Introduction
Liddle's syndrome (LS), first described by Liddle
et al (1) in 1963, is
characterized by high urinary potassium excretion, low urinary
sodium excretion and maintained hypokalemia and volume expansion,
resulting in hypertension and suppressed aldosterone excretion.
Liddle hypothesized that excessive sodium reabsorption in the
distal kidney tubules may be the reason for this clinical
presentation. LS is a hereditary disease caused by mutations of
epithelial sodium channels (ENaCs), which are located in kidney
distal convoluted tubules. ENaCs are constructed by three
homologous subunits. Each α-, β-, γ-ENaC subunit has a highly
conserved sequence termed the PY motif (Pro-Pro-Pro-X-Tyr motif)
that serves as a binding site for Nedd4-2 in the process of ENaC
ubiquitylation and endocytosis (2,3). LS is
genetically heterogeneous and arises from mutations in the
cytoplasmic C-terminus of either the β or γ subunit of the
amiloride-sensitive ENaC. Previous findings have indicated that
mutations in the α-subunit of ENaC genes are responsible for
multisystem pseudohypoaldosteronism type 1, which is a rare
autosomal recessive aldosterone unresponsiveness syndrome (4). Mutations in the β or γ subunits of ENaC
genes have been reported in a previous study and were strongly
associated with LS (5). However, LS
is a rare disease and can be easily overlooked or misdiagnosed.
Hypertension caused by LS presents as refractory and hard to
control. Inhibitors of sodium transport in the distal nephron,
including amiloride and triamterene, are effective treatment
options in patients with LS. Previous studies revealed that
mineralocorticoid antagonists, including spironolactone, are not
effective for patients with LS (6,7).
In the present study, a young man presented with
early-onset and refractory hypertension with hypokalemia and was
clinically suspected of having LS. His pedigree was surveyed and
molecular genetic studies were conducted.
Materials and methods
Clinical data
A 19-year-old male was admitted with early-onset
hypertension and hypokalemia in June 2012 to the Department of
Cardiology of Beijing Hospital (Beijing, China). The patient's
medical history revealed 1 year of hypertension, with intermittent
nausea and headache for 3 months. The patient had no history of
blurred vision, chest tightness, chest pain, proteinuria, hematuria
or edema. Furthermore, daily urine volume was normal. The basic
metabolic panel revealed that potassium level was 3.4 mmol/l. The
patient was followed up routinely by clinic visits and phone calls
for 3 years following the start of 5 mg per day of amiloride
treatment. In August 2015, the patient's clinical conditions were
re-evaluated. A total of 34 family members were recruited to
construct a pedigree. Clinical data were obtained from 29 family
members. All family members provided oral informed consent prior to
any procedure. Furthermore, the Ethics Committee of Beijing
Hospital approved the present study.
Genetic diagnosis
Genetic analysis was performed on the proband and
his family members. DNA was extracted from peripheral blood
leukocytes using a TIANamp Blood DNA kit (Tiangen Biotech Co.,
Ltd., Beijing, China). The reference sequences of SCNN1B and SCNN1G
were obtained from GenBank (https://www.ncbi.nlm.nih.gov/genbank/accession no.
NM_000336.2 for SCNN1B and NM_001039.3 for SCNN1G). Primers were
designed using Primer Premier 5.0 software (Premier Biosoft
International, Palo Alto, CA, USA). All the exons of SCNN1B and
SCNN1G were sequenced, but mutations were only identified in the
last exon of SCNN1B. Polymerase chain reaction (PCR) was used to
amplify the last exons of β and γ subunits of the ENaC based on the
following primers: β, forward, 5′-TGCTGTCCTCATCGAGTTTG-3′ and
reverse, 5′-CCTCCACCAGCTCGGCCACG-3′; and γ, forward,
5′-GCTTGGGTAGGAGGGAGA-3′ and reverse, 5′-CCGTAAAGAGCTGCATCAG-3′.
PCR products were purified using an Agarose Gel Purification kit
(Beijing Biomed Gene Technology Co., Ltd., Beijing, China). All
samples were sequenced in both forward and reverse directions with
an Applied Biosystems 3730/3730×l DNA Analyzers 3730 XL (Applied
Biosystems; Thermo Fisher Scientific, Inc., Waltham, MA, USA).
High resolution melting (HRM) was used for detection
of the mutation in other family members. Genotyping was performed
using a SYTO9 fluorescent dye (Thermo Fisher Scientific, Inc.) and
the HRM method on a Rotor-gene 6200 system (Qiagen, Inc., Valencia,
CA, USA), according to the manufacturer's protocol. In brief, a
short fragment containing the altered gene section was amplified
with the forward primer 5′-TGCTGTGCCTCATCGAGTTTG-3′ and a reverse
primer of 3′-CCTCCACCAGCTCGGCCACG-5′ using 5 µM of SYTO9
fluorescent dye. Subsequently, the PCR products were genotyped
using HRM analysis. DNA samples with known genotypes in the present
pedigree were used as positive controls and ddH2O was
used as a negative control.
In order to exclude single nucleotide polymorphisms
induced by the gene change, HRM analysis was performed on 500
normal control DNA samples obtained from the Human Genome Research
Center and College of Life Science and Technology of Huazhong
University of Science and Technology (Wuhan, China).
