Introduction
It has previously been reported that elevated
triglyceride levels are a major risk factor for residual
cardiovascular diseases (CVDs) (1).
Hypertriglyceridemia is a common issue worldwide (2), and great attention has been given to
identifying an appropriate treatment. Palmitic acid (PA), a major
component of triglyceride in the blood (3), has been reported to induce cell
dysfunction and death, particularly in nonadipose tissue cells,
including pancreatic β cells, cardiomyocytes and hepatocytes
(4). The lipotoxic effect of PA has
been implicated in the pathogenesis of numerous CVDs (5). In addition, endothelial cells are
important cellular components of the cardiovascular system, and
therefore endothelial dysfunction is usually one of the early signs
of CVD (6).
It is generally accepted that PA-induced cell death
can occur due to increased reactive oxygen species (ROS)
generation. A study recently reported that PA also serves an
important role in the initiation of autophagy (7). ROS and autophagy have been linked to a
number of pathophysiological mechanisms, and ROS at physiological
concentrations are known to regulate redox homeostasis and
kinase-driven signaling pathways (8). However, excessive ROS accumulation
leads to oxidative stress that contributes to various malignancies
and disorders (9). Macroautophagy,
commonly known as autophagy, typically serves as a cell survival
mechanism, although it can result in type II programmed cell death
under certain conditions (10).
Intracellular ROS are primarily generated as by-products in
mitochondria (11). Certain enzymes,
including nicotinamide adenine dinucleotide phosphate oxidases
(NOXs), xanthine oxidase, endoplasmic reticulum oxidoreductase 1
and myeloperoxidase, as well as a number of organelles, including
peroxisomes, are important sources of ROS generation (12,13). As
reported previously, excess ROS generation enhances autophagic
activity via multiple pathways, which in turn degrades impaired
mitochondria to restore normal ROS levels (14). However, exorbitant autophagy results
in lysosomal dysfunction and endoplasmic reticulum stress (15). Although autophagy inhibition
decreases ROS levels, the mechanism underlying this phenomenon
remains to be elucidated (16).
The aim of the present study was to investigate the
causal association between autophagy activation and ROS generation
following PA treatment, as well as the molecular mechanism
responsible for this effect in endothelial cells. The results
revealed that PA-induced lipotoxicity is associated with autophagy
activation, which enhances ROS generation via activating the
calcium ion/protein kinase Cα/nicotinamide adenine dinucleotide
phosphate oxidase 4 (Ca2+/PKCα/NOX4) pathway in
endothelial cells. These results provide an insight into the
potential of treating CVD by targeting autophagy.
Materials and methods
Cell culture
Human umbilical vein endothelial cells (HUVECs) at
passage 20 and 25 were used in all experiments (ATCC, Manassas, VA,
USA). Cells were grown in Dulbecco's modified Eagle's medium (DMEM;
Hyclone; GE Healthcare, Logan, UT, USA) supplemented with 10% fetal
bovine serum (FBS; Hyclone; GE Healthcare) and 1% penicillin and
streptomycin at 37°C in an atmosphere containing 5%
CO2.
PA treatment
A solution of 10% (w/v) bovine serum albumin (BSA;
Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) was used to dissolve
PA (Sigma-Aldrich; Merck KGaA) in order to obtain a final
concentration of 0.3 mM. The autophagy inhibitor 3-methyladenine
(3-MA; Selleck Chemicals, Houston, TX, USA) was dissolved in 0.3 mM
PA at 1 M (PA+3-MA group) and NOX4 inhibitor GKT137831 (Selleck
Chemicals) was dissolved in PA at 20 µM (PA+NOX4 inhibitor group).
BSA (10%) alone was used as the vehicle control. All groups were
treated at room temperature for 24 h.
Cell viability assessment
Cell viability was assessed using Cell Counting
Kit-8 (CCK-8; Dojindo Molecular Technologies, Inc., Kumamoto,
Japan). Briefly, cells were seeded at density of 1×104 cells/well
in 96-well plates. Cells were washed twice with PBS, following
which CCK-8 reagent pre-mixed with DMEM at a ratio of 1:10 was
added. Following incubation for 1 h, cell viability was measured by
reading the absorbance at 450 nm on a microplate reader (EnVision
XCite; PerkinElmer, Inc., Waltham, MA, USA).
ROS assay
Intracellular ROS production was measured using a
2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA) assay
according to the manufacturer's protocol (Beyotime Institute of
Biotechnology, Haimen, China). Cells were washed thrice with
serum-free DMEM, seeded in 6-well plates at a density of 2×105
cells/well and incubated for 30 min at 37°C with 1 ml DCFH-DA
(1:1,000). Subsequently, cells were washed thrice with serum-free
DMEM and observed under an inverted fluorescence microscope
(Olympus Corp., Tokyo, Japan).
