Introduction
Oxygen treatment is a life-saving therapy in infants
with hypoxic respiratory failure (1). However, excessive oxygen exposure
(hyperoxia) can interfere with the normal development by affecting
cell division and differentiation in the immature lung (2). Hyperoxia can result in alveolar damage,
pulmonary interstitial fibrosis and pulmonary vasculature dysplasia
(3). Normal lung development depends
on orderly proliferation of epithelial and mesenchymal cells, and
interruption of that process impacts lung structure and function.
Excessive oxygen exposure or hyperoxia may cause bronchopulmonary
dysplasia (BPD) in the immature lung of premature neonates
(4). BPD is the most common cause of
respiratory morbidity among preterm infants, affecting nearly
10,000 infants each year in the United States (5). The pathological characteristics of BPD
include simplified, enlarged alveoli, capillary deformation and
interstitial fibrosis (6). Although
much attention has been directed to lung epithelial and endothelial
cells, pulmonary interstitial fibroblasts are also active during
normal development and in response to hyperoxia (7). The development, repair and regeneration
of alveoli are affected by neighboring lung fibroblasts (LFs),
which further promote remodeling of the extracellular matrix (ECM)
and development of fibrosis (8).
Lung development is controlled by spatial and
temporal patterns of cell proliferation that are an important
determinant of the lung architecture. Pulmonary epithelial and
mesenchymal cells interact during localized growth, branching and
extension of primordial tubules (9).
Glandular and vascular development is associated with decreased
proliferation of LFs (10).
Disruption of the regulation of developmental patterns contributes
to BPD, which often occurs in premature infants exposed to
hyperoxia (5). BPD induces patchy
proliferation of mesenchymal derivatives, including fibroblasts
(8) and the regulation of LF
proliferation is of clinical and developmental interest.
The extracellular signal-regulated kinase (ERK)
cascade activates cell proliferation and experimental animal
studies have demonstrated that the ERK1/2 signaling pathway can be
activated by hyperoxia (11,12). The Mek gene that encodes for
ERKs is active in lung development and Mek mutations have
been associated with lung hypoplasia, tracheal defects and neonatal
deaths (13). LFs function as
secretory cells in the pulmonary ECM and mediate normal and
pathological remodeling. This study investigated cell proliferation
and the activation of the ERK1/2 signaling pathway in primary
cultures of LFs in a neonatal rat model of hyperoxia-induced lung
fibrosis.
Materials and methods
Animals and oxygen exposure
In the hyperoxia group, full-term newborn Wistar
rats (n=60, 3.7–4.2 g) and 3-month old mother rats (n=40, 220–240
g) were provided by the Department of Laboratory Animals, Shengjing
Hospital of China Medical University (Shenyang, China). Rats were
exposed to an atmosphere of 90% oxygen with CO2
maintained at <5% using soda lime. The temperature was kept at
25–27°C and the humidity at 50–70%. In the control group full-term
newborn Wistar rats (n=60) were kept in room air (normoxia, 21%
oxygen). To avoid oxygen toxicity, nursing mothers were rotated
every 24 h between the groups. All rats had free access to food and
water and were kept on a 12-h light/dark cycle. The current study
was approved by the Institutional Animal Care and Use Committee at
Shengjing Hospital of China Medical University (Shenyang, China).
Newborn rats were euthanized prior to the removal of lung tissue
with sodium pentobarbital (l00 mg/kg body weight; intraperitoneal
injection). Lung tissue samples were collected on day 3 for
saccular, day 7 for early alveolar or day 14 for bulk alveolar
stages (n=20 each).
Lung histology and
immunohistochemistry
Lung tissue samples were washed with PBS and fixed
in 4% paraformaldehyde overnight at 4°C prior to dehydration in a
graded alcohol series (80, 90, 95 and 100%) for 2 h at room
temperature and embedded in paraffin. Paraffin-embedded lung tissue
samples were cut into 4-µm slices and hematoxylin and eosin
(H&E) staining was performed. Briefly, samples were stained
with haematoxylin for 10 min at room temperature, followed by eosin
for 20 sec at room temperature and standard histological evaluation
was observed using a light microscope (magnification, ×400).
The alveolar developmental stage was determined by a
radial alveolar count (RAC), as previously described (14). A perpendicular line was drawn from
the center of the most peripheral bronchiole to the pleura or the
nearest interlobular septum. Alveolarization was evaluated by
counting the number of alveoli crossed by this line.
Fibrosis was scored as previously described by
Ashcroft et al (15). Each
successive field was individually assessed for severity of
interstitial fibrosis and given a score between 0 and 8. Normal
tissue received a score of 0, whilst a high score of 8 was given to
fields that were completely filled with fibrous tissue.
The immunohistochemical peroxidase-conjugated
streptavidin method was performed to detect the expression level of
phosphorylated (p)-ERK expression using an Histostain™-Plus kit
(cat. no. SP-0023; OriGene Technologies, Inc., Beijing, China),
according to the manufacturer's protocol. Briefly,
paraffin-embedded lung tissue samples were blocked for 20 min at
room temperature with 10% goat serum and incubated with the primary
antibody against p-ERK (1:100; cat. no. 4370; Cell Signaling
Technology, Inc., Danvers, MA, USA) overnight at 4°C. Following
incubation, tissue samples were incubated with biotinylated
secondary antibody for 20 min at room temperature followed by
horseradish peroxidase (HRP)-labeled streptavidin for 20 min at
room temperature. The tissue sample slides were incubated with
diaminobenzidine (DAB) solution (20X; cat. no. ZLI-9031; OriGene
Technologies, Inc.) for color development and observed using a
light microscope (magnification, ×400). Tissue samples were
subjected to morphometric computerized image analysis using
MetaMorph Software System (IPv6.0; Universal Imaging, Inc., Bedford
Hills, NY, USA), which was used to acquire images and quantify the
number of positively stained cells. DAB precipitates as a dark
brown pigment allowing easy visualization of positively stained
cells. For each slide, five high magnification (×400) fields in
total were randomly examined and average optical density score of
positively stained cells was calculated.
