal tubular epithelial cells in the control group.
In the hypoxia groups, the expression of HMGB1, in particular the
extracellular expression was increased, suggesting the upregulation
of nuclear translocation (from nucleus to cytoplasm) during
stimulation by hypoxia (Fig. 5).
mRNA expression of HMGB1 signaling
detected by RT-qPCR
Hypoxia stimulated the expression of HMGB1, TNF-α,
PPAR and RAGE in the rat renal tissues, and decreased the
expression of SIRT1 (Fig. 6;
Table II). Compared with the
control group (0.61±0.24), the expression of HMGB1 mRNA in the CH
(1.14±0.35), IH (1.30±0.40) and IHH (1.10±0.42) groups was
significantly upregulated (P<0.05). In addition, compared with
the control group (1.15±0.13), IH treatment (2.47+0.85) caused a
significant increase in RAGE mRNA expression (P<0.05). CH
(1.71+0.54) and IHH (1.30+0.47) treatments also resulted an
increase in RAGE mRNA expression, but the differences were not
statistically significant (P>0.05). The expression of PPAR and
TNF-α mRNA in the CH, IH and IHH groups was significantly increased
(P<0.05) compared with the control group. The IL-6 mRNA levels
in the CH, IH and IHH groups were also increased compared with the
control group, but the differences were not statistically
significant (P>0.05; Table II).
No significant differences in the expression of NF-κB and TLR4 mRNA
were observed in the hypoxia groups compared with the control group
(P>0.05). Levels of SIRT1 mRNA in the IHH group (0.73+0.27) were
significantly decreased compared with the control group (1.0±0.07;
P<0.05). No significant differences in SIRT1 mRNA expression in
the CH (0.88±0.21) and IH (0.94 ±0.29) groups compared with the
control group (P>0.05) were observed.
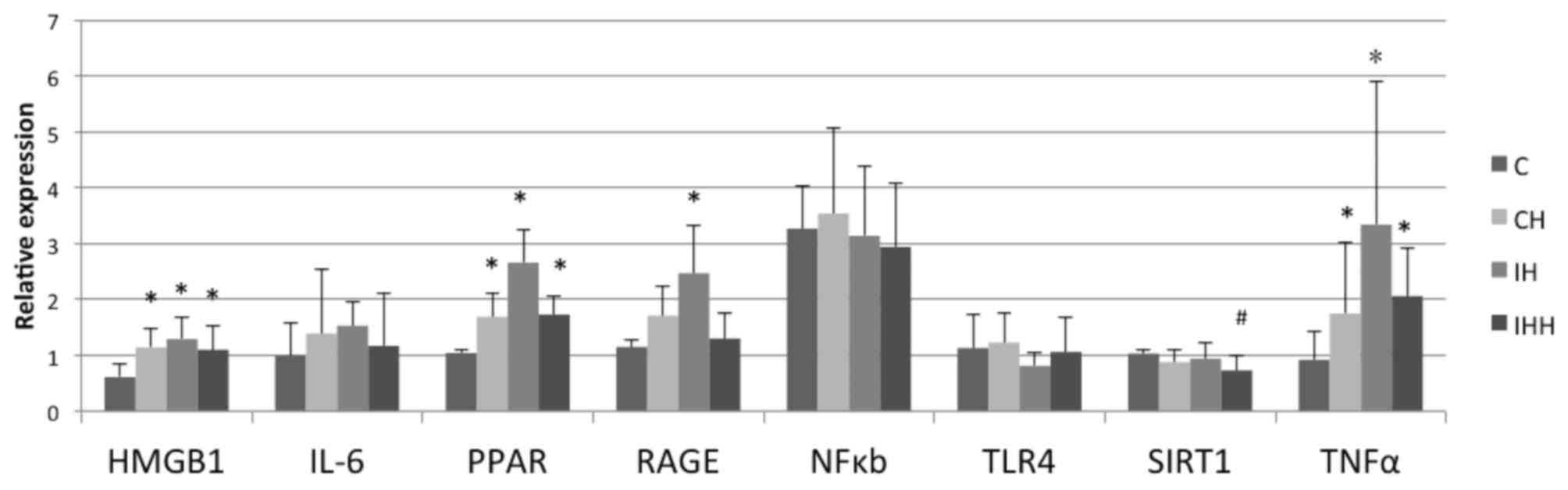 | Figure 6.Expression of HMGB1, IL-6, PPAR,
RAGE, TNF-α, SIRT1, TLR4 and NF-κB mRNA in kidney tissues. CH, IH
and IHH treatment increased the mRNA expression of HMGB1, TNF-α and
PPAR in kidney tissues. IH increased the expression of RAGE mRNA.