Cytosolic Ca2+
measurement
Fura-2 acetoxymethyl (AM; Beyotime Institute of
Biotechnology) was used to measure the cytosolic Ca2+
concentration. Briefly, cells were seeded in 96-well plates at a
density of 1×104 cells/well and seeded with 2 µM Fura-2 AM diluted
in Dulbecco's phosphate-buffered saline (DPBS). After incubation of
30 min, extracellular Fura-2 AM was then washed away three times by
DPBS. Cellular fluorescence intensity measured at excitation
wavelength of 340 and 380 nm. Cytosolic Ca2+ was
expressed as the ratio of emitted fluorescence at 340 and 380
nm.
Cell repair capability
measurement
A scratch-wound healing assay was performed to
assess the repair ability of endothelial cells. Briefly, cells were
seeded in 6-well plates at a density of 2×105 cells/well marked
with three equidistant parallel lines on the bottom of each well
and starved for 24 h before the experiment. Subsequently, scratch
wound was performed by a 10-µl max-volume pipette, the detached
cells were washed away by DPBS. Three fields were selected for
observation and imaging under a microscope (Olympus Corp.) when PA
was added, images were taken and evaluated as 0 h. After PA
treatment for 24 h, the same fields were assessed and marked as 24
h to evaluate the repair ability of endothelial cells.
Nitric oxide (NO) assay
NO generation in HUVECs was measured using a
Nitrate/Nitrite Assay kit (Beyotime Institute of Biotechnology).
Initially, cells were treated with PA for 24 h in 6-well plates.
Supernatants were collected after centrifugation for 5 min at 500 ×
g at room temperature for the NO assay (Beyotime Institute of
Biotechnology). Nitrate/Nitrite Assay reagents was added into these
supernatants according to the manufacturer's protocol. NO
expression was estimated by measuring the absorbance at 540 nm.
Matrigel tube formation assay
The formation of capillary-like structures by HUVECs
was assessed using the Matrigel Basement Membrane Matrix (Corning
Inc., Corning, NY, USA). Briefly, HUVECs were seeded in
Matrigel-coated 96-well plates at a density of 5×104 cells/cm2.
Tube formation was observed under an inverted phase fluorescent
microscope (Olympus Corp.), and images were captured with a digital
camera. The results were quantified by measuring the total length
of the tubules in each well.
Western blotting
Total protein was extracted from the cells using
radioimmunoprecipitation assay buffer (Beyotime Institute of
Biotechnology) and quantified by BCA (Beyotime Institute of
Biotechnology). Equal amounts of cellular proteins (20 µg) were
separated by SDS-PAGE on 10% gel and transferred to a
polyvinylidene membrane. After blocking with 5% non-fat milk at
room temperature for 2 h, the membranes were incubated overnight at
4°C with primary antibodies diluted at 1:1,000. The following
primary antibodies were used: Light chain 3 (LC3; cat. no. 12741;
Cell Signaling Technology, Inc., Danvers, MA, USA), PKCα (cat. no.
sc-8393; Santa Cruz Biotechnology, Inc., Dallas, TX, USA), p-PKCα
(cat. no. sc-377565; Santa Cruz Biotechnology, Inc.), p62 (cat. no.
sc-28359; Santa Cruz Biotechnology, Inc.), NOX4 (cat. no. ab133303;
Abcam, Cambridge, UK) and GAPDH (diluted at 1:2,000; cat. no.
sc-47724; Santa Cruz Biotechnology, Inc.). BeyoECL Plus (cat. no.
P0018; Beyotime Institute of Biotechnology) was used and the
detected bands were quantified by densitometric analysis and
normalized to those of the corresponding loading control GAPDH
using ImageJ software k 1.45 (National Institutes of Health,
Bethesda, MD, USA).
Transmission electron microscopy
Cells were harvested following 24 h of treatment,
washed with 0.1 M cacodylate buffer (cat. no. 37238-25, Nacalai
Tesque, Inc., Kyoto, Japan), fixed in 1% agarose in 0.1 M
cacodylate buffer and then post-fixed in 1% osmium tetroxide (cat.
no. 75632; Sigma-Aldrich; Merck KGaA) for 2 h. Following
dehydration in serially diluted ethanol solutions, the cells were
embedded in an epoxy resin. Ultrathin (60–80 nm) sections were cut
and placed on the slides. The sections on the slides were then
double stained with uranyl acetate and lead nitrate prior to
examining under a transmission electron microscope (HT-7700;
Hitachi, Ltd., Tokyo, Japan).
Statistical analysis
Data were analyzed using IBM SPSS software (version
20.0; IBM Corp., Armonk, NY, USA) and are expressed as the mean ±
standard error of the mean. Comparisons between multiple groups
were assessed by Student-Newman-Keuls test, and P<0.05 was
considered to indicate a statistically significant difference.
