Autosomal dominant polycystic kidney disease (ADPKD)
is the most frequently inherited kidney disease; its incidence has
been reported to be 1/400 to 1/2,500 worldwide and 1.5 million
individuals are affected in China (1–4). A
common cause of ADPKD is a mutation in polycystin (PC)-1, a
transient receptor potential channel which interacts with (PKD1)
and/or PKD2, and accounts for 85 and 15% of all detectable cases of
ADPKD, respectively. Approximately 10% of patients with ADPKD have
no detectable PKD1 or PKD2 mutation, which may be attributed to the
current lack of effective testing methods or to similar kidney
cysts caused by other gene mutations (5,6). ADPKD
is characterized by numerous enlarged cysts in the bilateral
kidneys and liver, in addition to specific rare manifestations in
other organs, including the pancreas, cerebral vasculature, aortic
arch and seminal vesicles (7–10). In
the case of the present study, a novel frameshift PKD1 mutation was
identified in ADPKD, which additionally caused epididymal cysts and
azoospermia.
In September 2017, a 33-year-old male patient
presented at the outpatient department of the First Affiliated
Hospital of Anhui Medical University (Hefei, China) and sought
medical advice due to the complaint of abdominal pain and
azoospermia. He was first diagnosed with ADPKD in 2014 by magnetic
resonance imaging, which identified numerous cysts in the kidneys
and liver, based on the standard of diagnosis established by Pei
et al (11). In 2016, the
patient was diagnosed with azoospermia by semen analysis following
long-term infertility and he additionally developed hypertension