Isolation, culture and identification
of LFs
Pups were randomly selected for euthanasia and
tissue collection on postnatal day 3, 7 or 14. Under sterile
conditions, lung tissue was removed, minced with fine seissors into
1 mm3 pieces and digested in 0.25% trypsin (Santa Cruz
Biotechnology, Inc., Dallas, TX, USA) for 30 min at 37°C in a
shaking water bath. The digested tissue was filtered through a
sterile 53 mm cell strainer and pellet cells at 400 × g for 5 min
at 4°C. The harvested cells were cultured with Dulbecco's modified
Eagle's medium (DMEM; HyClone; GE Healthcare Life Sciences, Logan,
UT, USA) supplemented with 10% fetal bovine serum (FBS; HyClone; GE
Healthcare Life Sciences) and maintained at 37°C in a 5%
CO2-humidified incubator. LFs were used in subsequent
experiments when confluent, elongated and spindle-shaped, and when
they exhibited actiniform or paliform appearance.
Immunofluorescence and immunocytochemistry was used
to detect LFs. Briefly, cultured LFs were fixed in 4% (w/v)
paraformaldehyde for 30 min at 37°C. Next, a primary antibody
against vimentin (1:100; cat. no. sc-5565; Santa Cruz
Biotechnology, Inc.) was added to the cells and incubated overnight
at 4°C, according to the manufacturer's protocol. Following primary
incubation, cells were stained with
fluorescein-isothiocyanate-conjugated goat anti-rat secondary
antibody (1:100; cat. no. ZF-0312; OriGene Technologies, Inc.) for
90 min at 37°C. LFs were stained using 50 µl DAPI solution (1:100;
cat. no. AR1176; Boster Biological Technology, Pleasanton, CA, USA)
for 10 min at 37°C and a fluorescence microscope (magnification,
×400) was used to analyze the results.
Immunocytochemical staining
The peroxidase-conjugated streptavidin method was
performed to detect the expression levels of vimentin or p-ERK1/2
in LFs using an Histostain™-Plus kit (OriGene Technologies, Inc.),
according to the manufacturer's protocol. Briefly, LFs were fixed
in cold 4% (w/v) paraformaldehyde for 30 min at 37°C and blocked
for 15 min at room temperature with 10% goat serum. LFs were
incubated with primary antibodies against vimentin (1:100; cat. no.
sc-5565; Santa Cruz Biotechnology) or p-ERK (1:100; cat. no. 4370;
Cell Signaling Technology, Inc.) overnight at 4°C. Following
primary incubation, LFs were incubated with biotinylated goat
anti-rat secondary antibodies for 20 min at 37°C followed by
HRP-labeled streptavidin for 20 min at 37°C. LFs were subsequently
stained with DAB solution (cat. no. ZLI-9031; OriGene Technologies,
Inc.) for 1 min at room temperature and observed under a light
microscope (magnification, ×400). LFs were subjected to
morphometric computerized image analysis using MetaMorph Software
System (version IPP6.0; Universal Imaging, Inc., Bedford Hills, NY,
USA), which was used to acquire images and quantify the average
optical density score of positively stained cells.
Cell Counting kit (CCK)-8 colorimetric
assay
LFs were cultured in 96-well plates
(5×105/well; 100 µl) in serum-free DMEM for 24 h at
37°C. CCK-8 solution (cat. no. C0038; Beyotime Institute of
Biotechnology, Haimen, China) was added to the wells (10 µl/well)
for 1 h at 37°C. Absorbance was measured at 450 nm using an ELISA
reader (UV-260; Shimadzu Corporation, Kyoto, Japan).
Flow cytometry cell cycle
analysis
To assess cells in G0 phase of the cell cycle, LFs
in logarithmic grow phase were collected and resuspended at density
of 1×106 cells/ml. Following centrifugation at 400 × g
for 5 min at 4°C, LFs were fixed in cold 75% ethanol overnight at
4°C. Subsequently, LFs were treated with 50 µg/ml RNase
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) for 30 min at 37°C
and LFs were stained with 100 µg/ml propidium iodide
(Sigma-Aldrich; Merck KGaA) in the dark for 30 min at 4°C. The cell
cycle assay was performed using a BD FACSCalibur™ flow
cytometer and CellQuest software (version 3.0; Becton Dickinson; BD
Biosciences, Franklin Lakes, NJ, USA).
ELISA
Collagen type I (Col-I) secreted by LFs in
logarithmic grow phase was determined by ELISA assay. Following
centrifugation (1,500 × g), cell culture supernatants were analyzed
for Col-I expression using the mouse Col-I ELISA kit (cat. no.
SEA571Mu; USCN Life Science Inc., Wuhan, China), according to the
manufacturer's protocol. Absorption was measured at 450 nm using a
microplate reader (ELX-800; BioTek Instruments, Inc., Winooski, VT,
USA).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Quantitative changes in mRNA expression of genes
encoding Col-I were assayed by RT-qPCR. Total RNA was extracted
from LFs using TRIzol® reagent (Takara Biotechnology
Co., Ltd., Dalian, China), according to the manufacturer's
protocol. Total RNA (500 ng/µl aliquots) were reverse-transcribed
into cDNA using a PrimeScript RT Reagent kit (cat. no. DRR037S;
Takara Biotechnology Co., Ltd.). qPCR was subsequently performed
using the SYBR Premix Ex Taq™ (cat. no. DRR041S; Takara
Biotechnology Co., Ltd.). The following gene-specific primers were
designed and synthesized by Takara Biotechnology Co., Ltd. and used
for the qPCR: Col-I forward, 5′-CTCCTGGCAAGAACGGAGATG-3′ and
reverse, 5′-CTGTTCCAGGCAATCCACGA-3′; β-actin forward,
5′-CCCGCGAGTACAACCTTCTT-3′ and reverse, 5′-TCATCCATGGCGAACTGGTG-3′.
The following thermocycling conditions were used for the qPCR:
Initial denaturation at 95°C for 10 sec, 40 cycles of 95°C for 5
sec and 60°C for 20 sec using the LightCycle 2.0 real-time PCR
System (Roche Diagnostics, Indianapolis, IN, USA). Relative
quantification of gene expression was determined using the
2−ΔΔCq method (16).