Results
PA induces endothelial dysfunction in
HUVECs
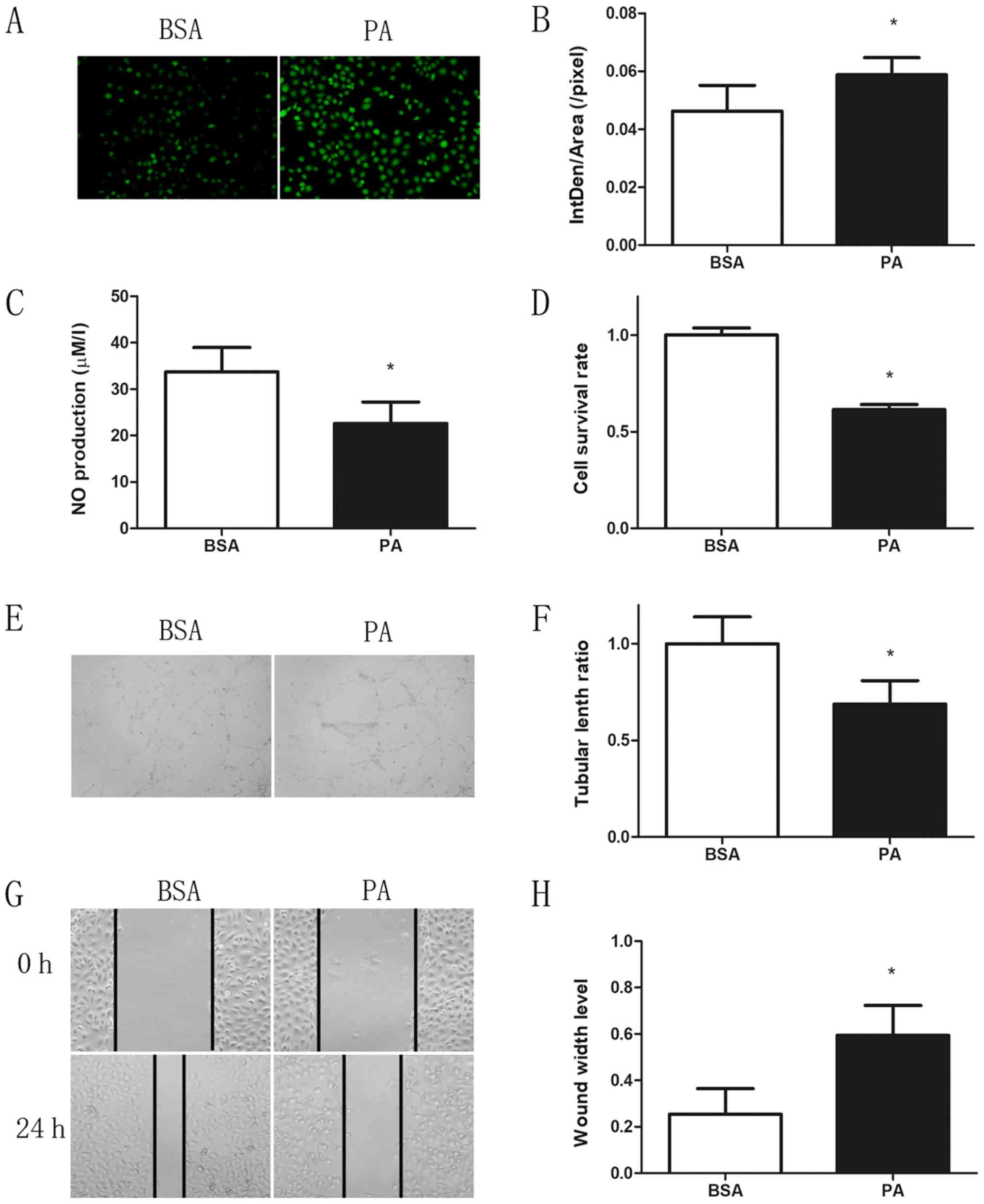
To assess whether ROS generation and the subsequent
decrease in NO synthesis serve a critical role in the pathogenesis
of endothelial dysfunction, ROS and NO production was assessed in
HUVECs treated with 0.3 mM PA for 24 h. As expected, treatment with
PA for 24 h resulted in a significant increase in ROS generation
and decrease in NO synthesis as compared with those observed in the
control group (Fig. 1A-C). Cell
viability was significantly decreased following PA treatment for 24
h, with ~60% of cells surviving (Fig.
1D). In addition, the ability to form capillary-like
structures, which is a crucial indicator of angiogenesis, was
markedly reduced following PA treatment (Fig. 1E and F). The repair ability of cells
was also significantly suppressed by PA treatment, as observed by
the reduced wound healing compared with the control group (Fig. 1G and H).