The expression of IL-6 mRNA exhibited an upward trend in all
hypoxia groups, but the change was not statistically significant.
IHH downregulated the expression of SIRT1 mRNA. There was no
significant difference in the expression of NF-κB and TLR4 mRNA
between the hypoxia groups and controls. *P<0.05 vs. control.
CH, Continuous hypoxia; IH, intermittent hypoxia; IHH, intermittent
hypoxia with hypercapnia; HMGB1, high mobility group box 1 protein;
RAGE, receptor for advanced glycosylation end products; TLR,
toll-like receptor; TNF-α, tumor necrosis factor α; IL,
interleukin; NF-κB, nuclear factor kappa-light-chain-enhancer of
active B cells; PPAR, peroxisome proliferator-activated receptor;
SIRT1, NAD-dependent protein deacetylase sirtuin-1. |
| Table II.Expression levels of HMGB1, IL-6,
PPAR, RAGE, NF-κB, TLR4, SIRT1 and TNF-α mRNA in kidney
tissues. |
Table II.
Expression levels of HMGB1, IL-6,
PPAR, RAGE, NF-κB, TLR4, SIRT1 and TNF-α mRNA in kidney
tissues.
|
| Relative
expression |
|---|
|
|
|
|---|
| Gene | C | CH | IH | IHH |
|---|
| HMGB1 | 0.61±0.24 |
1.14±0.35a |
1.30±0.40a |
1.10±0.42a |
| IL-6 | 0.99±0.59 | 1.39±1.16 | 1.53±0.44 | 1.17±0.93 |
| PPAR | 1.04±0.07 |
1.69±0.42a |
2.66±0.59a |
1.73±0.34a |
| RAGE | 1.15±0.13 | 1.71±0.54 |
2.47±0.85a | 1.30±0.47 |
| NF-κB | 3.27±0.77 | 3.54±1.54 | 3.14±1.25 | 2.94±1.15 |
| TLR4 | 1.13±0.59 | 1.23±0.52 | 0.81±0.25 | 1.06±0.62 |
| SIRT1 | 1.03±0.07 | 0.88±0.21 | 0.94±0.29 |
0.73±0.27b |
| TNF-α | 0.92±0.52 |
1.75±1.27a |
3.34±2.56a |
2.06±0.86a |
Expression of IL-6, TNF-α, soluble
(s)TLR2, sTLR4 and sPPAR in the peripheral blood detected by
ELISA
Hypoxia increased the expression of IL-6, TNF-α,
soluble TLR4, and decreased the expression of soluble TLR2 and PPAR
(Fig. 7). In the IHH group, the
expression of the sTLR2 (16.35±15.21 vs. 33.84±14.28) and sPPAR
(11.00±6.99 vs. 23.00±9.30) in the peripheral blood was
significantly decreased compared with the control group
(P<0.05). In addition, compared with the control group, no
significant difference in the expression of sTLR2 and sPPAR in CH
and IH were observed (P>0.05). It was also demonstrated that
hypoxia increased the levels of TNF-α, IL-6 and sTLR4 expression in
the peripheral blood. The expression of TNF-α in the CH
(3.48±0.69), IH (3.54±1.40) and IHH (3.52±1.39) groups was
significantly upregulated compared with the controls (2.29±0.78;
P<0.05).