Western blot analysis
Lung tissue and cultured LFs were washed three times
with cold PBS and total protein was extracted using
radioimmunoprecipitation assay buffer (cat. no. P0013; Beyotime
Institute of Biotechnology). Total protein was quantified using a
bicinchoninic acid assay kit (Sigma-Aldrich) and 50 µg protein/lane
was separated via SDS-PAGE on a 12% gel. Gels were run at 120 V for
2 h and separated proteins were transferred at 80 V for 2 h onto
nitrocellulose membranes. Membranes were blocked for 1 h at 37°C
for 1 h with 5% non-fat milk. The membranes were incubated with
primary antibodies against p-ERK (1;1,000; cat. no. 4370), ERK
(1:1,000: cat. no. 4696) and β-actin (1:1,000; cat. no. 3700; all
Cell Signaling Technology, Inc.) overnight at 4°C. Following
primary incubation, membranes were incubated with HRP-labeled goat
anti-rat secondary antibody (1:2,000; cat. no. ZB2305; OriGene
Technologies, Inc.) for 2 h at 37°C. Membranes were washed three
times in Tris-buffered saline containing 0.1% Tween® 20.
Proteins bands were visualized using the Amersham ECL Advanced
Western Blotting Detection kit (Amersham; GE Healthcare Life
Sciences, Changhai, China). Images were collected using a ChemiDoc
XRS system (Bio-Rad Laboratories, Inc., Hercules, CA, USA). Protein
expression was quantified using Image-Pro Plus software (version
6.0; National Institutes of Health, Bethesda, MD, USA).
Statistical analysis
All experiments were repeated at least five times
and data are presented as the mean ± standard deviation. All
statistical analyses were performed using SPSS software (version
17.0; SPSS, Inc., Chicago, IL, USA). Differences between the
hyperoxia and normoxia control groups were assessed by one-way
analysis of variance and a Student-Newman-Keuls test for multiple
comparisons. P<0.05 was considered to indicate a statistically
significant difference.
Results
Exposure to hyperoxia leads to
expanded alveoli and interstitial fibrosis in lung tissue
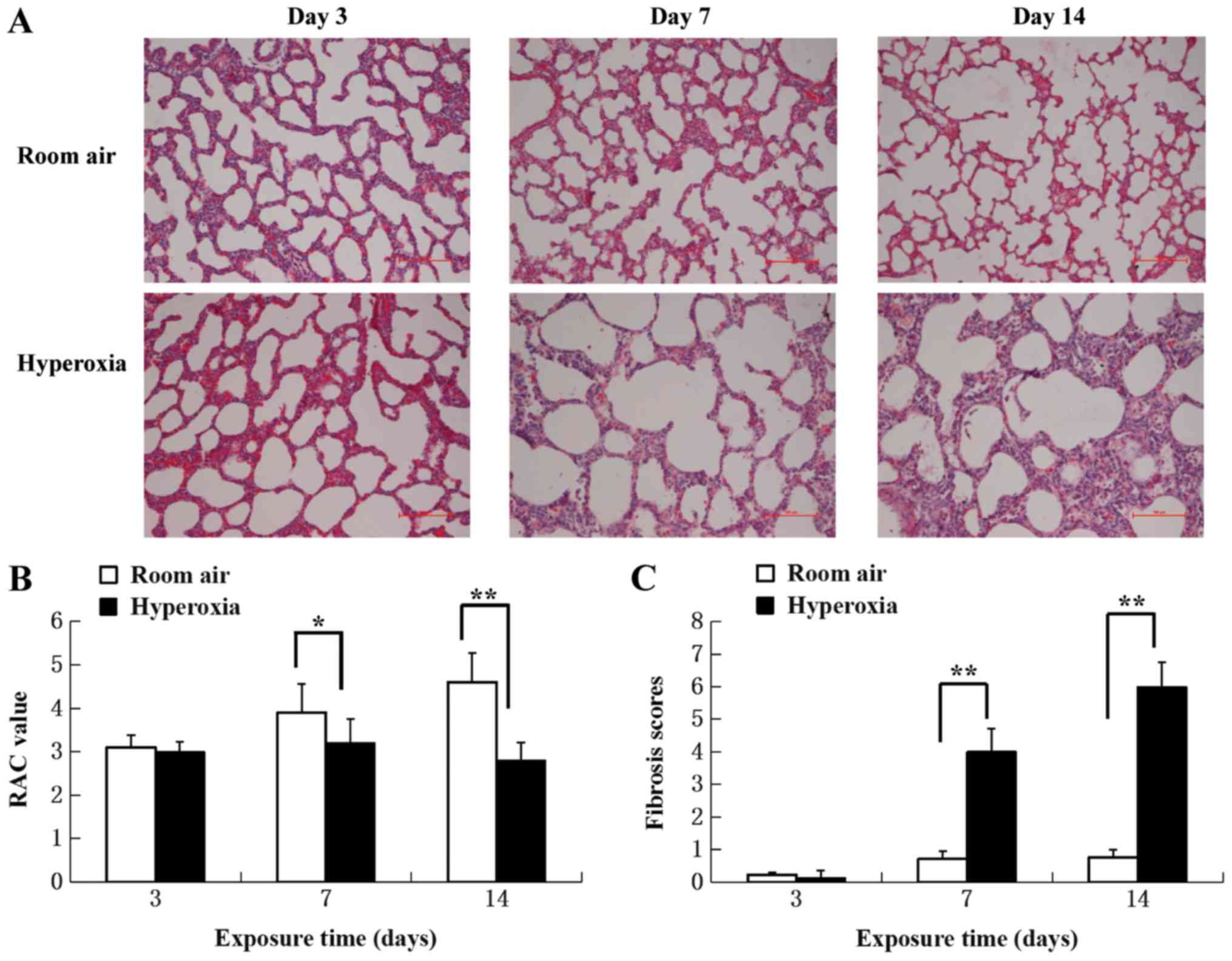
Pathological changes were evaluated by light
microscopy in H&E-stained sections of lung tissue from normoxia
and hyperoxia-exposed rats (Fig.
1A). No marked differences in pathology were apparent at day 3
between hyperoxia-exposed and normoxia control lung tissues.
Hyperoxia-associated differences were present on day 7 and
increased towards day 14. Most notably were the expansion and
dilatation of the alveoli, increased interstitial thickening and
pulmonary interstitial fibrosis.
The alveolar developmental stage and the number of
alveoli were determined by quantitative RAC. There were
significantly fewer alveoli in the lung tissues of the hyperoxia
group compared with the control group on day 7 and 14 (P<0.05;
Fig. 1B), suggesting the
interruption of alveoli formation.
Lung fibrosis scores were significantly increased in
the hyperoxia group at 7 and 14 day compared with the control group
(P<0.01; Fig. 1C), suggesting
that fibrosis started to develop between day 3 and 7 in hyperoxic
rats. The results demonstrated that hyperoxia disrupted normal lung
development and caused large, simplified alveolar structures and
matrix fibrosis.