Autophagy inhibition decreases
PA-induced ROS accumulation
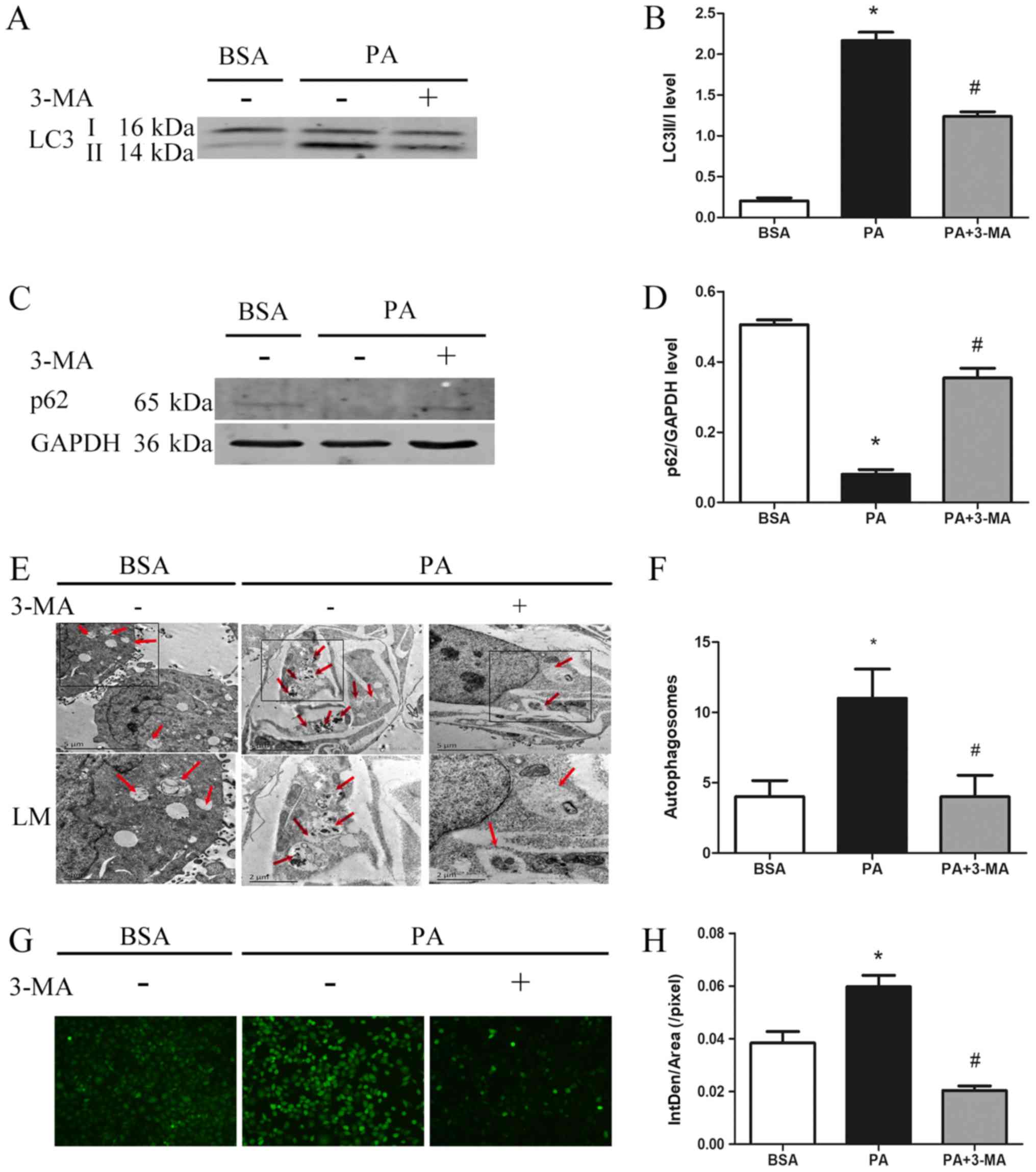
To assess the effect of autophagy on ROS production,
the autophagic activity was estimated by monitoring LC3 turnover
and p62 degradation. The results were in agreement with previous
observations (17). PA treatment was
observed to significantly increase the LC3II/LC3I ratio (Fig. 2A and B), markedly decrease p62
expression (Fig. 2C and D) and
significantly increase the number of autophagosomes (Fig. 2E and F). However, these effects of PA
were blocked by 3-MA, a specific autophagy inhibitor (Fig. 2A-F). Furthermore, PA-induced ROS
generation was significantly inhibited by 3-MA, suggesting that
PA-induced ROS generation is autophagy-dependent (Fig. 2G and H).