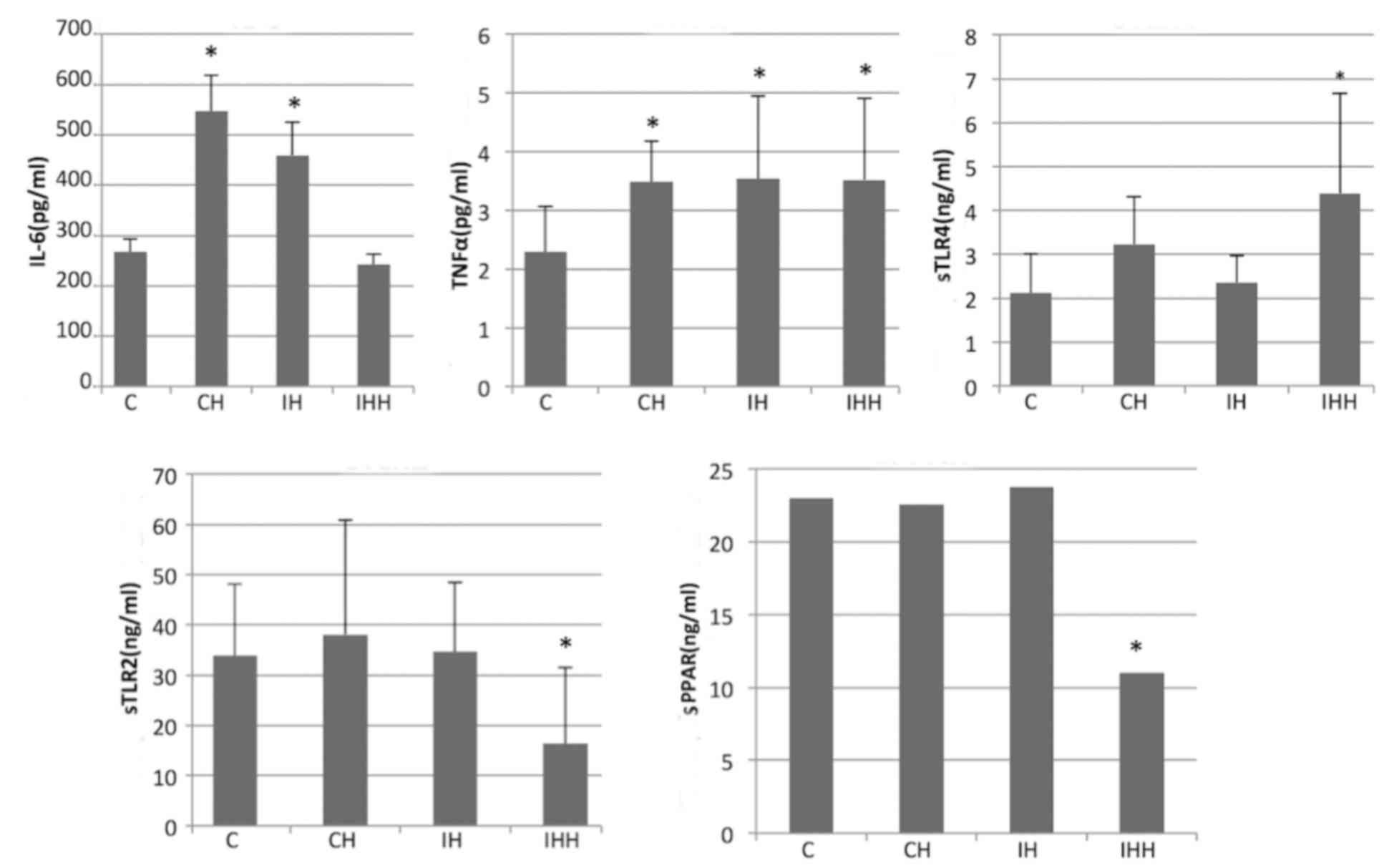 | Figure 7.Expression of IL-6, sPPAR, sTLR2,
sTLR4 and TNF-α in the peripheral blood of rats. CH, IH and IHH
increased the expression of TNF-α. CH and IH treatment increased
the expression of IL-6 and IHH increased the expression of sTLR4
significantly. IHH treatment decreased the expression of sTLR2 and
sPPAR-γ protein. *P<0.05 vs. controls. CH, continuous hypoxia;
IH, intermittent hypoxia; IHH, intermittent hypoxia with
hypercapnia; s, soluble; TLR, toll-like receptor; TNF-α, tumor
necrosis factor α; IL, interleukin; NF, nuclear factor
kappa-light-chain-enhancer of active B cells; PPAR, peroxisome
proliferator-activated receptor; SIRT1, NAD-dependent deacetylase
sirtuin-1. |
The expression of IL-6 in the CH (547.13±71.95) and
IH (459.54±65.13) groups was significantly upregulated compared
with the controls (267.11±26.65; P<0.05). However, there was no
significant difference in IL-6 expression between the IHH and
control groups (242.08±21.01 vs. 267.11±26.65; P>0.05).