LF identification
In primary cultures >95% of the cells were
identified as LFs, with typical stellate morphology and an
elongated spindle shape was observed under a light microscope. In
addition, positive cytoplasmic labeling using vimentin, a
mesenchymal cell marker was also observed (Fig. 2A and B).
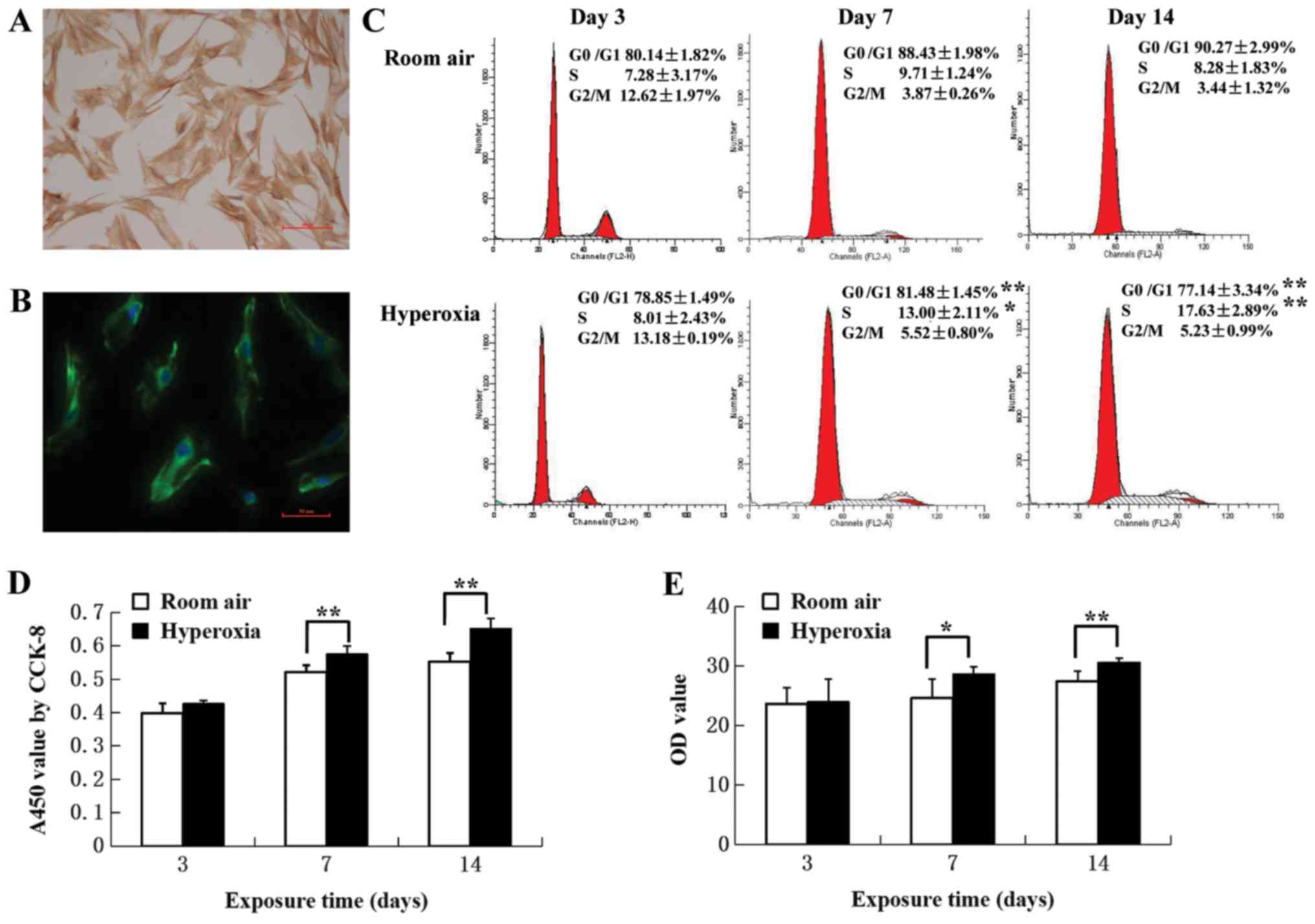 | Figure 2.Identification of LFs and
proliferation assays. (A) Immunocytochemical staining of LFs from
control rats. Primary LFs were stained with vimentin. LFs exhibited
stellate morphology, with an elongated spindle-like structure.
Scale bar, 100 µm. (B) Immunofluorescence staining of LFs. Primary
LFs from control rats were labeled with vimentin-specific
antibodies (green) and cell nuclei were labeled with
4′,6-diamidino-2-phenylindole (blue). Scale bar, 50 µm. (C)
Cell-cycle distribution. Cell populations in G0/G1, S and G2/M
phases were determined by calculating the mean of five independent
experiments. The proportion of cells in the G0/G1 phase decreased,
associated with an increase in the S phase in LFs from neonatal
rats in the hyperoxia group at postnatal day 7 and 14 compared with
the room air control group (n=5). (D) Effect of hyperoxia on cell
proliferation. CCK-8 assays were used to measure proliferation.
Hyperoxia promoted cell proliferation at postnatal day 7 and 14
(n=6). (E) Col-I secreted protein levels in LFs determined by
ELISA. An increase was observed in the hyperoxia group compared
with the room air control group (n=5). *P<0.05, **P<0.01. LF,
lung fibroblast; Col-I, collagen type I; CCK-8, cell counting
kit-8; OD, optical density. |
Exposure to hyperoxia induces
proliferation of LFs
The effect of hyperoxia on proliferation of LFs was
investigated by assaying cell cycle distribution by flow cytometry
and CCK-8. On day 3, most of the LFs in hyperoxia (78.85±1.49%) and
control (80.14±1.82%) groups were in G0/G1 phase (Fig. 2C). On day 7, the proportion of cells
in G0/G1 was decreased (81.48± 1.45 vs. 88.43±1.98%, respectively;
P<0.01) whilst the proportion of cells in S phase was increased
(13.00±2.11 vs. 9.71±1.24%, respectively; P<0.05) in LFs
isolated from hyperoxia group rats compared with control rats. On
day 14, there was a greater difference in the proportion of cells
in the S phase LFs isolated from hyperoxia group rats compared with
control rats (17.63±2.89 vs. 8.28±1.83%, respectively; P<0.01).