PA-induced autophagy activates the
Ca2+/PKCα/NOX4 pathway
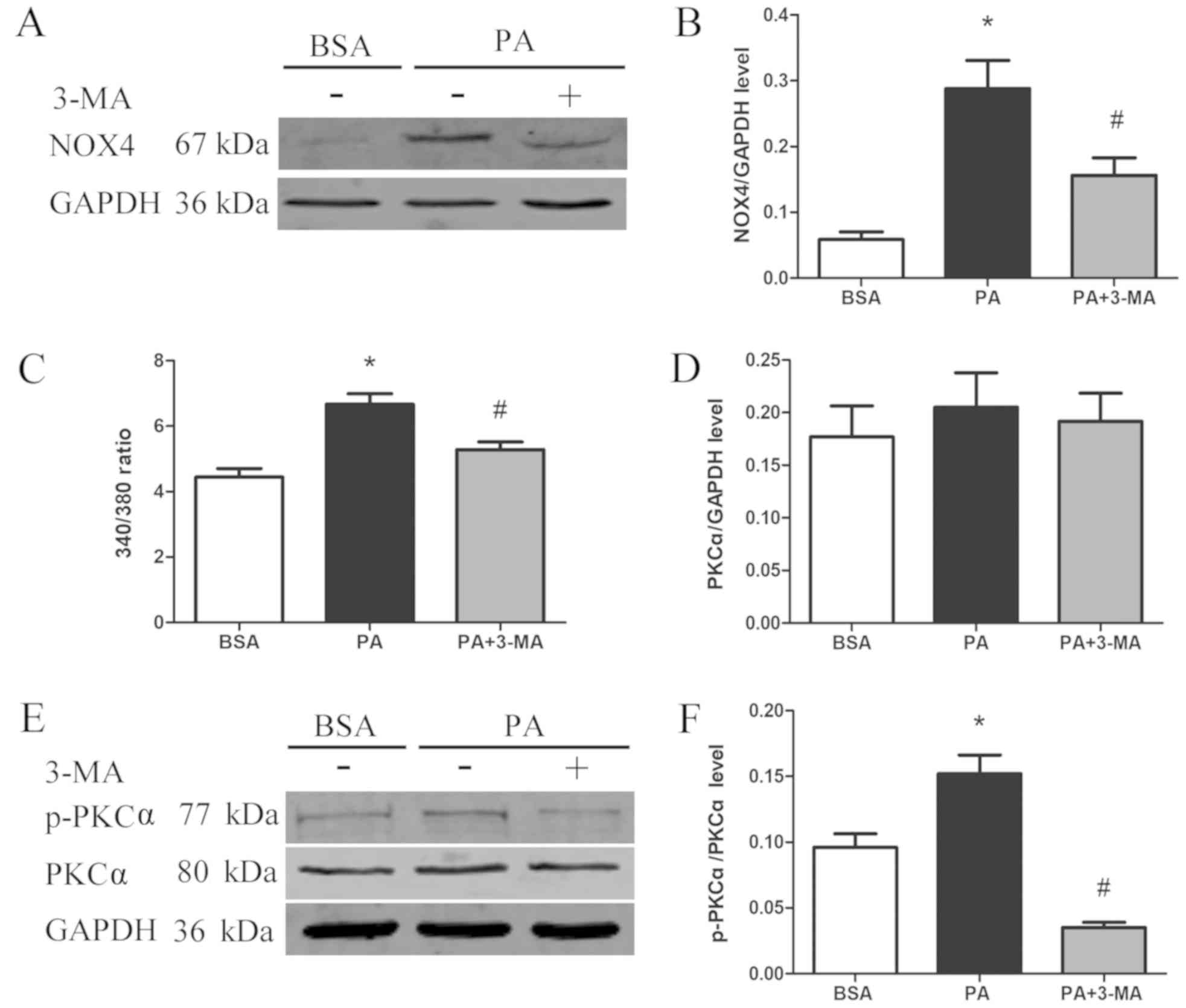
NOX4 is an important source of intracellular ROS
(18); therefore, the effect of
PA-induced autophagy on NOX4 protein expression was assessed in the
current study. NOX4 expression was significantly upregulated
following PA treatment; however, treatment with the autophagy
inhibitor 3-MA markedly blocked the PA-induced expression of NOX4
(Fig. 3A and B). In order to explore
the mechanism responsible for PA-induced NOX4 expression, the
cytosolic Ca2+ concentration and PKCα expression were
assessed using Fura-2 AM and western blotting analyses,
respectively. The results revealed that cytosolic Ca2+
content was significantly increased in PA-treated cells compared
with that in control cells, while 3-MA evidently reduced cytosolic
Ca2+ (Fig. 3C).
Similarly, PKCα activation was upregulated following PA treatment,
whereas it was significantly inhibited when 3-MA was also present
in the culture medium (Fig. 3D-F).
These results suggest that PA-induced autophagy was able to
activate the Ca2+/PKCα/NOX4 pathway.