The expression of sTLR4 was significantly increased
in the IHH group compared with the controls (4.39±2.28 vs.
2.12±0.88; P<0.05). In addition, compared to the controls, there
was an increase in sTLR4 expression in the CH and IH groups, but
the difference was not significant (P>0.05).
Discussion
The present study demonstrated that hypoxia
stimulation may cause early renal injury at the subcellular level
and increase the expression and translocation of HMGB1. Hypoxia
also upregulated the mRNA expression of factors of the
HMGB1-RAGE-TNF-α pathway in kidney tissues, and increased the
expression of sTLR4, TNF-α and IL-6 in the peripheral blood. This
suggested that the HMGB1-RAGE/TLR-TNF-α signaling pathway may
contribute to the molecular mechanisms of early renal injury
induced by hypoxia. This pathway may also be a marker for
OSA-associated early renal injury and serve as a future drug
intervention target.
Previously, increasing evidence has revealed a close
association between OSA and CKD. Intermittent hypoxia that occurs
in OSA may cause renal damage, which is an important risk factor
for CKD (3,4). However, at present, the molecular
mechanisms of kidney injury induced by intermittent hypoxia are not
fully understood. If the core molecular mechanism was identified,
and its central players used as intervention targets, it may serve
a vital role in the prevention and control of OSA-associated CKD.
The primary mechanism of OSA-induced disease complications is
considered to be immune inflammation (5). Hypoxia and oxidative stress in OSA may
cause direct cell damage, and the damaged cells and extracellular
matrix may release the decomposition products of proteins and
nucleic acid that serve as ‘danger signals’, known as DAMPs. HMGB1,
as the most important representative DAMP, has attracted much
attention (7). It is a highly
conserved nuclear protein that is abundant in mammalian cells,
which is transferred from the necrotic nucleus to the cytoplasm and
then released to the extracellular matrix. HMGB1 may promote
inflammation by combining with RAGE and TLR4 and TLR2 (10,11).
Inflammatory injury causes additional DAMPs to be released,
stimulating further inflammation.
In the present study, CH, IH and IHH rat models were
established. The effects of hypoxia stimulation on the renal
histopathological and ultrastructural changes, and the expression
of HMGB1 and its subsequent inflammatory pathway mediators in the
kidney tissues and peripheral bloods of rats, were
investigated.
The results of the present study suggested that
there was no severe histopathological injury, as indicated by HE
staining of the hypoxia groups; however, a certain degree of renal
tubular epithelial cell vacuolation was observed. By contrast,
ultrastructural changes, detected by electron microscopy, were more
significant in the hypoxia groups including foot process fusion,
increased secondary lysosomes and swelling and degeneration of
mitochondria. The subcellular structural changes of the kidney
tissue suggested early renal injury due to hypoxia.
Then, the expression of HMGB1 and its subsequent
signaling pathway mediators were detected by IHC, qPCR and ELISA,
in order to explore the underlying molecular mechanisms of early
renal injury. IHC staining demonstrated that the overall expression
and the expression in the cytoplasm of the HMGB1 protein was
increased in the hypoxia groups compared with the controls. This
suggested that hypoxia may upregulate the expression of HMGB1, and
may also increase its the nuclear translocation (from nucleus to
cytoplasm); this is where the HMGB1 protein is then acetylated to
its active form in the cytoplasm (19). An increased expression of HMGB1 in
the cytoplasm suggested an increase in HMGB1 activity.
