Introduction
Prostate cancer (PCa) is a common cause of
cancer-related mortality worldwide, and it is the second most
frequently diagnosed male cancer (1). Androgen-deprivation therapy (ADT) has
been used as the first-line treatment for PCa, and more than 90% of
PCa patients after surgical removal can be alleviated (2). Previous studies have shown that the
reasons of death in the PCa patients actually results from the
dissemination of cancer cells and the establishment of metastases
in lymph nodes or bones, but at present no treatments can inhibit
the metastatic spread of PCa effectively. Although the conventional
histological diagnosis can provide valuable information for
treatment, it is not sufficient to predict clinical outcomes
(3). Therefore, the appropriate
molecular biomarkers are urgently required to diagnose PCa patients
and predict their prognosis.
Due to the development of high-throughput sequencing
technology, abundant long non-coding RNAs (lncRNAs) have been
discovered in extensive biological processes and attracted wide
attention in the medical community. The lncRNAs belong to
non-protein-coding RNAs with the length of more than 200
nucleotides. Scholars have suggested that plenty of lncRNAs play
complex and critical roles in the progression and pathology of
tumors. Terracciano et al considered that the lncRNA
overexpression in human cancer cells could distinguish among
distinct cancer types unambiguously and could be used as prognostic
factors in various cancers (4). In
endometrial cancer, a comparison of lncRNAs between different
patients identified the lncRNAs with differential expression
(5). After functional analysis of
these lncRNAs, 5 lncRNAs were found to play key roles in the
development of cancer through regulating important related
biological processes. This is sufficient to prove that lncRNAs can
be regarded as the molecular biomarkers of cancer for diagnosis or
prognosis prediction.
The lncRNA functions of PCa are not well
characterized, thus the precise identification of lncRNA biomarkers
is difficult (6). The studies have
corroborated that lncRNAs regulate expression of other transcripts
to compete with endogenous RNAs (ceRNAs) (7,8). Recent
studies have confirmed that the ceRNA network was associated with
lncRNAs in many different cancers, and recognized the prognostic
lncRNAs by network analysis (9,10). Some
scholars have developed several ceRNA-related databases to
facilitate interference of lncRNA function. The databases uncovered
the significance of ceRNAs interactions with lncRNA, and declared
that the synthesis of network analysis and expression profiles
could recognize the risk lncRNAs and the underlying pathology of
cancer. Therefore, the functional lncRNA-mediated ceRNA network
(LMCN) associated with PCa was constructed by the multi-step
computational approach in this investigation.
This study applied a multi-step computational
approach to construct a functional LMCN associated with CRC, and
the relevant lncRNAs were identified based on the constructed
landscape map. Then functional enrichment analyses were carried out
for mRNAs which were significantly associated with lncRNAs, and
used the functions of the mature mRNAs to forecast the lncRNA
functions.
Materials and methods
The expression profiles of lncRNA and
mRNA in PCa
The gene expression profile was obtained from the
Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo/). The dataset is no.
GSE72220 which was deposited by Elai and Davicioni based on
GPL5175. It has a total of 147 data in the dataset, of which 90
samples are control group and 57 samples are the tumor group. The
information of dataset was changed into expressionSet by computer,
and stored to the working directory. The expressionSet was
preprocessed by the software after finding the relevant working
directory in the software analysis page, and a total of 14,451
genes were received.
Clinical characteristics of samples in
the profiles
The dataset consisted of 90 normal tissues and 57
prostate tumor tissues. The characteristic of all the samples is
RNA. The first step of sampling is dissecting a part of the tumor
or normal tissue in the corresponding area of the slide with a
sterile biopsy punch tool to extract RNAs. Then the biopsy was
scraped from the slide for nucleic acid extraction. Finally, the
extraction and purification of total RNA used the RNeasy FFPE kit
(Qiagen, Inc., Valencia, CA, USA), and the amplification and
marking of RNA used the Ovation WTA FFPE system (Nugen
Technologies, Inc., San Carlos, CA, USA).