with a peak blood pressure of 150/100 mmHg, which was controlled
with Valsartan tablets.
The patient had a height of 180 cm, weight of 75 kg
and body mass index of 23.1 kg/m2. Mild pain with a pain
score of 1 was determined using a visual analog scale, as
previously described (12). The
latest laboratory investigations demonstrated a normal serum
creatinine concentration (81.7 µmol/l; normal range: 61.9–114.9
µmol/l), blood urea nitrogen concentration (6.31 mmol/l; normal
range: 2.5–7.1 µmol/l) and estimated glomerular filtration rate
(eGFR; 106.78 ml/min/1.73 m2; normal range: 90–120
ml/min/1.73 m2), and an increased uric acid
concentration (506 µmol/l; normal range: 214–494 µmol/l). By
analyzing the patient's previous laboratory results, it was
identified that the eGFR was within the normal range from July 2015
to March 2018, without any abnormal result in December 2016 (86.91
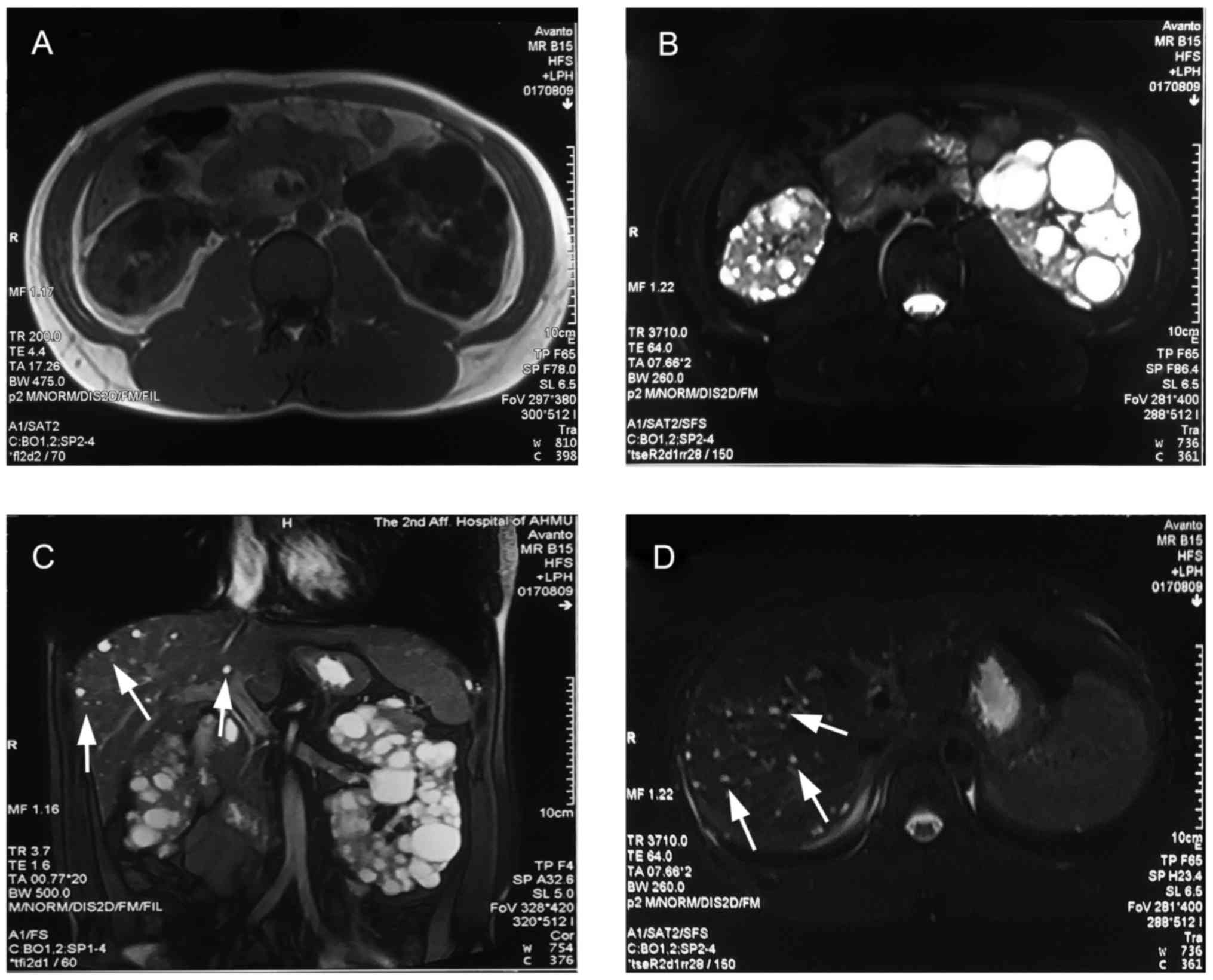
ml/min/1.73 m2). The most recent abdominal computed
tomography performed on the 14th of February 2016, demonstrated
that the kidneys were significantly enlarged with numerous cysts of
varying sizes with a total kidney volume of 1,127.21
cm3, while there were some isolated small cysts present
in the liver (Fig. 1). In the semen
analysis in July and August 2017, no sperm was detected, along with
a low volume of ejaculate (2 and 2.3 ml, respectively). In the
subsequent ultrasound examination, cysts were identified in the
bilateral epididymis, with a size of 6 mm on the left and 4 mm on
the right, in addition to dilated seminal vesicles. Of note, the
subsequent testicular biopsy identified numerous mature sperms with
progressive motility.