The results of the qPCR indicated that hypoxia
stimulation increased the expression of HMGB1, RAGE and TNF-α mRNA
(P<0.05). RAGE was the earliest identified HMGB1 receptor,
belonging to the immunoglobulin superfamily transmembrane receptor,
and is widely expressed in a variety of immune and endothelial
cells, and on the surface of other cells (2,13). In
combination with the respective ligands, these receptors may induce
intracellular signal transduction via the Ras-mitogen-activated
protein kinase pathway and NF-κB translocation and mediate an
inflammatory response. The increased expression of HMGB1-RAGE
signaling factors in kidney tissues suggested that hypoxia may
activate this pathway, which may serve a key role in early renal
inflammatory injury induced by hypoxia.
It has been established that sleep apnea may lead to
renal function damage, which is closely associated with proteinuria
and CKD (20,21). If the HMGB1-RAGE-TNF-α pathway serves
an important role in renal injury due to sleep apnea, the pathway
may be a key target for intervention. A previous clinical study
demonstrated that the HMGB1 level was increased in patients with
OSA compared with the controls, and that effective CPAP treatment
downregulated the HMGB1 level (22).
An additional study also indicated that dexmedetomidine may
downregulate the expression of the HMGB1-TLR-NF-κB signaling
pathway and decrease ischemia-reperfusion injury (23). Glycyrrhizin decreased
ischemia-reperfusion injury in a mouse brain by decreasing the
expression of HMGB1 and the subsequent inflammatory factors
(24).
The present study also suggested that hypoxia
stimulation decreased the expression of SIRT1 mRNA and increased
that of PPAR mRNA in kidney tissues (P<0.05). SIRT1 and PPAR are
two inhibitors of this pathway. SIRT1 protein is a NAD+ dependent
protein deacetylase and regulates gene transcription by
deacetylation (15,25). It is a key aging-associated gene and
is an important anti-inflammatory, antioxidant and inflammatory
repair factor. The expression of SIRT1 in renal tissue was
decreased in the IHH group in the present study, suggesting that
intermittent hypoxia with high CO2 may downregulate
SIRT1 expression and thereby weaken the effect of deacetylation,
which may result in increased acetylation and activation of HMGB1
and increased inflammatory injury. This may be a mechanism of renal
inflammation injury induced by hypoxia. PPAR, as a nuclear receptor
transcription factor superfamily, has attracted increasing
attention for its role in regulating target gene expression
(17). According to its receptor
structure, PPAR may be divided into three subtypes: α, β and γ.
PPARγ is abundantly distributed in adipose tissue, and also
expressed in vascular endothelial cells and mononuclear
macrophages. In the present study, hypoxia stimulation caused an
increase of PPAR mRNA expression in renal tissues, which may be a
compensatory and protective mechanism during hypoxia against
inflammation. The increased expression of PPARγ may inhibit the
transcription of HMGB1 and other inflammatory factors by inhibiting
signaling pathways, including the NF-κB pathway. Previous studies
have indicated that PPARγ agonists serve a role in protecting the
kidneys from injury (26,27).
The results of the ELISAs revealed that hypoxia
stimulation increased the expression of sTLR4, TNF-α and IL-6 in
the peripheral blood, and decreased the expression of sTLR2 and
PPARγ. The increase in sTLR4 expression during hypoxia suggested an
activation of TLR4 associated innate immunity, by which HMGB1
exerted its inflammatory effects. Previous studies have
demonstrated that HMGB1-TLR4 signaling may induce IL-17A secretion
and lead to brain ischemia/reperfusion injury (24,28). A
previous study indicated that TLR4 (−/-) mice were protected
against ischemia/reperfusion injury of the kidney (29). In the present study, TLR4 expression
was detected by RT-qPCR and ELISA. It was identified that there was
no significant increase in the levels of TLR4 mRNA, while the
levels of sTLR4 protein increased significantly during hypoxia. The
results may be due to the relatively short hypoxia time (2 weeks)
and the difference in TLR4 expression between kidney tissues and
peripheral blood requires additional investigation.