Results
The proband had early onset hypertension with a
blood pressure (BP) ranging from 140–230/80–140 mmHg. The patient
was asymptomatic when his BP was <180/100 mmHg. Blood
biochemistry parameters and urine tests were performed and plasma
renin and aldosterone concentration, cortisol circadian rhythm,
plasma catecholamine, and thyroid function were tested. Results
indicated that potassium level was 3.4 mmol/l and plasma renin and
aldosterone levels were normal while the subject was supine;
however, these levels were suppressed while the subject was
upright. Furthermore, renin and aldosterone levels decreased after
the patient stood up. Detailed clinical data and positive auxiliary
examination results of the proband (III-11) are presented in
Table I. The results of parameters
and tests not shown were all in the normal range. With the
exception of BP, no abnormalities were detected on physical
examination. Imaging was also normal, including imaging of the
abdomen, renal artery and pituitary gland. Notably, hypertension
was refractory following treatment with multiple drugs, including
calcium channel blockers, angiotensin-converting-enzyme inhibitors,
β-receptor blockers and diuretics. Based on the aforementioned
data, LS was suggested as a potential diagnosis. A DNA sample from
the patient was analyzed for detection of ENaC mutation. A novel
deletion mutation (c.1721delC) was identified, which was suspected
to be the cause of hypertension.
 | Table I.Clinical and biochemical
characteristics of the proband III-11. |
Table I.
Clinical and biochemical
characteristics of the proband III-11.
| Characteristic | Proband III-11 | Normal value |
|---|
| Sex |
| Male |
| Age (years) |
| 20 |
| Age at onset of HTN
(years) |
| 19 |
| BP before amiloride
treatment (mmHg) |
| 230/130 |
| Biochemical
parameters |
|
|
| Serum
potassium (mmol/l) | 3.5–5.0 | 3.4 |
| Serum
sodium (mmol/l) | 135–145 | 136.2 |
| Plasma
renin supine/upright (pg/ml) | 7-19/7–40 | 17/16 |
|
Aldosterone supine/upright
(pg/ml) | 60-174/68–300 | 34/44 |
|
Electrocardiogram |
| Sinus rhythm,
LVHV |
| Echocardiogram |
| LVPW, 13 mm; IVS, 12
mm; reduced LV diastolic function; |
|
|
| LVEF, 60% |
| Fundus
examination |
| Retinal artery
atherosclerosis in stage I |
| Nephrogram |
| Normal size, slightly
reduced GFR (left, 37.0 ml/min; right, 33.7 ml/min) |
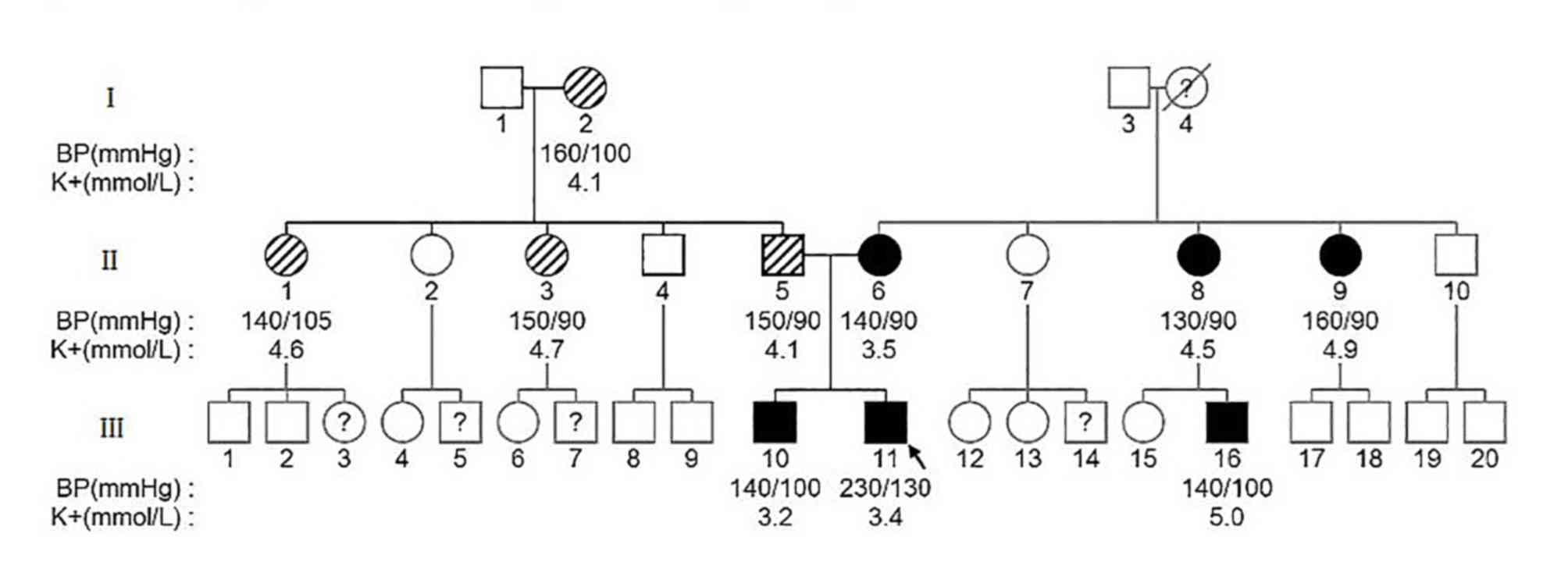
There were 34 members from three generations in the
current pedigree. As indicated in Fig.