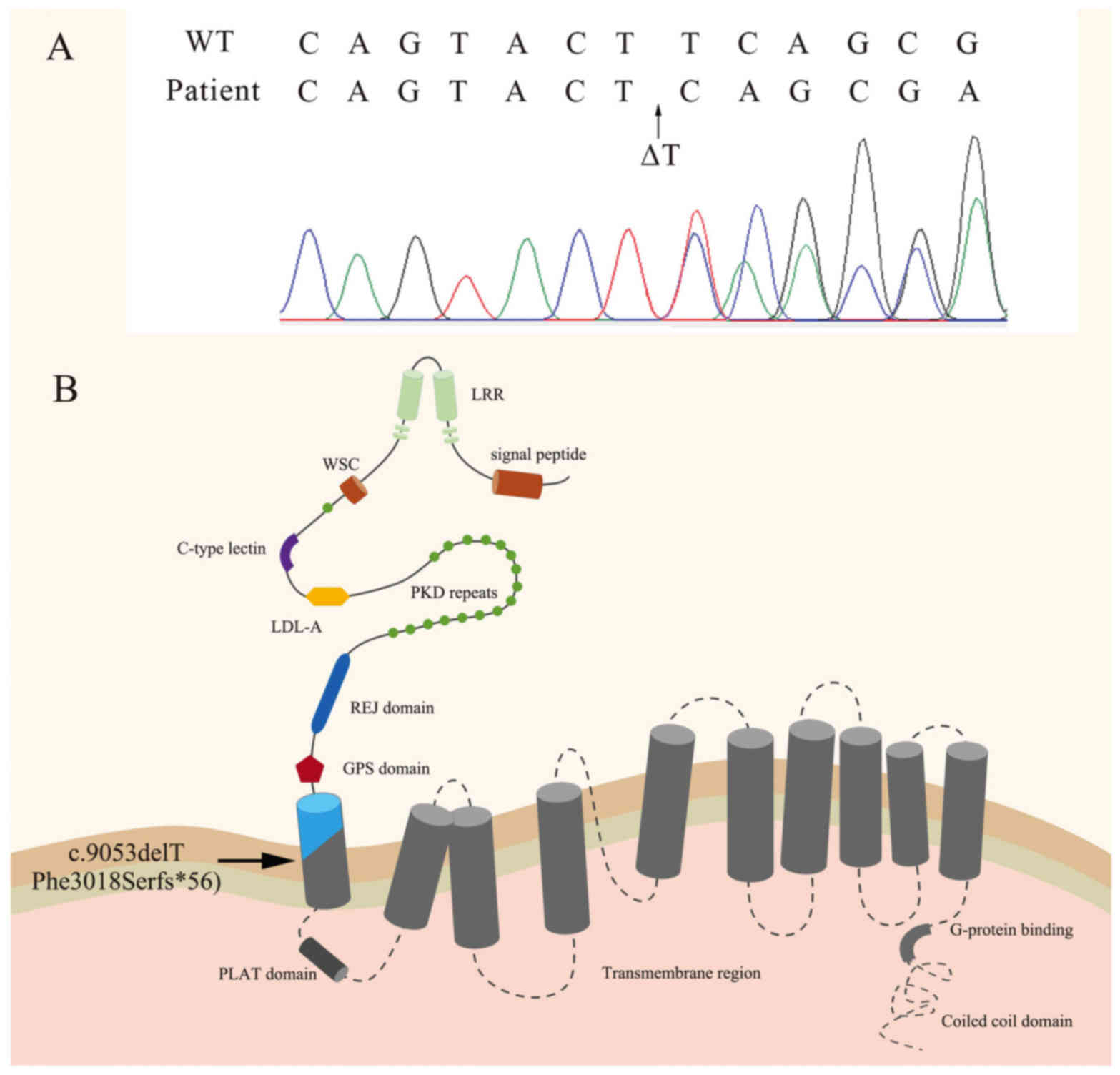
In order to make a definitive diagnosis of ADPKD,
the patient was subjected to genetic sequencing analysis of the
PKD1 and PKD2 genes. No mutation in the PKD2 gene was identified,
whereas the coding region of PKD1 in the patient exhibited a
c.9053delT frameshift mutation (p.Phe3018Serfs*56; Fig. 2), which has not been previously
reported, to the best of our knowledge, in the 1,000 Genomes
project, the Exome Aggregation Consortium or the PKD mutation
database (13–15). Prediction regarding the effects of
this mutation with Mutation Taster (www.mutationtaster.org) and PolyPhen-2 (genetics.bwh.harvard.edu/pph2) software
indicated that the protein variant is ‘disease-causing’ and
‘probably damaging’, respectively, which is in line with the
situation of the present case. Sanger sequencing of the mother of
the patient revealed no mutations in PKD1 or PKD2; renal function
was in the normal range and there were no kidney cysts observed.