The present study also demonstrated that hypoxia
decreased the expression of sTLR2 protein in the peripheral blood.
In a number of diseases, sTLRs are considered to be natural
regulators of TLRs, which may interact with cell-surface TLR2 and
maintain their levels within a reasonable range to avoid excessive
inflammation injury (30,31). sTLR2 released by stimulated immune
cells constitutes an important first-line negative regulatory
mechanism (31,32). In the present study, the expression
of sTLR2 protein in peripheral blood was decreased. This was
similar to unpublished results from our previous study, which
revealed that the expression of sTLR2 in the peripheral blood was
significantly decreased compared with that prior to sleep in 35
patients with OSA. This suggests that sTLR2 may be a negative
regulator in the inflammatory response.
Different hypoxia models were used in the present
study, including CH, IH and IHH. The results demonstrated that the
effects on the HMGB1 signal pathway were not identical between
these different models. For example, RAGE mRNA expression was
increased only in the IH group, but not in the CH and IHH groups.
The decrease of SIRT1 mRNA expression was only detected in IHH
group. For example, in the IHH group with high CO2,
CO2 serves its unique role: The effects of
CO2 and O2 stimulation on the expression of
relevant proteins are not identical. In some circumstance,
CO2 may also be a protective factor (33,34). For
example, kidney cell apoptosis was reduced in the hypercapnic
acidosis group compared with the normocapnia group in acute lung
injury rat models. It was suggested that hypercapnia exerted a
protect effect and reduced kidney cell apoptosis (33). In addition, hypercapnia has been
demonstrated to significantly decreased LPS-induced NF-κB
activation by suppression of IκB-α degradation, which in turn
attenuate neutrophil recruitment and lung injury (35). Additional studies are required to
clarify the specific differences in the effect of CO2
and O2.
In summary, the present study explored the effect of
hypoxia on the expression of HMGB1 and its subsequent inflammatory
pathway. Hypoxia stimulation caused renal injury at the subcellular
level, and an increase of HMrutGB1 expression and translocation. In
addition, hypoxia also upregulated the expression of the
HMGB1-RAGE/TLR-TNF pathway, suggesting that this pathway may be a
key molecular mechanism in OSA-associated renal injury and a target
for future drug development.
Limitations of the present study included: Only 2
weeks of hypoxia were applied to explore the mechanism of early
renal injury due to hypoxia. Long-term chronic hypoxia should be
studied to explore the effect of chronic hypoxia on kidney tissues.
Furthermore, CO2 serves a unique role in OSA-associated
kidney injury. A simple intermittent high CO2 group
should be established to explore the mechanism of CO2 in
this process. In addition, the present study only investigated the
expression levels of the HMGB1 signaling pathway, and an in
vitro intervention study should be performed to explore
additional molecular pathways.
Acknowledgements
The authors would like to thank Professor Jue Zhang
from the Academy for Advanced Interdisciplinary Studies, Peking
University (Beijing, China) for his help in developing the hypoxia
chamber. The authors would further like to thank Professor Dingfang
Bu of the Central Lab of Peking University First Hospital (Beijing,
China) for his support regarding qPCR.
Funding
The current study was supported by the National
Natural Science Foundation of China (grant no. 81341005).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
CZ and HD established the hypoxia model and
performed HE staining. CZ and FWC performed qPCR analysis. CZ and
YXW performed IHC staining. JM and GFW conceived and designed the
study and revised the manuscript. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Animal Ethics
Committee of Peking University First Hospital (Beijing, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Vijayan VK: Morbidities associated with
obstructive sleep apnea. Expert Rev Respir Med. 6:557–566. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wu X, Gu W, Lu H, Liu C, Yu B, Xu H, Tang
Y, Li S, Zhou J and Shao C: Soluble receptor for advanced glycation
end product ameliorates chronic intermittent hypoxia induced renal
injury, inflammation, and apoptosis via P38/JNK Signaling Pathways.