1, 29 family members were enrolled for genetic testing and the
4 family members could not be contacted and 1 family member was
deceased. The clinical data of all members in this pedigree are
presented in Table II. A total of
10/29 members in this pedigree were clinically diagnosed with
hypertension. III-10 (sibling of the proband) was diagnosed with
hypertension at the age of 20. Without regular treatment, the BP of
III-10 fluctuated between 150–160/90–110 mmHg and was poorly
controlled. Furthermore, the subject's potassium level was 3.2
mmol/l. In the proband's father's family, I-2, II-1, II-3 and II-5
(the proband's father) all developed hypertension in their 40s. In
the proband's mother's family, I-4 was died as a result of cerebral
vascular disease in her 70s. It was unknown whether this subject
had hypertension because she had never received a physical
examination. II-6 (the proband's mother) also exhibited early-onset
hypertension at the age of 30. Her BP was 140/90 mmHg and her
potassium level was 3.5 mmol/l. The maternal aunts of the proband
(II-8, II-9) had presented with high BP at the same age as II-6,
but without hypokalemia. The proband's cousin (III-16, son of II-8)
had not been diagnosed with hypertension, but a random BP check
revealed a BP of 140/100 mmHg with a serum potassium level of 5.0
mmol/l.
| Table II.Clinical data of the family members in
the pedigree. |
Table II.
Clinical data of the family members in
the pedigree.
| Subject | Mutation | Age (years) | HTN | Age of onset of HTN
(years) | BP (mmHg) | Medication before
amiloride | Comorbidity | K (mmol/l) | Na (mmol/l) |
|---|
| I-1 | No | 74 | UK | UK | 120/80, 140/90 |
| CVD | 4.4 | 138.9 |
| I-2 | No | 75 | Yes | 74 | 160/100,
150/80 |
| None | 4.1 | 142.3 |
| I-3 | No | 74 | No |
| 120/80, 146/80 |
| None | 4.1 | 141.5 |
| I-4 | UK | UK | UK | UK | UK | UK | UK | UK | UK |
| II-1 | No | 49 | Yes | 46 | 140/105,
160/100 | Reserpine,
captopril | None | 4.8 | 134.7 |
| II-2 | No | 51 | No |
| 130/90, 130/80 |
| None | 4.6 | 138.5 |
| II-3 | No | 46 | Yes | 46 | 125/85, 150/90 | Reserpine | None | 4.7 | 138.2 |
| II-4 | No | 43 | No |
| 120/80, 130/90 |
| T2DM | 4.4 | 137.5 |
| II-5 | No | 44 | Yes | 42 | 130/90,
144/100 | Nifedipine,
enalapril | None | 4.1 | 141.0 |
| II-6 | Yes | 55 | Yes | 30 | 140/90, 156/80 | Nifedipine,
reserpine | None | 4.6 | 137.0 |
| II-7 | No | 55 | No |
| 120/80, 150/90 |
| None | 4.6 | 137.0 |