The father of the patient died in a motor vehicle collision in 2005
at the age of 50 years, and therefore, it was not possible to
perform Sanger sequencing, however he had normal kidney function
and no family history of kidney disease.
ADPKD is a prevalent inherited kidney disease,
commonly caused by a mutation of PKD1 and/or PKD2 in the majority
of cases. The mutation in the remaining 10% of patients is
undetectable, which may be attributed to the current lack of
effective testing methods, or to similar kidney cysts caused by
other gene mutations (5,6); the glucosidase II α subunit gene, which
serves a role in protein folding and quality control, may be
another potential causal gene that induces ADPKD phenotypes
(16). Due to the advantages of low
cost and high-throughput, the technology of next-generation
sequencing is widely applied in the diagnosis and study of
hereditary disease, including ADPKD (17–20).
However, its short-read sequencing approaches are not able to
precisely recognize complex regions, including segmental
duplications, extreme GC content and gaps (21,22). The
PKD1 gene is ~50 kb long, contains 46 exons and encodes an open
reading frame of 12,909 bp, whereas the PKD2 gene is ~68 kb long,
contains 15 exons and encodes an open reading frame of 2,904 bp.
The sequencing of PKD1 is complex due to the six duplicated
pseudogenes adjacent to it, which are highly homologous with exons
1–33 of PKD1 (23). Due to the long
range and the six pseudogenes, single-molecule long-read sequencing
is more suitable for the detection of genetic variants in patients
with ADPKD (24–26). In the present study, long-range PCR
(LR-PCR) amplification and targeted next-generation sequencing, as
described previously (27), was used
to detect the variants of PKD1 and PKD2 in the 33-year-old male
patient. No PKD2 mutation was identified; however, a T-allele
deletion causing a frameshift mutation was identified at the
9,053th position of PKD1 complementary DNA. Subsequent to comparing
this alteration with the wild-type PKD1 gene, it was identified
that this frameshift mutation not only causes the 3,018th amino
acid of PC-1 to change from phenylalanine to serine, but also leads
to an early termination of protein translation. PC-1 is a membrane
protein composed of an N-terminal extracellular portion, 11
transmembrane (TM) domains and a short intracellular C-terminal
tail, which is able to directly bind PC-2 and G proteins to
initiate multiple signaling pathways (28–31). The
c.9053delT is a frameshift mutation, which leads to the formation
of a stop codon downstream of c.9053delT. This truncated
polypeptide consists of 3,017 intact and 56 unique amino acids,
without the TM region or the intracellular C-terminal tail.
Therefore, the c.9053delT mutation may be disease-causing.
For patients with ADPKD, the predominant
manifestation is numerous enlarged cysts in the bilateral kidneys.
However, the disease severity and renal survival times are
different between patients with mutant PKD1 and PKD2: Patients with
PKD1 mutations frequently have a shorter renal survival time and
suffer from end-stage renal disease (ESRD) ~20 years earlier
compared with patients with mutant PKD2 (32,33). The
greater disease severity associated with the PKD1 mutations is
caused by the earlier onset of cyst formation, not the faster cyst
growth (34). The additional renal
manifestations vary among patients; furthermore, cyst formation in
the liver is the most common sign associated with the disease,
detected in ~83% of patients with ADPKD (35). Intracranial aneurysms are
additionally associated with the disease, with a prevalence of
9–12%, and raise the risk of cerebrovascular accident (36). In the present study, the 33-year-old
patient sought medical advice for abdominal pain and azoospermia.
Chronic abdominal pain is a common complaint, and a study from the
HALT-PKD Trial documented complaints of abdominal fullness and pain
which were severe in patients with advanced disease, possibly due
to organ enlargement (37). The
total kidney volume (TKV) was identified as a critical biomarker to
predict the progression of ADPKD in recent studies; according to a
clinical evaluation model, an eGFR of <50 ml/min/1.73
m2 and a TKV of the bilateral kidneys of >1,000
cm3 is associated with an estimated median disease
course of ESRD of 4 years (38–40).