Oxid Med Cell Longev. 2016:10153902016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Adeseun GA and Rosas SE: The impact of
obstructive sleep apnea on chronic kidney disease. Curr Hypertens
Rep. 12:378–383. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Adams RJ, Appleton SL, Vakulin A, Hanly
PJ, McDonald SP, Martin SA, Lang CJ, Taylor AW, McEvoy RD, Antic
NA, et al: Chronic kidney disease and sleep apnea association of
kidney disease with obstructive sleep apnea in a population study
of men. Sleep. 40:Jan 1–2017.doi: 10.1093/sleep/zsw015. PubMed/NCBI
|
|
5
|
Wei Q, Bian Y, Yu F, Zhang Q, Zhang G, Li
Y, Song S, Ren X and Tong J: Chronic intermittent hypoxia induces
cardiac inflammation and dysfunction in a rat obstructive sleep
apnea model. J Biomed Res. 30:490–495. 2016.PubMed/NCBI
|
|
6
|
Zhu S, Li W, Ward MF, Sama AE and Wang H:
High mobility group box 1 protein as a potential drug target for
infection- and injury-elicited inflammation. Inflamm Allergy Drug
Targets. 9:60–72. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Piccinini AM and Midwood KS: DAMPening
inflammation by modulating TLR signalling. Mediators Inflamm.
2010:pii: 672395. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kent BD, Ryan S and McNicholas WT:
Obstructive sleep apnea and inflammation: Relationship to
cardiovascular co-morbidity. Respir Physiol Neurobiol. 178:475–481.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Andrassy M, Volz HC, Igwe JC, Funke B,
Eichberger SN, Kaya Z, Buss S, Autschbach F, Pleger ST, Lukic IK,
et al: High-mobility group box-1 in ischemia-reperfusion injury of
the heart. Circulation. 117:3216–3226. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hori O, Brett J, Slattery T, Cao R, Zhang
J, Chen JX, Nagashima M, Lundh ER, Vijay S and Nitecki D: The
receptor for advanced glycation end products (RAGE) is a cellular
binding site for amphoterin. Mediation of neurite outgrowth and
co-expression of rage and amphoterin in the developing nervous
system. J Biol Chem. 270:25752–25761. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tadie JM, Bae HB, Jiang S, Park DW, Bell
CP, Yang H, Pittet JF, Tracey K, Thannickal VJ, Abraham E and
Zmijewski JW: HMGB1 promotes neutrophil extracellular trap
formation through interactions with Toll-like receptor 4. Am J
Physiol Lung Cell Mol Physiol. 304:L342–L349. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chavakis T, Bierhaus A and Nawroth PP:
RAGE (receptor for advanced glycation end products): A central
player in the inflammatory response. Microbes Infect. 6:1219–1225.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yu SL, Wong CK, Szeto CC, Li EK, Cai Z and
Tam LS: Members of the receptor for advanced glycation end products
axis as potential therapeutic targets in patients with lupus
nephritis. Lupus. 24:675–686. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Schnare M, Barton GM, Holt AC, Takeda K,
Akira S and Medzhitov R: Toll-like receptors control activation of
adaptive immune responses. Nat Immunol. 2:947–950. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kozako T, Aikawa A, Shoji T, Fujimoto T,
Yoshimitsu M, Shirasawa S, Tanaka H, Honda S, Shimeno H, Arima N
and Soeda S: High expression of the longevity gene product SIRT1
and apoptosis induction by sirtinol in adult T-cell leukemia cells.
Int J Cancer. 131:2044–2055. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Michan S and Sinclair D: Sirtuins in
mammals: Insights into their biological function. Biochem J.
404:1–13. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Villapol S: Roles of peroxisome
proliferator-activated receptor gamma on brain and peripheral
inflammation. Cell Mol Neurobiol. 38:121–132. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Klune JR, Dhupar R, Cardinal J, Billiar TR
and Tsung A: HMGB1: Endogenous danger signaling. Mol Med.
14:476–484. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ahmed SB, Ronksley PE, Hemmelgarn BR, Tsai
WH, Manns BJ, Tonelli M, Klarenbach SW, Chin R, Clement FM and
Hanly PJ: Nocturnal hypoxia and loss of kidney function. PLoS One.