| II-8 | Yes | 50 | Yes | 30 | 130/90,
150/100 | Nifedipine,
reserpine | None | 4.5 | 137.1 |
| II-9 | Yes | 48 | Yes | 30 | 130/90,
150/100 | Nifedipine,
amiloride | None | 4.9 | 138.8 |
| II-10 | No | 45 | No |
| 120/80, 120/70 |
| None | 4.6 | 139.5 |
| III-1 | No | 23 | No |
| 120/80,
124/100 |
| None | UK | UK |
| III-2 | No | 25 | No |
| 124/60, 110/80 |
| None | UK | 122.9 |
| III-4 | No | 27 | No |
| 110/70, 120/70 |
| None | 4.0 | 142.0 |
| III-6 | No | 26 | No |
| 120/80, 120/80 |
| None | 3.9 | 140.0 |
| III-8 | No | 20 | No |
| 146/90, 140/90 |
| None | 4.8 | 142.0 |
| III-9 | No | 14 | No |
| 120/78, 110/70 |
| None | 4.6 | 140.0 |
| III-10 | Yes | 21 | Yes | 19 | 150/100,
160/110 |
| None | 3.2 | 142.8 |
| III-11 | Yes | 22 | Yes | 20 | 150/110,
150/90 | Irregular
medication | None | 3.4 | 136.2 |
| III-12 | No | 28 | No |
| 110/60, 104/62 |
| None | 4.1 | 138.0 |
| III-13 | No | 33 | No |
| 130/80, 124/80 |
| None | 4.2 | 143.0 |
| III-15 | No | 24 | No |
| 100/86, 130/80 |
| None | 3.6 | 141.0 |
| III-16 | Yes | 25 | Yes | 25 | 140/100,
150/100 |
| None | 5.0 | 141.4 |
| III-17 | No | 21 | No |
| 124/86, 130/80 |
| None | 4.6 | 142.0 |
| III-18 | No | 25 | No |
| 134/90, 120/90 |
| None | 4.8 | 141.1 |
| III-19 | No | 24 | No |
| 140/90, 130/80 |
| None | 4.2 | 142.0 |
| III-20 | No | 21 | No |
| 124/80, 134/80 |
| None | 5.4 | 139.9 |
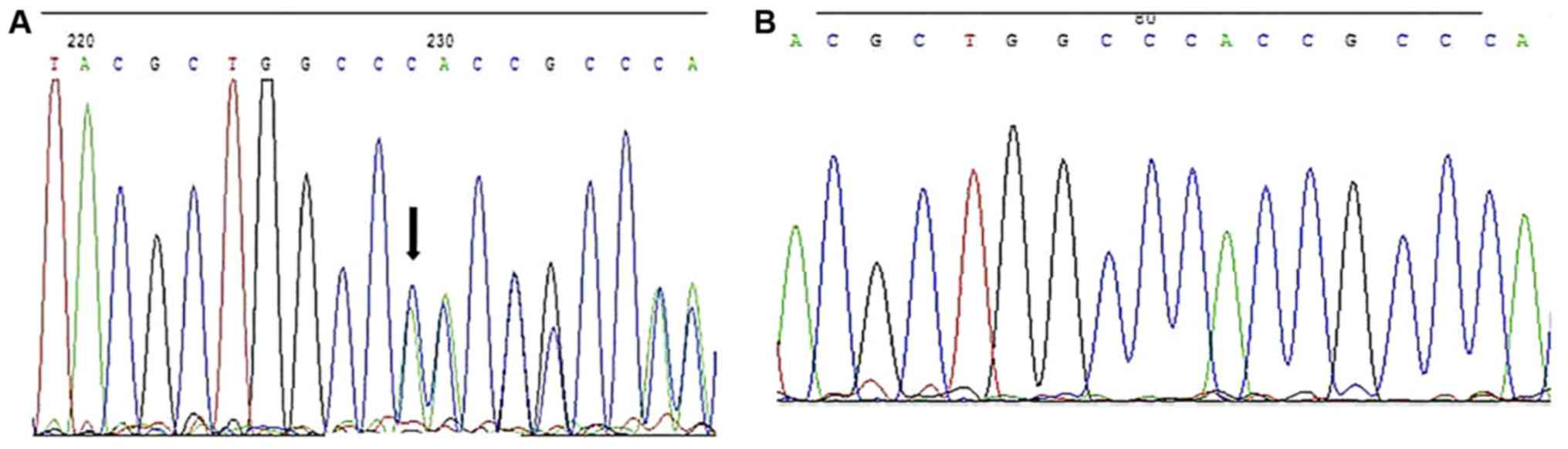
DNA sequencing revealed a deletion of cytosine at
nucleotide 1721 of the coding DNA structure (c.1721delC) compared
with the wild-type sequence in SCNN1B in the proband III-11, as
indicated in Fig. 2. This deletion
can cause a length extension of the SCNN1B coding sequence from
1,923 to 2,025 bp, leading to the modification of the open reading
frame after the proline at position 574 and introduction of a new
stop codon at position 675 (p.Pro574HisfsX675). Family members
III-10, III-16, II-6, II-8, and II-9 shared the same mutation.
However, no 1721delC mutation was found in the other family
members. No mutation of SCNN1G coding sequences was detected in the
proband. The deletion mutation was identified in 6 people in the
pedigree.
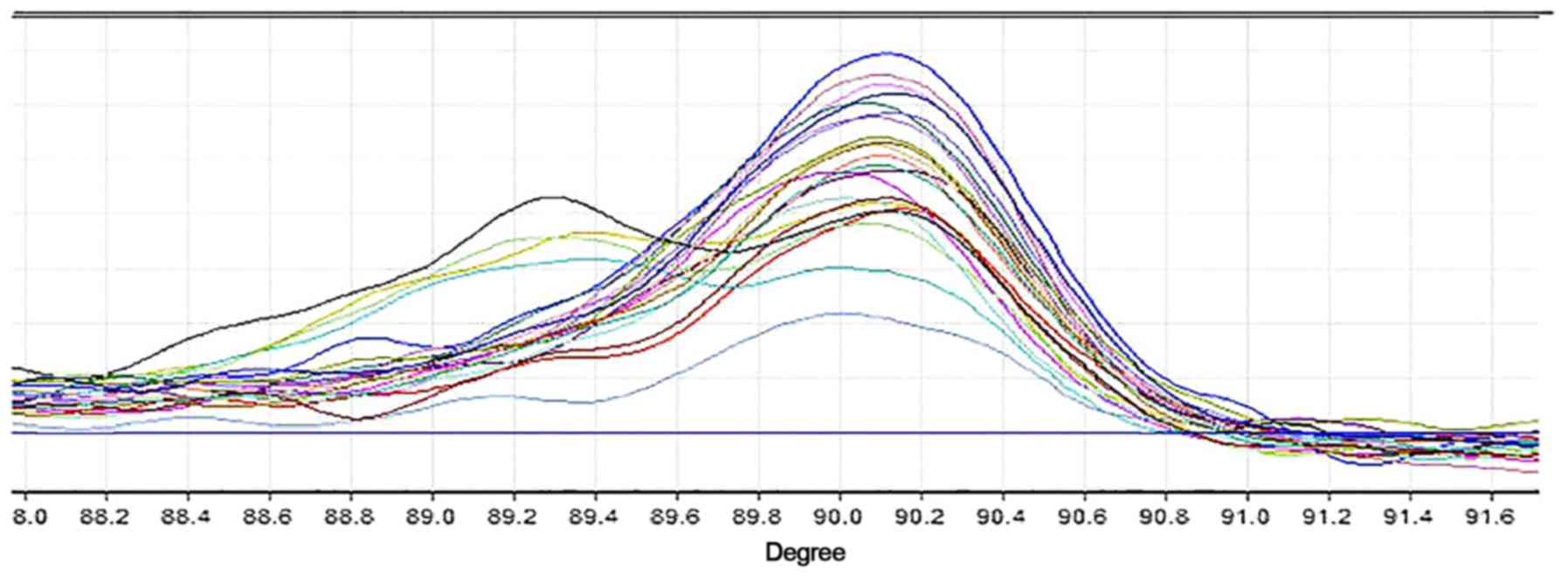
The HRM curve was compared between patients with and
without c.1721delC mutation in the pedigree. The bimodal HRM curves
of II-6, II-8, II-9 and III-10, who were discovered to be mutation
carriers, are presented in Fig. 3.