Clarification of the interaction
relationship between lncRNA, miRNA and mRNA
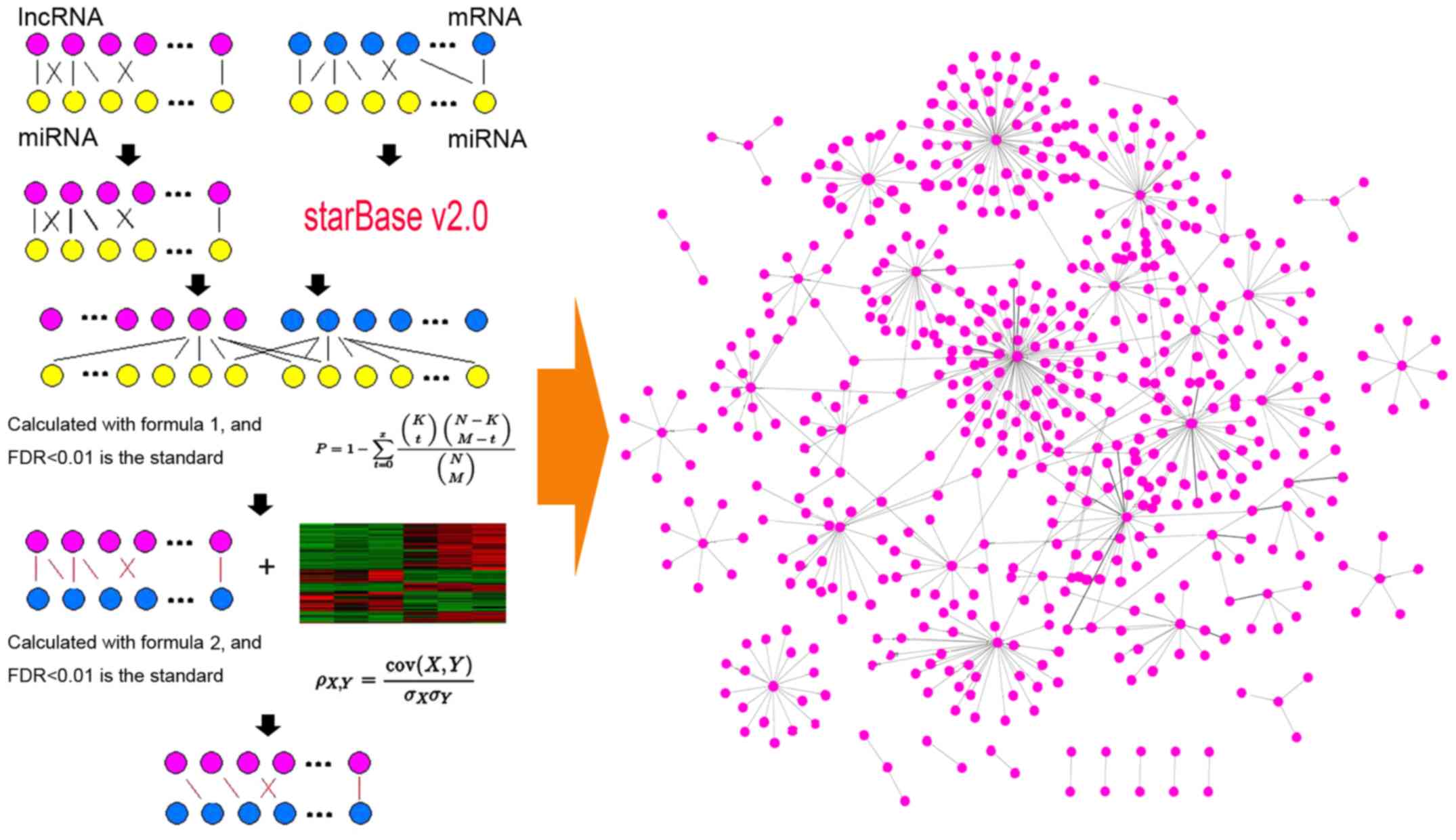
The LMCN was constructed by integrating the
expression profiles between lncRNA and mRNA and analyzing the
interactions of miRNA-target. The first step is to download
miRNA-mRNA interactions and lncRNA-miRNA pinsections from starBase
v2.0. The starBase 2.0 is a network which decodes miRNA-ceRNA,
miRNA-lncRNA and protein-RNA interactions from 108 sets of CLIP-Seq
data generated from 37 independent studies. In the second step, the
overlap on the mRNA and lncRNA between the expression profile and
the interactions of miRNA-mRNA and lncRNA-miRNA were obtained. A
new expression profile was obtained which had a total of 8,522
genes including 8,471 mRNAs and 51 lncRNAs. Finally, new
interactions were collected from aforesaid miRNA-mRNA interactions
and lncRNA-miRNA intersections which had 265,782 pairs of
miRNA-mRNA interactions and 598 pairs of lncRNA-miRNA.