6:e190292011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hanly PJ and Ahmed SB: Sleep apnea and the
kidney: Is sleep apnea a risk factor for chronic kidney disease?
Chest. 146:1114–1122. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wu KM, Lin CC, Chiu CH and Liaw SF: Effect
of treatment by nasal continuous positive airway pressure on serum
high mobility group box-1 protein in obstructive sleep apnea.
Chest. 137:303–309. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang JJ, Peng K, Zhang J, Meng XW and Ji
FH: Dexmedetomidine preconditioning may attenuate myocardial
ischemia/reperfusion injury by down-regulating the
HMGB1-TLR4-MyD88-NF-кB signaling pathway. PLoS One.
12:e01720062017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhang J, Wu Y, Weng Z, Zhou T, Feng T and
Lin Y: Glycyrrhizin protects brain against ischemia-reperfusion
injury in mice through HMGB1-TLR4-IL-17A signaling pathway. Brain
Res. 1582:176–186. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lee IC, Ho XY, George SE, Goh CW, Sundaram
JR, Pang KKL, Luo W, Yusoff P, Sze NSK and Shenolikar S: Oxidative
stress promotes SIRT1 recruitment to the GADD34/PP1α complex to
activate its deacetylase function. Cell Death Differ. 25:255–267.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chen DZ, Chen LQ, Lin MX, Gong YQ, Ying BY
and Wei DZ: Esculentoside A inhibits LPS-induced acute kidney
injury by activating PPAR-γ. Microb Pathog. 110:208–213. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wu SH, Wang MJ, Lü J and Chen XQ: Signal
transduction involved in lipoxin A4-induced protection of tubular
epithelial cells against hypoxia/reoxygenation injury. Mol Med Rep.
15:1682–1692. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhu H, Li J, Wang S, Liu K, Wang L and
Huang L: Hmgb1-TLR4-IL-23-IL-17A axis promote ischemia-reperfusion
injury in a cardiac transplantation model. Transplantation.
95:1448–1454. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wu H, Chen G, Wyburn KR, Yin J, Bertolino
P, Eris JM, Alexander SI, Sharland AF and Chadban SJ: TLR4
activation mediates kidney ischemia/reperfusion injury. J Clin
Invest. 117:2847–2859. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Dulay AT, Buhimschi CS, Zhao G, Oliver EA,
Mbele A, Jing S and Buhimschi IA: Soluble TLR2 is present in human
amniotic fluid and modulates the intraamniotic inflammatory
response to infection. J Immunol. 182:7244–7253. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Henrick BM, Yao XD, Taha AY, German JB and
Rosenthal KL: Insights into soluble Toll-Like receptor 2 as a
downregulator of virally induced inflammation. Front Immunol.
7:2912016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Raby AC, Le Bouder E, Colmont C, Davies J,
Richards P, Coles B, George CH, Jones SA, Brennan P, Topley N and
Labéta MO: SolubleTLR2 reduces inflammation without compromising
bacterial clearance by disrupting TLR2 triggering. J Immunol.
183:506–517. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Nardelli LM, Rzezinski A, Silva JD,
Maron-Gutierrez T, Ornellas DS, Henriques I, Capelozzi VL, Teodoro
W, Morales MM, Silva PL, et al: Effects of acute hypercapnia with
and without acidosis on lung inflammation and apoptosis in
experimental acute lung injury. Respir Physiol Neurobiol. 205:1–6.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chonghaile Ni M, Higgins BD, Costello JF
and Laffey JG: Hypercapnic acidosis attenuates severe acute
bacterial pneumonia-induced lung injury by a neutrophil-independent
mechanism. Crit Care Med. 36:3135–3144. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Takeshita K, Suzuki Y, Nishio K, Takeuchi
O, Toda K, Kudo H, Miyao N, Ishii M, Sato N, Naoki K, et al:
Hypercapnic acidosis attenuates endotoxin-induced nuclear
factor-[kappa]B activation. Am J Respir Cell Mol Biol. 29:124–132.
2003. View Article : Google Scholar : PubMed/NCBI
|