NOX4 inhibition improves PA-induced
HUVEC dysfunction
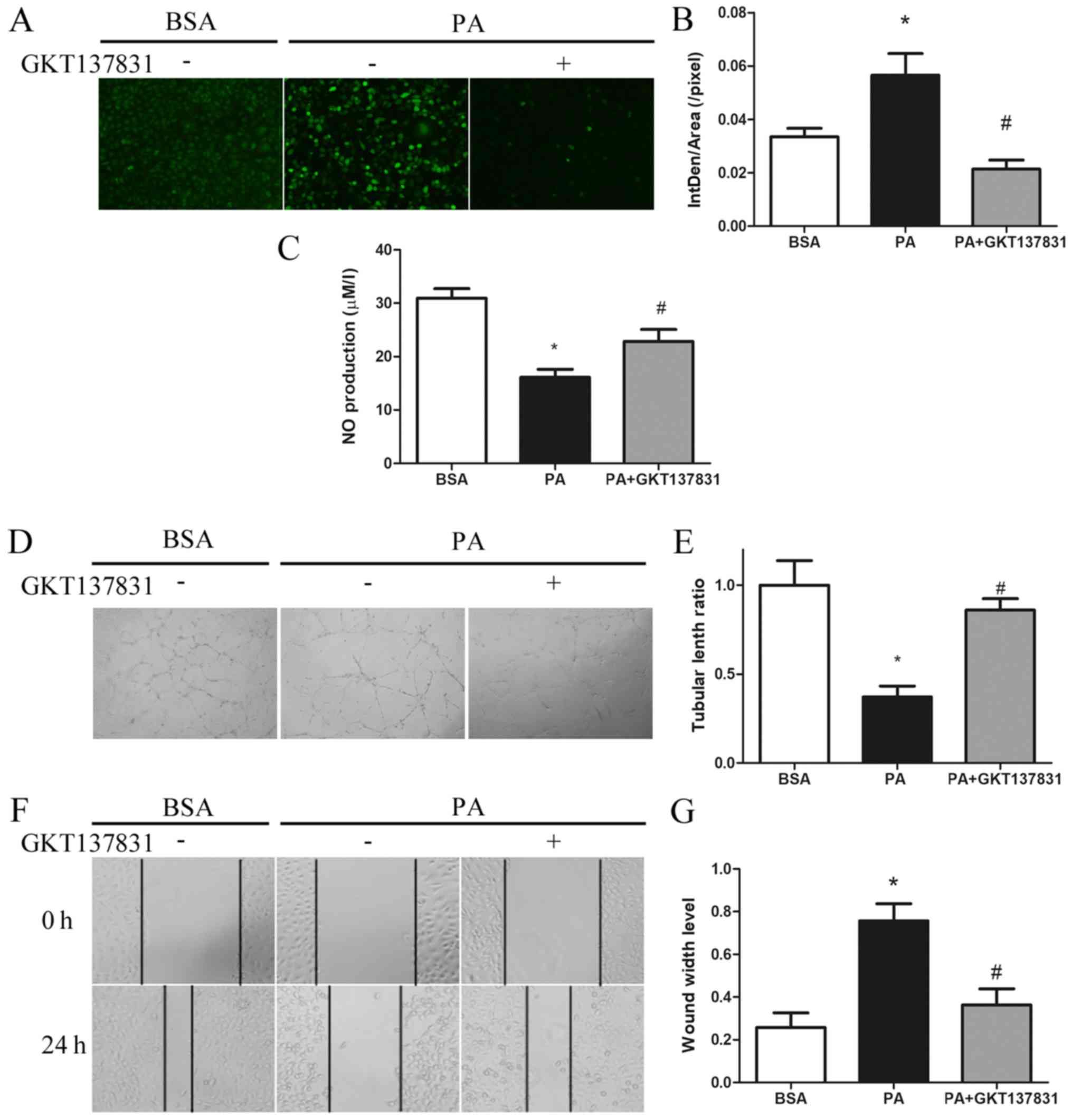
To determine the role of PA-mediated activation of
the Ca2+/PKCα/NOX4 pathway in endothelial dysfunction,
the specific NOX4 inhibitor GKT137831 was used. The results
revealed that GKT137831 significantly reduced PA-induced ROS
generation (Fig. 4A and B).
Furthermore, NOX4 inhibition reversed the PA-induced decrease in NO
synthesis, as indicated by the increased intracellular NO levels
(Fig. 4C). The PA-mediated damage in
the formation of capillary-like structures was also alleviated by
NOX4 inhibition (Fig. 4D and E).
Finally, the repair ability of cells was significantly improved, as
indicated by a narrower scratch wound at 24 h in cells treated with
a combination of PA and GKT137831, as compared with those treated
with PA alone (Fig. 4F and G). These
results suggest that the PA-induced endothelial dysfunction is
mediated through autophagy-dependent activation of the
Ca2+/PKCα/NOX4 pathway.
Discussion
In the present study a novel mechanism of PA-induced
endothelial dysfunction in HUVECs was identified. Autophagy
inhibition was demonstrated to downregulate PA-induced ROS
generation by interfering with the Ca2+/PKCα/NOX4
pathway. Alleviating ROS and oxidative stress affects numerous
signaling pathways associated with cell survival, proliferation,
vasodilation (NO generation) and angiogenesis (19–23).
A number of studies have linked autophagic activity
to ROS generation (24–26). Generally, ROS activates autophagy,
which inhibits excessive ROS generation in a negative feedback
manner. ROS accumulation promotes autophagic activity via multiple
mechanisms, including activation of adenosine
5′-monophosphate-activated protein kinase (27), promotion of high mobility group box 1
translocation from the nucleus to the cytoplasm (28,29), and
stabilization of hypoxia-inducible factor-1α (30,31).