CCK-8 assays revealed that hyperoxia significantly promoted cell
proliferation when comparing day 7 with day 14 (P<0.01; Fig. 2D). These results suggested that LF
proliferation was associated with increasing proportions of cells
entering the S phase.
Exposure to hyperoxia induces Col-I
expression of LFs
Col-I protein was assayed in the supernatants of
primary LF cultures to evaluate the effect of hyperoxia on
fibrosis. ELISA assay revealed that hyperoxia resulted in the
increased production of Col-I protein on day 7 (P<0.05) and 14
(P<0.01) compared with the room air control (Fig. 2E). RT-qPCR revealed that Col-I mRNA
levels on day 3 (ΔCq, −5.36±0.35) were significantly increased by
1.93-fold compared with the control group (ΔCq, −4.51±0.16;
P<0.01). On day 7, Col-I mRNA expression in the hyperoxia group
(ΔCq, −6.73±0.27) was significantly increased by 2.62-fold compared
with the control group (ΔCq, −5.34±0.09; P<0.01). Col-I mRNA
levels in the hyperoxia group at day 14 (ΔCq, −7.16±0.21) were
significantly increased by 2.77-fold compared with that in the room
air control group (ΔCq, −5.69±0.37; P<0.01).
Hyperoxia induces time-dependent
activation of ERK in lung tissue
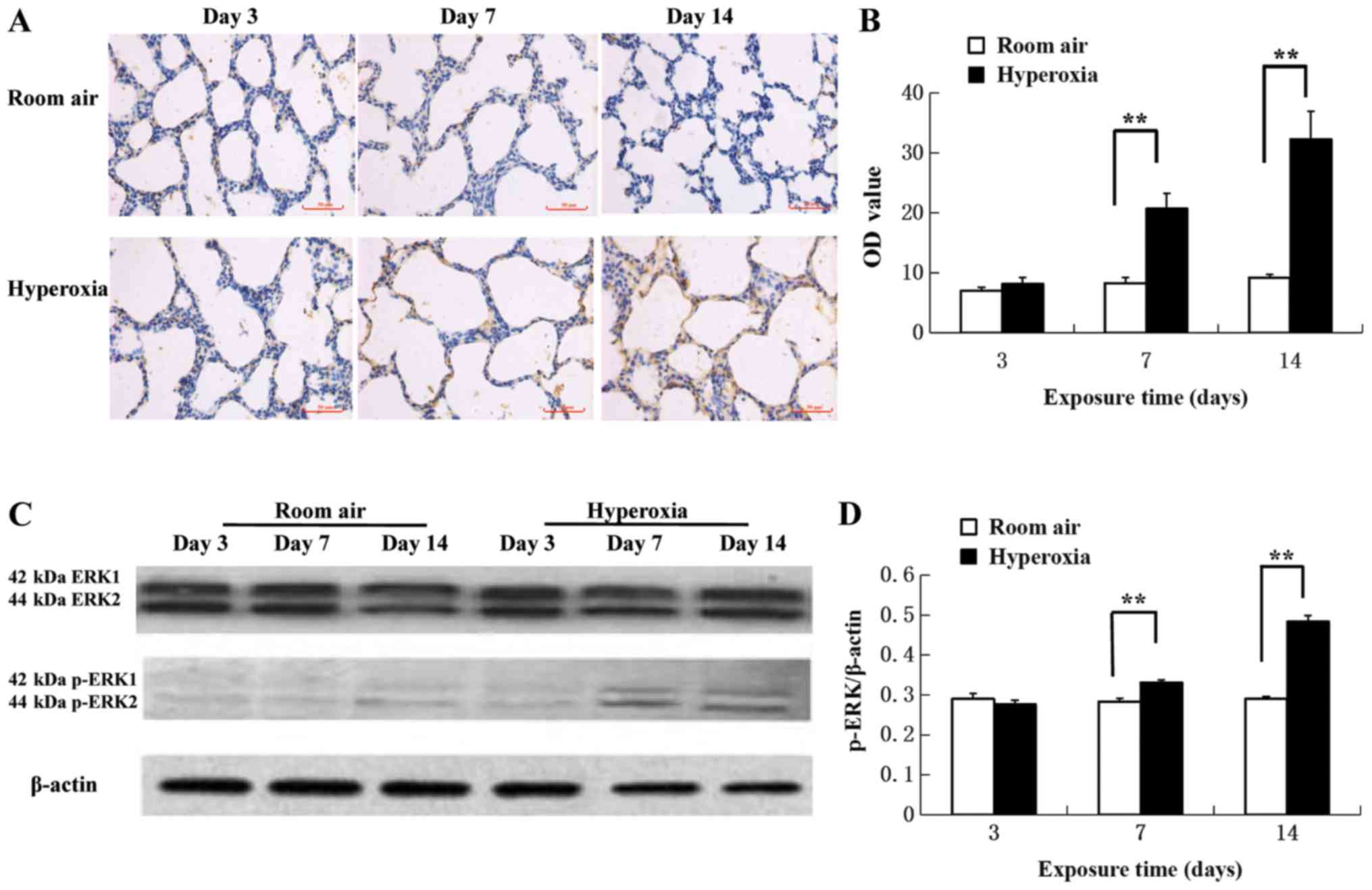
Immunohistochemical assays of p-ERK protein levels
demonstrated increased p-ERK1/2 antigen response in the cytoplasm
and nuclei of pulmonary epithelial and mesenchymal cells in
hyperoxic rats on day 7 and 14 compared with the room air control
(Fig. 3A). The difference in
expression in the two groups at day 3 was not significant
(P>0.05; Fig. 3B). However, the
protein levels of p-ERK detected significantly increased on day 7
in the hyperoxia group compared with the air control (20.80±2.52
vs. 8.32±0.88, respectively; P<0.01; Fig. 3B). Additionally, the protein levels
of p-ERK detected significantly increased on day 14 in the
hyperoxia group compared with the air control (32.39±4.64 vs.
9.24±0.54, respectively; P<0.01; Fig.
3B). Western blot analysis revealed that hyperoxia
significantly increased ERK1/2 phosphorylation on day 7 and 14
compared with the room air control (Fig.
3C and D).