Therefore, the 33-year-old patient was advised to maintain good
renal function to prolong the renal survival time, by having a low
sodium diet, drinking more water and avoiding strenuous exercise.
In patients with ADPKD, seminal vesicle cysts represent a common
additional renal manifestation with a prevalence of 39–60%, as
opposed to a prevalence of 5% in the general population; however,
these cysts are rarely associated with infertility in male patients
with ADPKD (41,42). A number of previous case reports
regarding infertility and azoospermia in patients with ADPKD have
been published, which suggest that the lack of PC-1 or PC-2 may
result in the obstruction or atonicity of seminal vesicles or
ejaculatory duct cysts and lead to ejaculation failure (43–47). In
the present study, the ultrasound examination detected the presence
of epididymis cysts and dilated seminal vesicles; however, the
testicular biopsy identified mature sperm. This suggested that the
cysts of the epididymis and the dilated seminal vesicles may
obstruct the ejaculation of semen. May result in the obstruction or
atonicity of seminal vesicles or ejaculatory duct cysts and lead to
ejaculation failure
In summary, the patient presented in this case study
suffered from ADPKD and azoospermia due to a novel mutation in the
PKD1 gene, c.9053delT. Assisted reproductive technology coupled
with pre-implantation genetic diagnosis of ADPKD may be used to
eliminate the inheritance of this mutation to produce a healthy
embryo (48,49).
The authors would like to thank Dongyue Ma, Qingsong
Niu and Zhengming Bai, Anhui Province PKD Center, The First
Affiliated Hospital of Anhui Medical University, (Hefei, China),
for their help in recording the medical history of the patient.
The present study was supported by a grant from the
Sci-Tech Planning Projects of Anhui Province, China (grant no.
1704e1002230 to CL).
The datasets generated and/or analyzed during the
current study are available from the corresponding author on
reasonable request.
CZL performed the clinical examination and diagnosed
the patient with ADPKD, as well as reviewed the manuscript. JLM
recorded and analyzed the results from the clinical examinations.
JLM and SXF obtained the blood samples, extracted the total DNA,
completed the long-range PCR (LR-PCR) amplification and the
targeted next-generation sequencing. JLM and YCX recorded and
analyzed the results from the clinical examinations. All authors
read and approved the final manuscript.
Informed consent for participation in the study was
obtained.
The patient provided written informed consent
regarding the publication of his data and images.
The authors declare that they have no competing
interests.
|
1
|
Dalgaard OZ: Bilateral polycystic disease
of the kidneys; a follow-up of two hundred and eighty-four patients
and their families. Acta Med Scand Suppl. 328:1–255.
1957.PubMed/NCBI
|
|
2
|
Wilson PD: Polycystic kidney disease. N
Engl J Med. 350:151–164. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Willey CJ, Blais JD, Hall AK, Krasa HB,
Makin AJ and Czerwiec FS: Prevalence of autosomal dominant
polycystic kidney disease in the European Union. Nephrol Dial
Transplant. 32:1356–1363. 2017.PubMed/NCBI
|
|
4
|
Xue C, Zhou CC, Wu M and Mei CL: The
clinical manifestation and management of autosomal dominant
polycystic kidney disease in China. Kidney Dis (Basel). 2:111–119.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pei Y and Watnick T: Diagnosis and
screening of autosomal dominant polycystic kidney disease. Adv
Chronic Kidney Dis. 17:140–152. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cornecle GE, Audrézet MP, Renaudineau E,
Hourmant M, Charasse C, Michez E, Frouget T, Vigneau C, Dantal J,
Siohan P, et al: PKD2-related autosomal dominant polycystic kidney
disease: Prevalence, clinical presentation, mutation spectrum, and
prognosis. Am J Kidney Dis. 70:476–485. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Reig B, Blumenfeld J, Donahue S and Prince
MR: Seminal megavesicle in autosomal dominant polycystic kidney
disease. Clin Imaging. 39:289–292. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Vikrant S and Parashar A: Autosomal
dominant polycystic kidney disease: Study of clinical
characteristics in an Indian population. Saudi J Kidney Dis
Transpl. 28:115–124. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yoshida H, Higashihara E, Maruyama K,
Nutahara K, Nitatori T, Miyazaki I and Shiokawa Y: Relationship
between intracranial aneurysms and the severity of autosomal
dominant polycystic kidney disease. Acta Neurochir (Wien).