ROS-induced activation of autophagy clears damaged mitochondria and
subsequently decreases ROS generation (24). Conversely, it has been reported that
autophagy promotes ROS generation rather than suppressing it
(16). In the present study,
PA-induced ROS generation was significantly decreased when cells
were simultaneously treated with an inhibitor, suggesting that
autophagy possibly regulates ROS production. Based on these
findings, the mechanism by which autophagy may promote ROS
production was further explored, and it was demonstrated that 3-MA
treatment reduced PA-induced cytosolic Ca2+
upregulation. Given that elevated cytosolic Ca2+
concentrations have been reported to activate NOXs via PKC
activation (32), PKCα activation
and NOX4 expression were examined in cells treated with PA and/or
3-MA. The results revealed that the inhibition of autophagy
significantly decreased the PA-induced expression levels of p-PKCα
and NOX4. Although NOX4 has been reported to activate
phosphoinositide 3-kinase (33),
3-MA still effectively inhibited autophagy in HUVECs, as
illustrated by the western blotting and electron microscopy results
in the current study. In addition, using the specific NOX4
inhibitor GKT137831, it was confirmed that NOX4 suppression
improved PA-induced endothelial dysfunction. Taken together, these
findings suggest that PA-induced ROS generation is achieved by
activation autophagy via the Ca2+/PKCα/NOX4 pathway.
In conclusion, the results of the present study
suggested that PA-induced autophagy activates the
Ca2+/PKCα/NOX4 pathway to promote ROS generation. The
novel pathway identified in the present study may help to improve
our understanding of PA lipotoxicity, therefore providing a novel
strategy that may have potential as a treatment for CVDs caused by
hypertriglyceridemia.
Acknowledgements
The authors would like to thank Dr Wei Chen for
guiding Matrigel tube formation assay and Dr Hui Peng for guiding
the NO assay (both Center for Experimental Medical Research, The
Third Xiangya Hospital of Central South University, Changsha,
China).
Funding
The present study was supported by funding from the
National Natural Science Foundation of China (grant nos. 81870352
and 81470593), the National Basic Research Program of China (grant
no. 2014CB542400) and the Key Research and Development Project of
Hunan Province (grant no. 2017SK2024).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
All authors made substantial contributions to this
research. Experiments were primarily performed by PC and HLiu, and
were in charge of article editing and revision of the study. HX,
ZZ, and SZ assisted in performing the experiment. Data were
analyzed by JX, ZS and RC. JZ ensured that all questions related to
accuracy and integrity of any part of the work are appropriately
investigated and resolved. HLu and SC made substantial
contributions to the conception and design of this research. HLu
approved final version to be published.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Fruchart JC, Sacks FM, Hermans MP, Assmann
G, Brown WV, Ceska R, Chapman MJ, Dodson PM, Fioretto P, Ginsberg
HN, et al: The residual risk reduction initiative: A call to action
to reduce residual vascular risk in dyslipidaemic patient. Diab
Vasc Dis Res. 5:319–335. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Miller M, Stone NJ, Ballantyne C, Bittner
V, Criqui MH, Ginsberg HN, Goldberg AC, Howard WJ, Jacobson MS,
Kris-Etherton PM, et al: Triglycerides and cardiovascular disease:
A scientific statement from the American Heart Association.
Circulation. 123:2292–2333. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Borradaile NM and Schaffer JE:
Lipotoxicity in the heart. Curr Hypertens Rep. 7:412–417. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Brookheart RT, Michel CI and Schaffer JE:
As a matter of fat. Cell Metab. 10:9–12. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Titov VN: The excess of palmitic fatty
acid in food as main cause of lipoidosis of insulin-dependent
cells: Skeletal myocytes, cardio-myocytes, periportal hepatocytes,
kupffer macrophages and b-cells of pancreas. Klin Lab Diagn.
61:68–77. 2016.(In Russian). PubMed/NCBI
|
|
6
|
Sena CM, Pereira AM and Seiça R:
Endothelial dysfunction-a major mediator of diabetic vascular
disease. Biochim Biophys Acta. 1832:2216–2231. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Khan MJ, Rizwan Alam M, Waldeck-Weiermair
M, Karsten F, Groschner L, Riederer M, Hallström S, Rockenfeller P,
Konya V, Heinemann A, et al: Inhibition of autophagy rescues
palmitic acid-induced necroptosis of endothelial cells. J Biol
Chem. 287:21110–21120. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gough DR and Cotter TG: Hydrogen peroxide:
A Jekyll and Hyde signalling molecule. Cell Death Dis. 2:e2132011.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Schieber M and Chandel NS: ROS function in
redox signaling and oxidative stress. Curr Biol. 24:R453–R462.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mariño G, Niso-Santano M, Baehrecke EH and
Kroemer G: Self-consumption: The interplay of autophagy and
apoptosis. Nat Rev Mol Cell Biol. 15:81–94. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Huang S, Van Aken O, Schwarzländer M, Belt
K and Millar AH: The roles of mitochondrial reactive oxygen species
in cellular signaling and stress response in plants. Plant Physiol.
171:1551–1559. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Arnhold J and Flemmig J: Human
myeloperoxidase in innate and acquired immunity. Arch Biochem
Biophys. 500:92–106. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fransen M, Nordgren M, Wang B and
Apanasets O: Role of peroxisomes in ROS/RNS-metabolism:
Implications for human disease. Biochim Biophys Acta.
1822:1363–1373. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Scherz-Shouval R and Elazar Z: Regulation
of autophagy by ROS: Physiology and pathology. Trends Biochem Sci.