Identification of ceRNA
interactions
To evaluate the statistical significance of the
shared miRNAs between each lncRNA and mRNA, the hypergeometric test
was used for identifying competing lncRNA-mRNA interactions.
Because the hypergeometric test assessment and enrichment of miRNAs
have interactions of lncRNA and mRNA could achieve this goal. The
formula of calculation is as follows (formula 1).
P=1-∑t=0x(Kt)(N-KM-t)(NM),
In formula 1, N is the total number of miRNAs, of
which K and M are the numbers of miRNAs connected with the current
lncRNA and mRNA, and × is the number of common miRNA shared by the
lncRNA and mRNA. The calculated P-value is used to evaluate the
enrichment of the function. It is noteworthy that the false
discovery rate (FDR) is used to correct the P-value and the FDR
<0.01 regarded as the threshold. After correction, the LMCN with
P-value <0.01 contained 51 lncRNAs, 8,125 mRNAs and 34,586
interactions.
Screening the subnetworks of LMCN
(sub-LMCN) regulated by the highly competitive lncRNA
To analyze the co-expression of the screened
lncRNA-mRNA interactions, Pearson's correlation coefficients of the
control and disease groups were calculated. The formula of
calculation is as follows (formula 2).
ρX,Y=cov(X,Y)σXσY,
In formula 2, cov (X,Y) is the covariance of
variables X and Y, of which σX and σY are the
standard deviations for X and Y, respectively. FDR <0.01 was
regarded as the threshold. The difference value of Pearson's
correlation coefficient was >0.45 and considered as the
significant coexpression of the ceRNA interactions.
Prediction of lncRNA function
The prediction of lncRNA function used the mature
function of mRNA though functional enrichment analysis of the mRNAs
which were significantly correlated with lncRNA. First of all, the
Gene Ontology (GO) analysis was used for prediction. Using the
P=0.01 as the threshold, the enrichment analysis of mRNAs in the
sub-LMCN was based on the BP classification of GO analysis. The
pathway was subsequently predicted. In this study, the pathway
database was KEGG, and the Fisher test was used to identify the
enrichment pathway of lncRNA. After eliminating the pathway with
enrichment P-value <0.05, the other results were the pathway
which may be regulated by lncRNA.
Results
Structure of LMCN
This study constructed an LMCN to assess the
lncRNA-mediated ceRNA interaction landscape. A multi-step approach
was used to accomplish this. lncRNA transcripts that compete with
endogenous mRNAs to bind miRNAs could be identified by the present
miRNA target prediction methods. To identify the interactions with
miRNA-lncRNA and miRNA-mRNA of PCa, starBase v2.0 was applied to
decode and collect data sets in large-scale CLIP-seq data. Unlike
the results predicted by current software, the data set obtained
from starBase v2.0 provided high confidence miRNA-target
interactions supported by the experiments. The new expression
profiles of 8,522 genes were obtained, which contained 8,471 mRNAs
and 51 lncRNAs after assessment of intersection between the
obtained data set and the genes of the chip platform HuEx-1_0-st
(Affymetrix Human Exon 1.0 ST Array), as well as and combining mRNA
and lncRNA in the data set. In this new expression profile, there
were 265,782 pairs of miRNA-mRNA interactions and 598 pairs of
lncRNA-miRNA intersections. Then the LMCN model was constructed
with the significantly co-expressed lncRNA-mRNA ceRNA pairs
(Fig. 1).