All other individuals involved in the study produced a unimodal
curve. Notably, HRM curves of III-11 and III-16 were not observed
due to technical failure of extracting DNA. Furthermore, HRM
analysis was performed on 500 normal control DNA samples. Smooth
unimodal curves were shown in accordance with non-mutation carriers
in this family.
Irbesartan, hydrochlorothiazide, spironolactone,
amlodipine, bisoprolol and nifedipine treatment were all prescribed
before LS was diagnosed; however, the proband's BP remained poorly
controlled. After genetic testing, treatment was switched to
amiloride and low-sodium diet (2 g NaCl/day). BP was gradually
decreased to 110/80 mmHg and potassium level increased to 4.4
mmol/l after 4 weeks of treatment. The proband was followed up with
phone calls and re-evaluated after amiloride treatment for 3 years.
BP was successfully controlled at the 3-year follow up. The
patient's left ventricular hypertrophy (LVH), fundus artery and
kidney function was improved at his second evaluation, as indicated
in Table III. Notably, family
members II-6, II-8, II-9 and III-10 also had their BP controlled by
amiloride therapy.
| Table III.Clinical data for the proband before
and after amiloride treatment for 3 years. |
Table III.
Clinical data for the proband before
and after amiloride treatment for 3 years.
| Variable | Before
amiloride | After
amiloride |
|---|
| Mean BP (mmHg) | 153/102 | 129/69 |
| Serum potassium
(mmol/l) | 3.4 | 3.6 |
| Echocardiogram | IVS, 12 mm; PWT, 13
mm; LVID, 53 mm; LVEDV, 135 ml | IVS, 11 mm; PWT, 11
mm; LVID, 48 mm; LVEDV, 108 ml |
| 24 h urine protein
(g/24 h) | 0.10 | 0.09 |
| GFR | Left, 37.0 ml/min;
right, 33.7 ml/min | Left, 45.5 ml/min;
right, 49.8 ml/min |
| Fundus
examination | Retinal artery
atherosclerosis in stage I | No retinal artery
atherosclerosis |
Comparison of the proband's clinical data and
auxiliary examination results before and after amiloride treatment
for 3 years revealed an improvement in the proband's condition:
High BP was decreased to a normal range, serum potassium was
increased, and LVH and fundus conditions were improved.
Discussion
In 1994, LS was confirmed by the discovery of a
molecular defect that was an activating mutation in the subunit of
ENaC (8). Numerous mutations in ENaC
have now been reported (Table IV)
(8–28). The majority of these were missense
mutations, but deletion and insertion mutations have also been
reported. Regardless of the mutation in SCNN1B and SCNN1G, a
frameshift mutation may cause the LS phenotype. Genetic defects
frequently affect a highly conserved sequence known as the PY motif
(starting from p.616 in SCNN1B and p.623 in SCNN1G), which serves
as a binding site for Nedd4-2. Nedd4-2 acts as a bridge that
connects the PY motif of the ENaC at one side and combines with
ubiquitin ligase on the other side. Notably, ubiquitylation is
important for degradation of the ENaC to maintain a constant number
of ENaCs. In the process of ENaC ubiquitylation and endocytosis,
the mutated PY motif fails to bind with Nedd4-2, resulting in an
excessive number and overactivation of ENaCs on the cell surface,
which leads to an increase of sodium and fluid absorption in the
distal convoluted tubules (29).
| Table IV.Mutations in β and γ subunits of
epithelial sodium channel identified in Liddle's syndrome. |
Table IV.
Mutations in β and γ subunits of
epithelial sodium channel identified in Liddle's syndrome.