159:2325–2330. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhang W, Han Q, Liu Z and Zhou W, Cao Q
and Zhou W: Whole exome sequencing reveals a stop-gain mutation of
PKD2 in an autosomal dominant polycystic kidney disease family
complicated with aortic dissection. BMC Med Genet. 19:192018.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Pei Y, Obaji J, Dupuis A, Paterson AD,
Magistroni R, Dicks E, Parfrey P, Cramer B, Coto E, Torra R, et al:
Unified criteria for ultrasonographic diagnosis of ADPKD. J Am Soc
Nephrol. 20:205–212. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Huskisson EC: Measurement of pain. Lancet.
2:1127–1131. 1974. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lek M, Karczewski KJ, Minikel EV, Samocha
KE, Banks E, Fennell T, O'Donnell-Luria AH, Ware JS, Hill AJ,
Cummings BB, et al: Analysis of protein-coding genetic variation in
60,706 humans. Nature. 536:285–291. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gout AM, Martin NC, Brown AF and Ravine D:
PKDB: Polycystic kidney disease mutation database-a gene variant
database for autosomal dominant polycystic kidney disease. Hum
Mutat. 28:654–659. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Moller M, Jöud M, Storry JR and Olsson ML:
Erythrogene: A database for in-depth analysis of the extensive
variation in 36 blood group systems in the 1,000 genomes project.
Blood Adv. 1:240–249. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Porath B, Gainullin VG, Cornec-Le GE,
Dillinger EK, Heyer CM, Hopp K, Edwards ME, Madsen CD, Mauritz SR,
Banks CJ, et al: Mutations in GANAB, encoding the glucosidase IIα
subunit, cause autosomal-dominant polycystic kidney and liver
disease. Am J Hum Genet. 98:1193–1207. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Codinasolà M, Rodríguezsantiago B, Homs A,
Santoyo J, Rigau M, Aznar-Laín G, Del Campo M, Gener B, Gabau E,
Botella MP, et al: Integrated analysis of whole-exome sequencing
and transcriptome profiling in males with autism spectrum
disorders. Mol Autism. 6:212015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Oliver GR, Hart SN and Klee EW:
Bioinformatics for clinical next generation sequencing. Clin Chem.
61:124–135. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Edrees BM, Athar M, Al-Allaf FA, Taher MM,
Khan W, Bouazzaoui A, Al-Harbi N, Safar R, Al-Edressi H, Alansary
K, et al: Next-generation sequencing for molecular diagnosis of
autosomal recessive polycystic kidney disease. Gene. 591:214–226.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ranjzad F, Aghdami N, Tara A, Mohseni M,
Moghadasali R and Basiri A: Identification of three novel
frameshift mutations in the PKD1 gene in iranian families with
autosomal dominant polycystic kidney disease using efficient
targeted next-generation sequencing. Kidney Blood Press Res.
43:471–478. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Steinberg KM, Schneider VA, Graves-Lindsay
TA, Fulton RS, Agarwala R, Huddleston J, Shiryev SA, Morgulis A,
Surti U, Warren WC, et al: Single haplotype assembly of the human
genome from a hydatidiform mole. Genome Res. 24:2066–2076. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chaisson MJ, Huddleston J, Dennis MY,
Sudmant PH, Malig M, Hormozdiari F, Antonacci F, Surti U, Sandstrom
R, Boitano M, et al: Resolving the complexity of the human genome
using single-molecule sequencing. Nature. 517:608–611. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bogdanova N, Markoff A, Gerke V, McCluskey
M, Horst J and Dworniczak B: Homologues to the first gene for
autosomal dominant polycystic kidney disease are pseudogenes.