36:30–38. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Almaguel FG, Liu JW, Pacheco FJ, De Leon
D, Casiano CA and De Leon M: Lipotoxicity mediated cell dysfunction
and death involves lysosomal membrane permeabilization and
cathepsin L activity. Brain Res. 1318:133–143. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gong J, Muñoz AR, Chan D, Ghosh R and
Kumar AP: STAT3 down regulates LC3 to inhibit autophagy and
pancreatic cancer cell growth. Oncotarget. 5:2529–2541. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Klionsky DJ, Abdalla FC, Abeliovich H,
Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M,
Agostinis P, Aguirre-Ghiso JA, et al: Guidelines for the use and
interpretation of assays for monitoring autophagy. Autophagy.
8:445–544. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Evangelista AM, Thompson MD, Bolotina VM,
Tong X and Cohen RA: Nox4- and Nox2-dependent oxidant production is
required for VEGF-induced SERCA cysteine-674 S-glutathiolation and
endothelial cell migration. Free Radic Biol Med. 53:2327–2334.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Song P and Zou MH: Redox regulation of
endothelial cell fate. Cell Mol Life Sci. 71:3219–3239. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Fandy TE, Jiemjit A, Thakar M, Rhoden P,
Suarez L and Gore SD: Decitabine induces delayed reactive oxygen
species (ROS) accumulation in leukemia cells and induces the
expression of ROS generating enzymes. Clin Cancer Res.
20:1249–1258. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Borradaile NM, Han X, Harp JD, Gale SE,
Ory DS and Schaffer JE: Disruption of endoplasmic reticulum
structure and integrity in lipotoxic cell death. J Lipid Res.
47:2726–2737. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Xu MJ, Song P, Shirwany N, Liang B, Xing
J, Viollet B, Wang X, Zhu Y and Zou MH: Impaired expression of
uncoupling protein 2 causes defective postischemic angiogenesis in
mice deficient in AMP-activated protein kinase alpha subunits.
Arterioscler Thromb Vasc Biol. 31:1757–1765. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jansen F, Yang X, Hoelscher M, Cattelan A,
Schmitz T, Proebsting S, Wenzel D, Vosen S, Franklin BS,
Fleischmann BK, et al: Endothelial microparticle-mediated transfer
of MicroRNA-126 promotes vascular endothelial cell repair via
SPRED1 and is abrogated in glucose-damaged endothelial
microparticles. Circulation. 128:2026–2038. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yan Y and Finkel T: Autophagy as a
regulator of cardiovascular redox homeostasis. Free Radic Biol Med.
109:108–113. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Li L, Tan J, Miao Y, Lei P and Zhang Q:
ROS and autophagy: Interactions and molecular regulatory
mechanisms. Cell Mol Neurobiol. 35:615–621. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Filomeni G, De Zio D and Cecconi F:
Oxidative stress and autophagy: The clash between damage and
metabolic needs. Cell Death Differ. 22:377–388. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Papandreou I, Lim AL, Laderoute K and
Denko NC: Hypoxia signals autophagy in tumor cells via AMPK
activity, independent of HIF-1, BNIP3, and BNIP3L. Cell Death
Differ. 15:1572–1581. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tang D, Kang R, Cheh CW, Livesey KM, Liang
X, Schapiro NE, Benschop R, Sparvero LJ, Amoscato AA, Tracey KJ, et
al: HMGB1 release and redox regulates autophagy and apoptosis in
cancer cells. Oncogene. 29:52992010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tang D, Kang R, Livesey KM, Cheh CW,
Farkas A, Loughran P, Hoppe G, Bianchi ME, Tracey KJ, Zeh HJ III
and Lotze MT: Endogenous HMGB1 regulates autophagy. J Cell Biol.
190:881–892. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Guzy RD, Hoyos B, Robin E, Chen H, Liu L,
Mansfield KD, Simon MC, Hammerling U and Schumacker PT:
Mitochondrial complex III is required for hypoxia-induced ROS
production and cellular oxygen sensing. Cell Metab. 1:401–408.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bellot G, Garcia-Medina R, Gounon P,
Chiche J, Roux D, Pouysségur J and Mazure NM: Hypoxia-induced
autophagy is mediated through hypoxia-inducible factor induction of
BNIP3 and BNIP3L via their BH3 domains. Mol Cell Biol. 6:2570–2581.
2009. View Article : Google Scholar
|
|
32
|
Lob H, Rosenkranz AC, Breitenbach T,
Berkels R, Drummond G and Roesen R: Antioxidant and nitric
oxide-sparing actions of dihydropyridines and ACE inhibitors differ
in human endothelial cells. Pharmacology. 76:8–18. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhang C, Lan T, Hou J, Li J, Fang R, Yang
Z, Zhang M, Liu J and Liu B: NOX4 promotes non-small cell lung
cancer cell proliferation and metastasis through positive feedback
regulation of PI3K/Akt signaling. Oncotarget. 5:4392–4405. 2014.
View Article : Google Scholar : PubMed/NCBI
|