| A, Mutations in
SCNN1B |
|---|
|
|---|
| Author, year |
Mutationa |
Consequenceb |
Phenotypec | Initially
termedd | (Refs.) |
|---|
| Rayner et
al, 2003 | c.1688G>A | p.Arg563Gln | HT, R↓, A↓, K↓ | p.Arg563Gln | (9) |
| Schild et
al, 1995; | c.1696C>T | p.Arg566X | HT, R↓, A↓, K↓,
SD | p.Arg564X | (10,11) |
| Shimkets et
al, 1994 |
| Gong et al,
2014 | c.1698C>T | p.Arg566X | HT, R↓, A↓, K↓ | p.Arg566X | (12) |
| Jeunemaitre et
al, 1997 |
c.1735_1766del32 |
p.Ala579LeufsX582 | HT, R↓, A↓, K↓,
SD |
p.Ala579_589Glydel | (13) |
| Shimkets et
al, 1994 | c.1771C>T | p.Arg591X | HT, R↓, A↓, K↓ | p.Gln589X | (11) |
| Shimkets et
al, 1994 | c.1781dupC |
p.Thr594HisfsX607 | HT, R↓, A↓, K↓ |
p.Thr592ThrfsX605 | (11) |
| Gong et al,
2014 |
c.1784_1789insC |
p.Arg597ProfsX607 | HT, R↓, A↓, K↓ |
p.Arg597ProfrX607 | (12) |
| Shimkets et
al, 1994 | c.1789delC |
p.Arg597AlafsX675 | HT, R↓, A↓, K↓ |
p.Arg595AlafsX673 | (11) |
| Jackson et
al, 1998; | c.1789dupC |
p.Arg597ProfsX607 | HT, R↓, A↓, K↓ |
p.Arg595ProfsX605 | (14,15) |
| Nakano et
al, 2002 |
| Hiltunen et
al, 2002; |
c.1800_1801insG |
p.Thr601AspfsX607 | HT, R↓, A↓, K↓ |
p.Thr601AspfsX607 | (16,17) |
| Ma et al,
2001 |
| Sawathiparnich
et al, 2009 | c.1850C>A | p.Pro617His | HT, R↓, A↓ | p.Pro615His | (18) |
| Uehara et
al, 1998 | c.1852C>T | p.Pro618Ser | HT, R↓, A↓, K↓ | p.Pro616Ser | (19) |
| Hansson et
al, 1995 | c.1853C>T | p.Pro618Leu | HT, R↓, A↓, K↓,
SD | p.Pro616Leu | (20) |
| Wang et al,
2012 | c.1853C>A | p.Pro618His | HT, R↓, A↓, K↓ | p.Pro616Ser | (21) |
| Furuhashi et
al, 2005 | c.1853C>G | p.Pro618Arg | HT, R↓, A↓, K↓ | p.Pro616Arg | (22) |
| Yang et al,
2015 | c.1854dupC |
p.Asn619GlnfsX621 | HT, R↓, A↓, K↓ |
p.Asn619GlnfsX3 | (23) |
| Tamura et
al, 1996 | c.1858T>C | p.Try620His | HT, R↓, A↓, K↓ | p.Try618His | (24) |
| Present study | c.1721delC |
p.Pro574HisfsX675 | HT, R↓, A↓, K↓ |
| – |
|
| B, Mutations in
SCNN1G |
|
| Author,
year |
Mutationa |
Consequenceb |
Phenotypec | Initially
termedd | (Refs.) |
|
| Hiltunen et
al, 2002 | c.1589A>G | p.Asn530Ser | HT, R↓, A↓, K↓ | p.Asn530Ser | (16) |
| Shi et al,
2010 | c.1699C>T | p.Gln567X | HT, R↓, A↓, K↓ | p.Gln567X | (25) |
| Hansson et
al, 1995 | c.1718G>A | p.Trp573X | HT, R↓, A↓, K↓ | p.Trp574X | (26) |
| Yamashita et
al, 2001 | c.1724G>A | p.Trp575X | HT, R↓, A↓, K↓ | p.Trp576X | (27) |
| Wang et al,
2007 |
c.1749_1753del5 |
p.Glu583GlufsX585 | HT, R↓, A↓, K↓ |
p.Glu583GlufsX585 | (28) |
The proband in the current case was characterized by
early-onset refractory hypertension with features of familial
aggregation. For this patient, signs of primary aldosteronism (PA)
were looked for based on his hypertension and hypokalemia, but no
mass was found in the adrenal gland. The Endocrinology Society
guidelines recommend the use of aldosterone-to-renin ratio as the
most reliable test for detecting PA (30). Notably, the levels of renin in the
current patient were very low without hyperaldosteronemia and serum
aldosterone was low. For the present case there were several other
possibilities, including congenital adrenal hyperplasia (CAH),
apparent mineralocorticoid excess (AME), LS and renal tubular
acidosis (RTA) (31). CAH is a group
of autosomal recessive disorders encompassing enzyme deficiencies
in the adrenal steroidogenesis pathway, which lead to impaired
cortisol biosynthesis. A lack of 17α-hydroxylase and 21-hydroxylase
is the most common type of CAH, which can lead to notable sex
character changes, including masculinization, precocious puberty
and testicular tumors (32,33). AME is a syndrome associated with the
absence or impaired activity of the enzyme 11β-hydroxysteroid
dehydrogenase (34). Diagnosis
relies on a triad of hypertension, hypokalemia and suppressed
plasma aldosterone levels, plus an abnormal urinary cortisol to
cortisone ratio and electrolyte disturbance (34). It is difficult to differentiate from
LS in certain cases. However, AME patients are normally sensitive
to spironolactone treatment. Furthermore, certain RTA patients can
present with hypertension and hypokalemia, but their symptoms are
often accompanied with other features, including dehydration,
obtundation, restricted skeletal growth and urinary tract stones.
Such cases are usually detected in infancy. However, milder
versions of the disease typically present later in childhood
(35). All of the clinical features
and auxiliary examination results in the present case indicated the
possibility of LS. The genetic diagnosis for LS is an indispensable
method alongside typical clinical presentation. Genetic analysis of
the current case indicated a mutated β ENaC subunit, c.1721delC,
which was located before the PY coding sequence, and caused a
length extension of SCNN1B coding sequence from 1,923 to 2,025 bp.
This may affect the combination of Nedd4-2 and the ubiquitylation
of ENaC. This mutation was also detected in 5 other family members
who presented with hypertension in early adulthood. A notable
finding of the present study was that hypokalemia was only present
in the proband's immediate family, but not in other mutation
carriers. This result indicated that differences of penetrance are
possible, even in the same pedigree and among carriers of the same
mutation. Furthermore, the present results revealed that the
mutation had an autosomal dominant inheritance pattern and
presented with features of pedigree co-segregation.