Genomics. 74:333–341. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Eisenberger T, Decker C, Hiersche M,
Hamann RC, Decker E, Neuber S, Frank V, Bolz HJ, Fehrenbach H, Pape
L, et al: An efficient and comprehensive strategy for genetic
diagnostics of polycystic kidney disease. PLoS One.
10:e01166802015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Carrera P, Calzavara S, Magistroni R, den
Dunnen JT, Rigo F, Stenirri S, Testa F, Messa P, Cerutti R, Scolari
F, et al: Deciphering variability of PKD1 and PKD2 in an italian
cohort of 643 patients with autosomal dominant polycystic kidney
disease (ADPKD). Sci Rep. 6:308502016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Borràs DM, Vossen RHAM, Liem M, Buermans
HPJ, Dauwerse H, van Heusden D, Gansevoort RT, den Dunnen JT,
Janssen B, Peters DJM, et al: Detecting PKD1 variants in polycystic
kidney disease patients by single-molecule long-read sequencing.
Hum Mutat. 38:870–879. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Xu D, Ma Y, Gu X, Bian R, Lu Y, Xing X and
Mei C: Novel mutations in the PKD1 and PKD2 genes of Chinese
patients with autosomal dominant polycystic kidney disease. Kidney
Blood Press Res. 43:297–309. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Qian F, Germino FJ, Cai Y, Zhang X, Somlo
S and Germino GG: PKD1 interacts with PKD2 through a probable
coiled-coil domain. Nat Genet. 16:179–183. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Delmas P, Nomura H, Li X, Lakkis M, Luo Y,
Segal Y, Fernández-Fernández JM, Harris P, Frischauf AM, Brown DA
and Zhou J: Constitutive activation of G-proteins by polycystin-1
is antagonized by polycystin-2. J Biol Chem. 277:11276–11283. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Dere R, Wilson PD, Sandford RN and Walker
CL: Carboxy terminal tail of polycystin-1 regulates localization of
TSC2 to repress mTOR. PLoS One. 5:e92392010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kim H, Kang AY, Ko AR, Park HC, So I, Park
JH, Cheong HI, Hwang YH and Ahn C: Calpain-mediated proteolysis of
polycystin-1 C-terminus induces JAK2 and ERK signal alterations.
Exp Cell Res. 320:62–68. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Cornec-Le Gall E, Audrezet MP, Chen JM,
Hourmant M, Morin MP, Perrichot R, Charasse C, Whebe B, Renaudineau
E, Jousset P, et al: Type of PKD1 mutation influences renal outcome
in ADPKD. J Am Soc Nephrol. 24:1006–1013. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kurashige M, Hanaoka K, Imamura M, Udagawa
T, Kawaguchi Y, Hasegawa T, Hosoya T, Yokoo T and Maeda S: A
comprehensive search for mutations in the PKD1 and PKD2 in Japanese
subjects with autosomal dominant polycystic kidney disease. Clin
Genet. 87:266–272. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chapman AB, Devuyst O, Eckardt KU,
Gansevoort RT, Harris T, Horie S, Kasiske BL, Odland D, Pei Y,
Perrone RD, et al: Autosomal-dominant polycystic kidney disease
(ADPKD): Executive summary from a kidney disease: Improving global
outcomes (KDIGO) controversies conference. Kidney Int. 88:17–27.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Bae KT, Zhu F, Chapman AB, Torres VE,
Grantham JJ, Guay-Woodford LM, Baumgarten DA, King BF Jr, Wetzel
LH, Kenney PJ, et al: Magnetic resonance imaging evaluation of
hepatic cysts in early autosomal-dominant polycystic kidney
disease: The consortium for radiologic imaging studies of
polycystic kidney disease cohort. Clin J Am Soc Nephrol. 1:64–69.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Vlak MH, Algra A, Brandenburg R and Rinkel
GJ: Prevalence of unruptured intracranial aneurysms, with emphasis