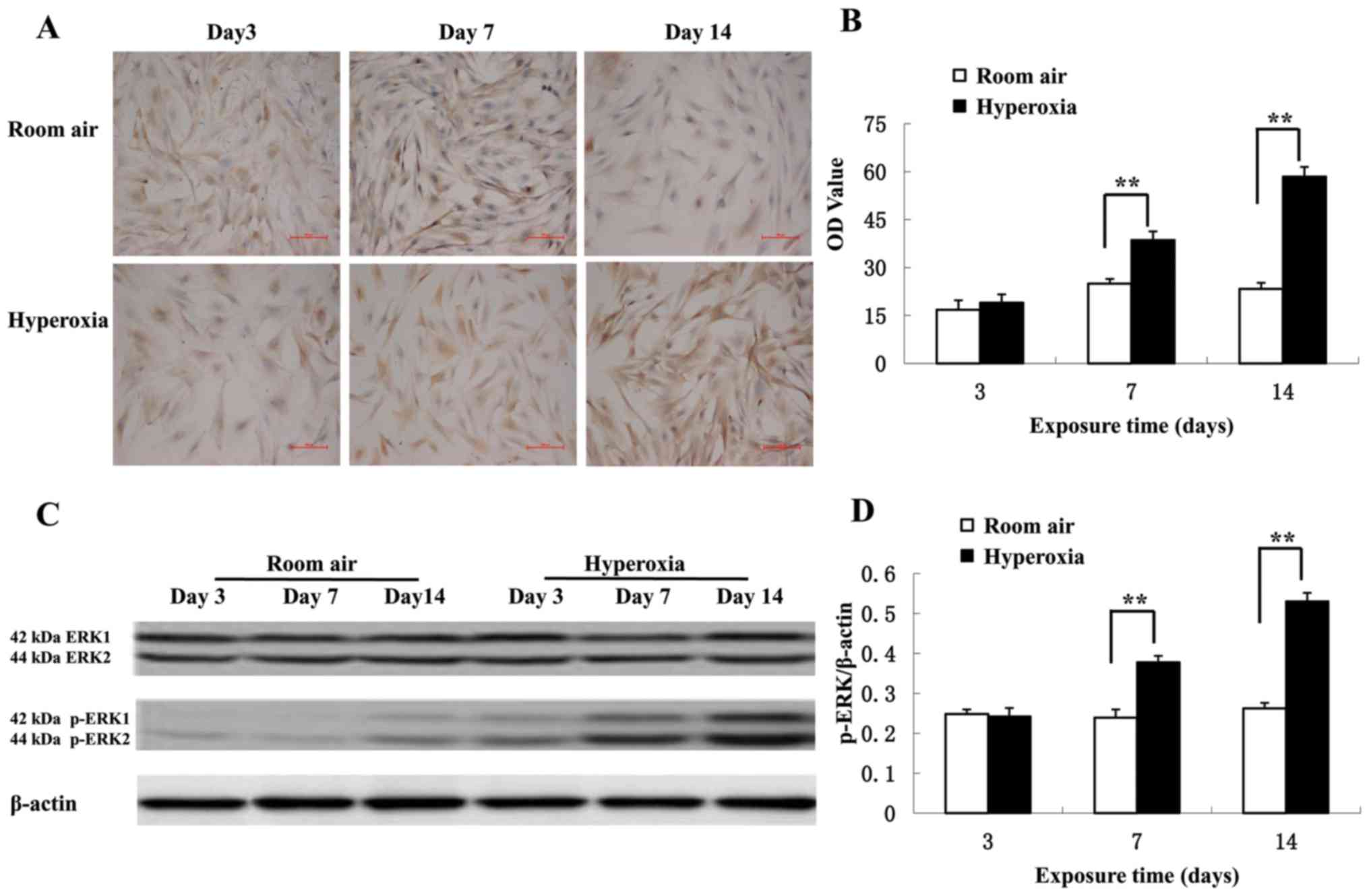
Immunocytochemical staining of LFs from primary
cultures demonstrated that p-ERK levels are increased in the
cytoplasm in the hyperoxia group compared with the control group
(Fig. 4A). On day 3, there was no
significant difference in p-ERK levels between the hyperoxic and
control groups (P>0.05; Fig. 4B).
However, the p-ERK levels significantly increased on day 7 in the
hyperoxia group compared with the control group (38.64±2.68 vs.
24.93±1.51, respectively; P<0.01). Additionally, the p-ERK
levels significantly increased on on day 14 in the hyperoxia group
compared with the control group (58.53±2.97 vs. 23.33±1.90,
respectively; P<0.01). Western blot analysis revealed that
hyperoxia significantly increased ERK1/2 phosphorylation on day 7
and 14 compared with the room air control (P<0.01; Fig. 4C and D) confirming the
immunocytochemistry results. There was no change in the total
expression of ERK1/2 in lung tissues and primary cultured LFs (data
not shown).
Discussion
BPD is a chronic lung disease commonly seen in
premature infants and the associated morbidity has increased with
the use of pulmonary surfactants, assisted ventilation and oxygen
treatment (4,17). BPD is a consequence of long-term
oxygen therapy threatening the health and life of premature infants
(18). The pathology of the
pulmonary response to hyperoxia is characterized by remodeling of
the airway development and parenchymal, and the vascular structure.
This situation is further influenced by the immaturity of specific
lung cells in newborn rats that develop BPD (19,20). LFs
are present in the ECM and serve essential roles during pulmonary
epithelial injury and in response to the fibrosis development in
BPD. It has been observed that in vitro the direct effect of
hyperoxia on LFs over 48 h inhibited proliferation and decreased
collagen expression (21). A
previous study demonstrated that hyperoxia on lung fibroblasts for
48 h in vitro led to the inhibition of cellular
proliferation and decreased collagen expression (6). The direct effect of LFs to hyperoxic
exposure was contradictory to the pathological interstitial
proliferation character of BPD (22). Primary cultured LFs from newborn rats
with BPD are a promising technique to investigate mechanisms of
hyperoxia-induced fibrosis. In the current study, hyperoxia
disrupted the normal lung development by exhibiting large,
simplified alveoli and pulmonary fibrosis. Primary cultured LFs
from hyperoxic rats demonstrated a proliferative status with
increased levels of Col-I secretion. Hyperoxia upregulated the
phosphorylation of ERK1/2 in lung tissues and LFs from rats on
postnatal day 7 and 14. These findings indicated that hyperoxia
promoted an increase of proliferation of LFs in newborn rats
linearly over time and the phosphorylation of ERK1/2 may have
served a role in this process.