Treatment for LS includes a sodium-restricted diet,
inhibition of ENaCs and potassium supplementation. Amiloride is a
typical ENaC inhibitor, which combines with ENaCs before the PY
motif site to block the ENaC. By inhibiting
Na+-K+ exchange and
Na+-H+ exchange, amiloride can alleviate
sodium and water retention to further improve hypertension and
hypokalemia. In the current case, the patient had poorly controlled
BP with a range of 140–220/80-140 mmHg and had developed gingival
hyperplasia subsequent to high doses of nifedipine. However, after
single amiloride treatment for 3 years, BP was well controlled and
end-organ damage had been reversed. The antihypertensive benefit of
amiloride for LS was also verified in other family members who
carried the same mutation, providing further supporting evidence
for the diagnosis of LS.
In conclusion, a patient with LS was diagnosed
clinically and genetically in the present study. The patient's
clinical presentation included early onset and refractory
hypertension, familial aggregation, hypokalemia and
hypoaldosteronemia. To the best of our knowledge, the deletion
mutation (c.1721delC) has not been reported in previous literature.
The present findings indicated that genetic analysis is helpful in
the diagnosis of hypertension in a patient who is clinically
suspected of LS. Furthermore, the results suggested that it is also
useful to screen the proband's family members. These findings
demonstrate the benefits of genetic testing and tailored
treatment.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
XD and CZ acquired or analyzed data. NJ assisted in
data collection and analysis. YZ and QH designed the current study
and performed follow up. DD designed the experiments and performed
genetic testing. YZ and CX performed genetic testing and HRM
analysis. JC and QW also designed the experiments of the current
study and performed genetic testing.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of Beijing Hospital (Beijing, China).
Patient consent for publication
All family members provided oral informed consent
prior to publication.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
LS
|
Liddle's syndrome
|
|
PCR
|
polymerase chain reaction
|
|
HRM
|
high resolution melting
|
|
PA
|
primary aldosteronism
|
|
CAH
|
congenital adrenal hyperplasia
|
|
AME
|
apparent mineralocorticoid excess
|
|
RTA
|
renal tubular acidosis
|
References
|
1
|
Liddle GW, Bledsoe T and Coppage WS: A
familial renal disorder simulating primary aldosteronism but with
negligible aldosterone secretion. Trans Assoc Am Physicians.
76:199–213. 1963.
|
|
2
|
Canessa CM, Schild L, Buell G, Thorens B,
Gautschi I, Horisberger JD and Rossier BC: Amiloride-sensitive
epithelial Na+ channel is made of three homologous subunits.
Nature. 367:463–467. 1994. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Schild L: The ENaC channel as the primary
determinant of two human diseases: Liddle syndrome and
pseudohypoaldosteronism. Nephrologie. 17:395–400. 1996.PubMed/NCBI
|
|
4
|
Edelheit O, Hanukoglu I, Gizewska M,
Kandemir N, Tenenbaum-Rakover Y, Yurdakök M, Zajaczek S and
Hanukoglu A: Novel mutations in epithelial sodium channel (ENaC)
subunit genes and phenotypic expression of multisystem
pseudohypoaldosteronism. Clin Endocrinol (Oxf). 62:547–553. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Schild L, Lu Y, Gautschi I, Schneeberger
E, Lifton RP and Rossier BC: Identification of a PY motif in the
epithelial Na channel subunits as a target sequence for mutations
causing channel activation found in Liddle syndrome. EMBO J.
15:2381–2387. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yang KQ, Xiao Y, Tian T, Gao LG and Zhou
XL: Molecular genetics of Liddle's syndrome. Clin Chim Acta.
436:202–206. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhou R, Patel SV and Snyder PM: Nedd4-2
catalyzes ubiquitination and degradation of cell surface ENaC. J
Biol Chem. 282:20207–20212. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Botero-Velez M, Curtis JJ and Warnock DG:
Brief report: Liddle's syndrome revisited-a disorder of sodium
reabsorption in the distal tubule. N Engl J Med. 330:178–181. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rayner BL, Owen EP, King JA, Soule SG,
Vreede H, Opie LH, Marais D and Davidson JS: A new mutation, R563Q,
of the beta subunit of the epithelial sodium channel associated
with low-renin, low-aldosterone hypertension. J Hypertens.
21:921–926. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Schild L, Canessa CM, Shimkets RA,
Gautschi I, Lifton RP and Rossier BC: A mutation in the epithelial
sodium channel causing Liddle disease increases channel activity in
the Xenopus laevis oocyte expression system. Proc Natl Acad Sci
USA. 92:5699–5703. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Shimkets RA, Warnock DG, Bositis CM,
Nelson-Williams C, Hansson JH, Schambelan M, Gill JR Jr, Ulick S,
Milora RV, Findling JW, et al: Liddle's syndrome: Heritable human
hypertension caused by mutations in the beta subunit of the
epithelial sodium channel. Cell. 79:407–414. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gong L, Chen J, Shao L, Song W, Hui R and
Wang Y: Phenotype-genotype analysis in two Chinese families with
Liddle syndrome. Mol Biol Rep. 41:1569–1575. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jeunemaitre X, Bassilana F, Persu A,
Dumont C, Champigny G, Lazdunski M, Corvol P and Barbry P:
Genotype-phenotype analysis of a newly discovered family with
Liddle's syndrome. J Hypertens. 15:1091–1100. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jackson SN, Williams B, Houtman P and
Trembath RC: The diagnosis of Liddle syndrome by identification of
a mutation in the beta subunit of the epithelial sodium channel. J
Med Genet. 35:510–512. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Nakano Y, Ishida T, Ozono R, Matsuura H,
Yamamoto Y, Kambe M, Chayama K and Oshima T: A frameshift mutation
of beta subunit of epithelial sodium channel in a case of isolated
Liddle syndrome. J Hypertens. 20:2379–2382. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hiltunen TP, Hannila-Handelberg T,
Petäjäniemi N, Kantola I, Tikkanen I, Virtamo J, Gautschi I, Schild
L and Kontula K: Liddle's syndrome associated with a point mutation
in the extracellular domain of the epithelial sodium channel gamma
subunit. J Hypertens. 20:2383–2390. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ma X, Tian Y, Gao Y and Guo X: A study of
mutation(s) of the epithelial sodium channel gene in a Liddle's
syndrome family. Zhonghua Nei Ke Za Zhi. 40:390–393. 2001.(In
Chinese). PubMed/NCBI
|
|
18
|
Sawathiparnich P, Sumboonnanonda A,
Weerakulwattana P and Limwongse C: A novel mutation in the
beta-subunit of the epithelial sodium channel gene (SCNN1B) in a
Thai family with Liddle's syndrome. J Pediatr Endocrinol Metab.