on sex, age, comorbidity, country, and time period: A systematic
review and meta-analysis. Lancet Neurol. 10:626–636. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Miskulin DC, Abebe KZ, Chapman AB, Perrone
RD, Steinman TI, Torres VE, Bae KT, Braun W, Winklhofer FT, Hogan
MC, et al: Health-related quality of life in patients with
autosomal dominant polycystic kidney disease and CKD stages 1–4: A
cross-sectional study. Am J Kidney Dis. 63:214–226. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Perrone RD, Mouksassi MS, Romero K,
Czerwiec FS, Chapman AB, Gitomer BY, Torres VE, Miskulin DC,
Broadbent S and Marier JF: Total kidney volume is a prognostic
biomarker of renal function decline and progression to end-stage
renal disease in patients with autosomal dominant polycystic kidney
disease. Kidney Int Rep. 2:442–450. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Yu ASL, Shen C, Landsittel DP, Harris PC,
Torres VE, Mrug M, Bae KT, Grantham JJ, Rahbari-Oskoui FF, Flessner
MF, et al: Baseline total kidney volume and the rate of kidney
growth are associated with chronic kidney disease progression in
autosomal dominant polycystic kidney disease. Kidney Int.
93:691–699. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Xue C, Zhou C and Mei C: Total kidney
volume: The most valuable predictor of autosomal dominant
polycystic kidney disease progression. Kidney Int. 93:540–542.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Torra R, Sarquella J, Calabia J, Martí J,
Ars E, Fernández-Llama P and Ballarin J: Prevalence of cysts in
seminal tract and abnormal semen parameters in patients with
autosomal dominant polycystic kidney disease. Clin J Am Soc
Nephrol. 3:790–793. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Vora N, Perrone R and Bianchi DW:
Reproductive issues for adults with autosomal dominant polycystic
kidney disease. Am J Kidney Dis. 51:307–318. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Kanagarajah P, Ayyathurai R and Lynne CM:
Male infertility and adult polycystic kidney disease-revisited:
Case report and current literature review. Andrologia. 44:838–841.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Grenha V, Pereira BJ, Retroz E, Coelhoa H,
Godinhoa R, Temidoa P and Motaa A: Mechanisms of male infertility
in autosomal dominant polycystic kidney disease. Acta Urológica
Portuguesa. 31:41–44. 2014. View Article : Google Scholar
|
|
45
|
Shefi S, Levron J, Nadu A and Raviv G:
Male infertility associated with adult dominant polycystic kidney
disease: A case series. Arch Gynecol Obstet. 280:457–460. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Manno M, Marchesan E, Tomei F, Cicutto D,
Maruzzi D, Maieron A and Turco A: Polycystic kidney disease and
infertility: Case report and literature review. Arch Ital Urol
Androl. 77:25–28. 2005.PubMed/NCBI
|
|
47
|
Hendry WF, Rickards D, Pryor JP and Baker
LR: Seminal megavesicles with adult polycystic kidney disease. Hum
Reprod. 13:1567–1569. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Li W, Ma Y, Yu S, Sun N, Wang L, Chen D,
Yang G, Lu S, Li Y, Yang B and Mei C: The mutation-free embryo for
in vitro fertilization selected by MALBAC-PGD resulted in a healthy
live birth from a family carrying PKD 1 mutation. J Assist Reprod
Genet. 34:1653–1658. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Murphy EL, Droher ML, Dimaio MS and Dahl
NK: Preimplantation genetic diagnosis counseling in autosomal
dominant polycystic kidney disease. Am J Kidney Dis. Mar
30–2018.(Epub ahead of print). View Article : Google Scholar
|