The regulation of LF proliferation via the ERK
signaling pathway was in line with the pathologic changes observed
in BPD. Hyperoxic exposure has been demonstrated to lead to
myofibroblast accumulation, patchy areas of interstitial thickening
and increased lung Col-I and α-smooth muscle actin (SMA) levels
(23–26). The findings of the current study
suggested that the ERK signaling pathway may serve an important
role in regulating primary cultured LF proliferation and is in
accordance with previous studies (27–30).
Exposure of newborn Sprague-Dawley rat pups, started within 12 h of
birth, for 1 week with 95% oxygen followed by two weeks with 60%
oxygen resulted in an increase of total collagen, Col-I and α-SMA
expression and activated ERK in lung tissues on postnatal day 7 and
21, consistent with the development of pulmonary fibrosis (27). Hyperoxia has further been reported to
promote alveolar interstitial fibroblast-to-myofibroblast
transdifferentiation (28).
Increased p-ERK1/2, α-SMA and Col-I expression was further promoted
in human fetal LFs exposed to 95% oxygen for 48 h (29). Following bleomycin administration to
produce acute lung injury in rats, high tidal volume ventilation
induced lung fibrosis that was partly dependent on the ERK1/2
signaling pathway activation and was attenuated by PD98059, an
ERK1/2 inhibitor (30).
The ERK signaling pathway may be important in BPD
due to its participation in normal lung development. However,
further research is required to validate these finding. The
Mek gene encodes ERKs and loss of the Mek1 gene in
rats resulted in placental defects and embryonic death (31). Mek2-mutant rats did not
express an abnormal phenotype as a result of a protein threshold
effect (13). Inactivation of both
Mek genes in epithelial cells causes lung agenesis and may
be fatal, and mutant Mek genes in mesenchymal cells present
as lung hypoplasia, tracheal defects and neonatal death, but not
kyphosis and omphalocele (31).
Expression studies have indicated potential interactions of the ERK
and Wnt signaling pathways during lung development (32). Dermo1Cre-deletion
rats are deficient in the mesenchyme-specific Mek gene
function, which is associated with intrauterine growth restriction,
giant omphalocele, kyphosis, lung hypoplasia, altered trachea
patterning and still birth (33).
Lung hypoplasia is accompanied by reduced branching, decreased
mesenchymal cell proliferation and increased apoptosis (6).
ERK also appears to promote fibrosis through
transduction signals that involve matrix metalloproteinases (MMPs)
and transforming growth factor (TGF)-β (34–38).
MS80 is a novel, sulfated oligosaccharide extracted from seaweed
and it inhibits bleomycin-induced pulmonary fibrosis in rats by
arresting TGF-β1-induced proliferation of human embryonic LFs,
collagen deposition and MMP activity (34). TGF-β1 has also been suggested to
induce α-SMA expression and collagen production in human LFs via
ERK1/2 activation (35), and MMP
mRNA and p-ERK1/2 expression increased following TGF-β stimulation
in primary cultured human LFs (36).
Collectively, the evidence indicates that the promotion of fibrosis
by MMPs and TGF-β is closely associated with the function of
ERK1/2. However, there is no direct evidence to suggest whether
ERK1/2 is an up- or downstream regulator of TGF-β or MMPs. Previous
studies have demonstrated that hydrogen sulfide and rosiglitazone
suppress migration, proliferation and phenotypic differentiation of
human LFs in vitro through inhibition of ERK activation and
that these compounds may be effective in treating pulmonary
interstitial fibrosis (37,38). ERK1/2 may become a target of measures
to prevent and treat pulmonary hypoplasia and fibrosis associated
with BPD in premature infants.
Acknowledgements
The authors would like to thank Dr Xueyan Liu
(Shengjing Hospital of China Medical University) for her
encouragement and support of this study.
Funding
This work was funded by the Natural Science
Foundation of China (grant no. 30801245).
Availability of data and materials
All datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YH performed the experiments. JF collected and
analyzed the data. XX designed the study and was the major
contributor in writing the manuscript. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
The current study was approved by the Medical Ethics
Committee at Shengjing Hospital of China Medical University (no.
2018PS209K).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
ERK
|
extracellular signal-regulated
kinase
|
|
BPD
|
bronchopulmonary dysplasia
|
|
Col-I
|
collagen type I
|
|
LFs
|
lung fibroblasts
|
References
|
1
|
Kayton A, Timoney P, Vargo L and Perez JA:
A review of oxygen physiology and appropriate management of oxygen
levels in premature neonates. Adv Neonatal Care. 18:98–104. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Vogel ER, Britt RD Jr, Trinidad MC, Faksh
A, Martin RJ, MacFarlane PM, Pabelick CM and Prakash YS: Perinatal
oxygen in the developing lung. Can J Physiol Pharmacol. 93:119–127.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Perrone S, Bracciali C, Di Virgilio N and
Buonocore G: Oxygen use in neonatal care: A two-edged sword. Front
Pediatr. 9:1432017.
|
|
4
|
Jobe AH: Mechanisms of lung injury and
bronchopulmonary dysplasia. Am J Perinatol. 33:1076–1078. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Thekkeveedu Kalikkot R, Guaman MC and
Shivanna B: Bronchopulmonary dysplasia: A review of pathogenesis
and pathophysiology. Respir Med. 132:170–177. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Coalson JJ: Pathology of bronchopulmonary
dysplasia. Semin Perinatol. 30:179–184. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sheppard D: Epithelial-mesenchymal
interactions in fibrosis and repair. Transforming growth factor-β
activation by epithelial cells and fibroblasts. Ann Am Thorac Soc.
1 Suppl 12:S21–S23. 2015. View Article : Google Scholar
|
|
8
|
McGowan S: Understanding the developmental
pathways pulmonary fibroblasts may follow during alveolar
regeneration. Cell Tissue Res. 367:707–719. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Murray MJ: The role of netrins and their
receptors in epithelial mesenchymal plasticity during development.
Cells Tissues Organs. 203:71–81. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hayes D Jr, Feola DJ, Murphy BS, Shook LA
and Ballard HO: Pathogenesis of bronchopulmonary dysplasia.
Respiration. 79:425–436. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Porzionato A, Sfriso MM, Mazzatenta A,
Macchi V, De Caro R and Di Giulio C: Effects of hyperoxic exposure
on signal transduction pathways in the lung. Respir Physiol
Neurobiol. 209:106–114. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Rubinfeld H and Seger R: The ERK cascade:
A prototype of MAPK signaling. Mol Biotechnol. 31:151–174. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bélanger LF, Roy S, Tremblay M, Brott B,
Steff AM, Mourad W, Hugo P, Erikson R and Charron J: Mek2 is
dispensable for mouse growth and development. Mol Cell Biol.