22:85–89. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Uehara Y, Sasaguri M, Kinoshita A, Tsuji
E, Kiyose H, Taniguchi H, Noda K, Ideishi M, Inoue J, Tomita K and
Arakawa K: Genetic analysis of the epithelial sodium channel in
Liddle's syndrome. J Hypertens. 16:1131–1135. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hansson JH, Schild L, Lu Y, Wilson TA,
Gautschi I, Shimkets R, Nelson-Williams C, Rossier BC and Lifton
RP: A de novo missense mutation of the beta subunit of the
epithelial sodium channel causes hypertension and Liddle syndrome,
identifying a proline-rich segment critical for regulation of
channel activity. Proc Natl Acad Sci USA. 92:11495–11499. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang LP, Gao LG, Zhou XL, Wu HY, Zhang L,
Wen D, Li YH, Liu YX, Tian T, Fan XH, et al: Genetic diagnosis of
Liddle's syndrome by mutation analysis of SCNN1B and SCNN1G in a
Chinese family. Chin Med J (Engl). 125:1401–1404. 2012.PubMed/NCBI
|
|
22
|
Furuhashi M, Kitamura K, Adachi M, Miyoshi
T, Wakida N, Ura N, Shikano Y, Shinshi Y, Sakamoto K, Hayashi M, et
al: Liddle's syndrome caused by a novel mutation in the
proline-rich PY motif of the epithelial sodium channel
beta-subunit. J Clin Endocrinol Metab. 90:340–344. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yang KQ, Lu CX, Xiao Y, Liu YX, Jiang XJ,
Zhang X and Zhou XL: A novel frameshift mutation of epithelial
sodium channel β-subunit leads to Liddle syndrome in an isolated
case. Clin Endocrinol (Oxf). 82:611–614. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tamura H, Schild L, Enomoto N, Matsui N,
Marumo F and Rossier BC: Liddle disease caused by a missense
mutation of beta subunit of the epithelial sodium channel gene. J
Clin Invest. 97:1780–1784. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Shi JY, Chen X, Ren Y, Long Y and Tian HM:
Liddle's syndrome caused by a novel mutation of the gamma-subunit
of epithelial sodium channel gene SCNN1G in Chinese. Zhonghua Yi
Xue Yi Chuan Xue Za Zhi. 27:132–135. 2010.(In Chinese). PubMed/NCBI
|
|
26
|
Hansson JH, Nelson-Williams C, Suzuki H,
Schild L, Shimkets R, Lu Y, Canessa C, Iwasaki T, Rossier B and
Lifton RP: Hypertension caused by a truncated epithelial sodium
channel gamma subunit: Genetic heterogeneity of Liddle syndrome.
Nat Genet. 11:76–82. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yamashita Y, Koga M, Takeda Y, Enomoto N,
Uchida S, Hashimoto K, Yamano S, Dohi K, Marumo F and Sasaki S: Two
sporadic cases of Liddle's syndrome caused by De novo ENaC
mutations. Am J Kidney Dis. 37:499–504. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang Y, Zheng Y, Chen J, Wu H, Zheng D and
Hui R: A novel epithelial sodium channel gamma-subunit de novo
frameshift mutation leads to Liddle syndrome. Clin Endocrinol
(Oxf). 67:801–804. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhou R, Patel SV and Snyder PM: Nedd4-2
catalyzes ubiquitination and degradation of cell surface ENaC. J
Biol Chem. 282:20207–20212. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sabbadin C and Fallo F:
Hyperaldosteronism: Screening and dagnostic tests. High Blood Press
Cardiovasc Prev. 23:69–72. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ding X: A review for the reasons of
hypertension and hypokelamia. Chin J Gen Pract. 1:42015.(In
Chinese).
|
|
32
|
Bhimji SS and Sinha V: Adrenal, congenital
hyperplasia. StatPearls StatPearls Publishing. StatPearls
Publishing LLC; Treasure Island (FL): 2018
|
|
33
|
El-Maouche D, Arlt W and Merke DP:
Congenital adrenal hyperplasia. Lancet. 390:2194–2210. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Funder JW: Apparent mineralocorticoid
excess. J Steroid Biochem Mol Biol. 165:151–153. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yaxley J and Pirrone C: Review of the
Diagnostic Evaluation of Renal Tubular Acidosis. Ochsner J.
16:525–530. 2016.PubMed/NCBI
|