23:4778–4787. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jakkula M, Le Cras TD, Gebb S, Hirth KP,
Tuder RM, Voelkel NF and Abman SH: Inhibition of angiogenesis
decreases alveolarization in the developing rat lung. Am J Physiol
Lung Cell Mol Physiol. 279:L600–L607. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ashcroft T, Simpson JM and Timbrell V:
Simple method of estimating severity of pulmonary fibrosis on a
numerical scale. J Clin Pathol. 41:467–470. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C (T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Coalson JJ: Experimental models of
bronchopulmonary dysplasia. Biol Neonate. 1 Suppl 71:S35–S38. 1997.
View Article : Google Scholar
|
|
18
|
Baraldi E and Filippone M: Chronic lung
disease after premature birth. N Engl J Med. 357:1946–1955. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chen Y, Whitney PL and Frank L:
Comparative responses of premature versus full-term newborn rats to
prolonged hyperoxia. Pediatr Res. 35:233–237. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cox AM, Gao Y, Perl AT, Tepper RS and
Ahlfeld SK: Cumulative effects of neonatal hyperoxia on murine
alveolar structure and function. Pediatr Pulmonol. 52:616–624.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hussain N, Wu F, Christian C and Kresch
MJ: Hyperoxia inhibits fetal rat lung fibroblast proliferation and
expression of procollagens. Am J Physiol. 273:L726–L732.
1997.PubMed/NCBI
|
|
22
|
Silva DM, Nardiello C, Pozarska A and
Morty RE: Recent advances in the mechanisms of lung alveolarization
and the pathogenesis of bronchopulmonary dysplasia. Am J Physiol
Lung Cell Mol Physiol. 309:L1239–L1272. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Porzionato A, Zaramella P, Macchi V,
Grisafi D, Salmaso R, Baraldi M, Fornaro E, Tassone E, Masola V,
Onisto M, et al: Fluoxetine may worsen hyperoxia-induced lung
damage in neonatal rats. Histol Histopathol. 27:1599–1610.
2012.PubMed/NCBI
|
|
24
|
Porzionato A, Zaramella P, Macchi V,
Sarasin G, Di Giulio C, Rigon A, Grisafi D, Dedja A, Chiandetti L
and De Caro R: Cyclosporine and hyperoxia-induced lung damage in
neonatal rats. Respir Physiol Neurobiol. 187:41–46. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Grisafi D, Pozzobon M, Dedja A, Vanzo V,
Tomanin R, Porzionato A, Macchi V, Salmaso R, Scarpa M, Cozzi E, et
al: Human amniotic fluid stem cells protect rat lungs exposed to
moderate hyperoxia. Pediatr Pulmonol. 48:1070–1080. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Grisafi D, Tassone E, Dedja A, Oselladore
B, Masola V, Guzzardo V, Porzionato A, Salmaso R, Albertin G,
Artusi C, et al: L-citrulline prevents alveolar and vascular
derangement in a rat model of moderate hyperoxia-induced lung
injury. Lung. 190:419–430. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jiang JS, Lang YD, Chou HC, Shih CM, Wu
MY, Chen CM and Wang LF: Activation of the renin-angiotensin system
in hyperoxia-induced lung fibrosis in neonatal rats. Neonatology.
101:47–54. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Li Z, Choo-Wing R, Sun H, Sureshbabu A,
Sakurai R, Rehan VK and Bhandari V: A potential role of the JNK
pathway in hyperoxia-induced cell death, myofibroblast
transdifferentiation and TGF-β1-mediated injury in the developing
murine lung. BMC Cell Biol. 12:542011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lang YD, Hung CL, Wu TY, Wang LF and Chen
CM: The renin-angiotensin system mediates hyperoxia-induced
collagen production in human lung fibroblasts. Free Radic Biol Med.
49:88–95. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Li LF, Liao SK, Huang CC, Hung MJ and
Quinn DA: Serine/threonine kinase-protein kinase B and extracellar
signal-regulated kinase regulate ventilator-induced pulmonary
fibrosis after bleomycin-induced acute lung injury: A prospective,
controlled animal experiment. Crit Care. 12:R1032008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Aoidi R, Maltais A and Charron J:
Functional redundancy of the kinases MEK1 and MEK2: Rescue of the
Mek1 mutant phenotype by Mek2 knock-in reveals a protein threshold
effect. Sci Signal. 9:ra92016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Boucherat O, Landry-Truchon K, Aoidi R,
Houde N, Nadeau V, Charron J and Jeannotte L: Lung development
requires an active ERK/MAPK pathway in the lung mesenchyme. Dev
Dyn. 246:72–82. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Boucherat O, Nadeau V, Bérubé-Simard FA,
Charron J and Jeannotte L: Crucial requirement of ERK/MAPK
signaling in respiratory tract development. Development.
142:38012015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Jiang HD and Guan HS: MS80, a novel
sulfated oligosaccharide, inhibites pulmonary fibrosis by targeting
TGF-beta1 both in vitro and in vivo. Acta Pharmacol Sin.
30:973–979. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Caraci F, Gili E, Calafiore M, Failla M,
La Rosa C, Crimi N, Sortino MA, Nicoletti F, Copani A and Vancheri
C: TGF-beta1 targets the GSK-3beta/beta-catenin pathway via ERK
activation in the transition of human lung fibroblasts into
myofibroblasts. Pharmacol Res. 57:274–282. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Asano K, Shikama Y, Shoji N, Hirano K,
Suzaki H and Nakajima H: Tiotropium bromide inhibits TGF-β-induced
MMP production from lung fibroblasts by interfering with Smad and
MAPK pathways in vitro. Int J Chron Obstruct Pulmon Dis. 5:277–286.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Fang LP, Lin Q, Tang CS and Liu XM:
Hydrogen sulfide suppresses migration, proliferation and
myofibroblast transdifferentiation of human lung fibroblasts. Pulm
Pharmacol Ther. 22:554–561. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lin Q, Fang LP, Zhou WW and Liu XM:
Rosiglitazone inhibits migration, proliferation, and phenotypic
differentiation in cultured human lung fibroblasts. Exp Lung Res.
36:120–128. 2010. View Article : Google Scholar : PubMed/NCBI
|