Biological and topological properties
revealed by LMCN
The hypergeometric test was used to evaluate the
enrichment of miRNAs, and 34,586 pairs of interactions which
included 51 lncRNAs and 8,125 mRNAs after screened by FDR
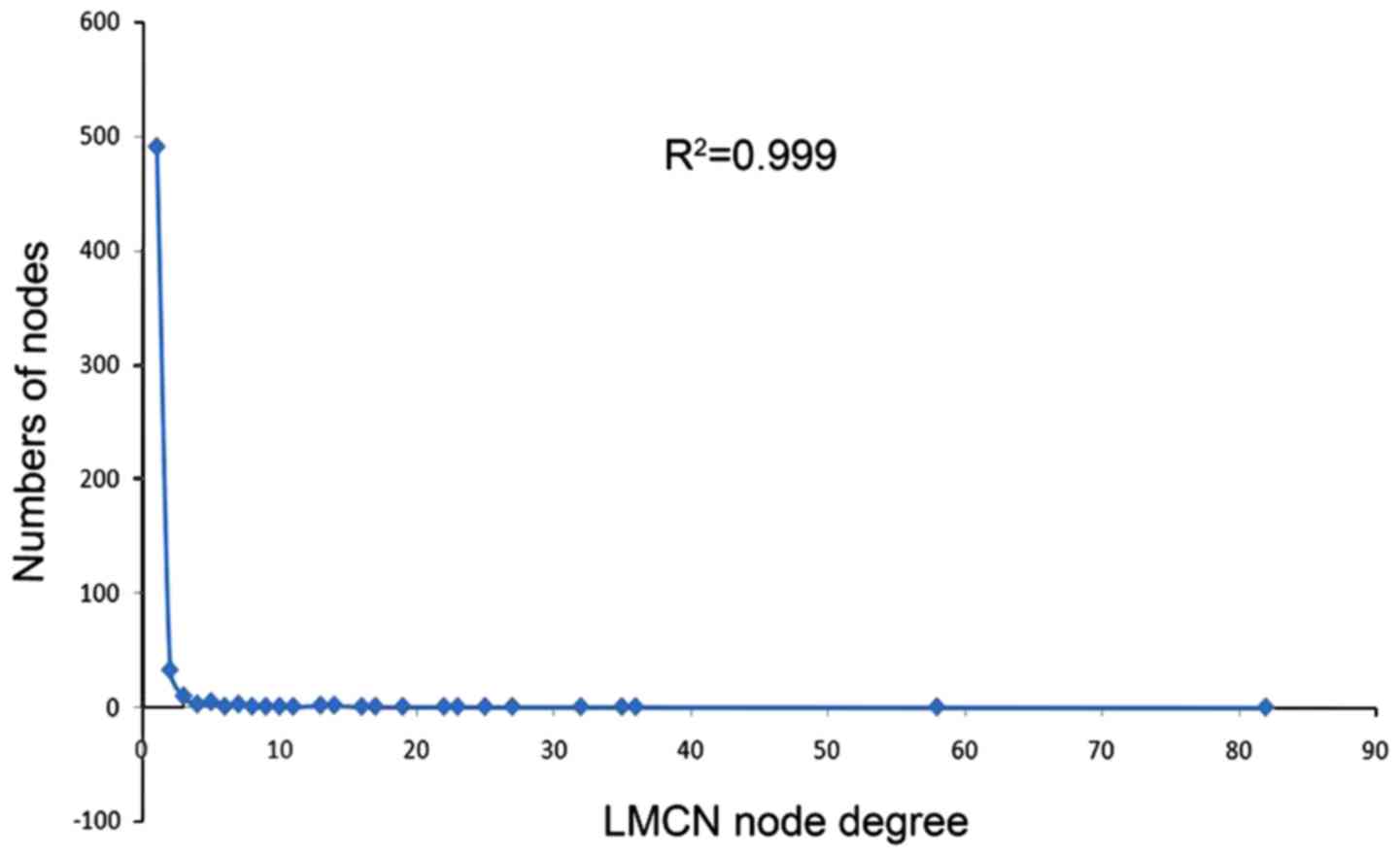
correction were obtained. The statistics of nodes degree were
obtained to reveal power law distribution, and the
R2=0.999 is the PCa-associated LMCN, a scale-free
network (Fig. 2). Then the node
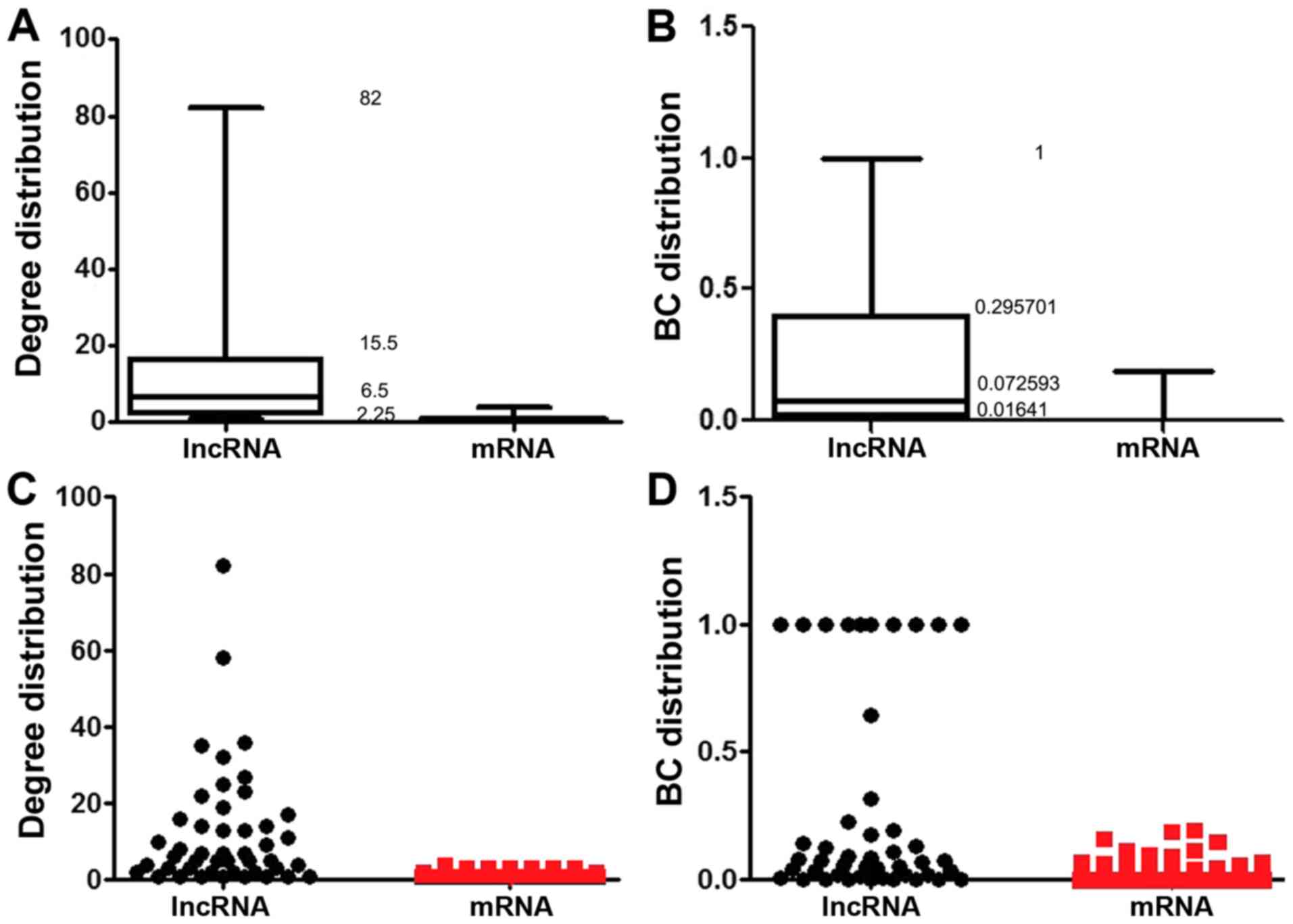
degree and betweenness centrality (BC) of LMCN were analyzed, which
are topological attributes. The box plots (Fig. 3A and B) show the statistics of the
degree and BC value which included the maximum value, quartile
value and median value. The scatter plots (Fig. 3C and D) show the distribution of
lncRNAs and mRNAs. A higher degree demonstrated that the node was a
hub, which was involved in more ceRNA interactions. A higher BC
showed that the node was a bottleneck, which acted as bridges
connecting different network modules. Compared to mRNA nodes, the
lncRNA nodes show more specific topological properties because of
their greater degrees and BC values.
Critical lncRNAs of PCa
The Pearson correlation coefficient of the screened
lncRNA-mRNA interactions was calculated between the control and PCa
groups using co-expression analysis. The difference value (D-value)
of Pearson correlation coefficient was calculated for the same
interaction between the control and PCa groups, and the
interactions with D-value >0.45 were regarded as the significant
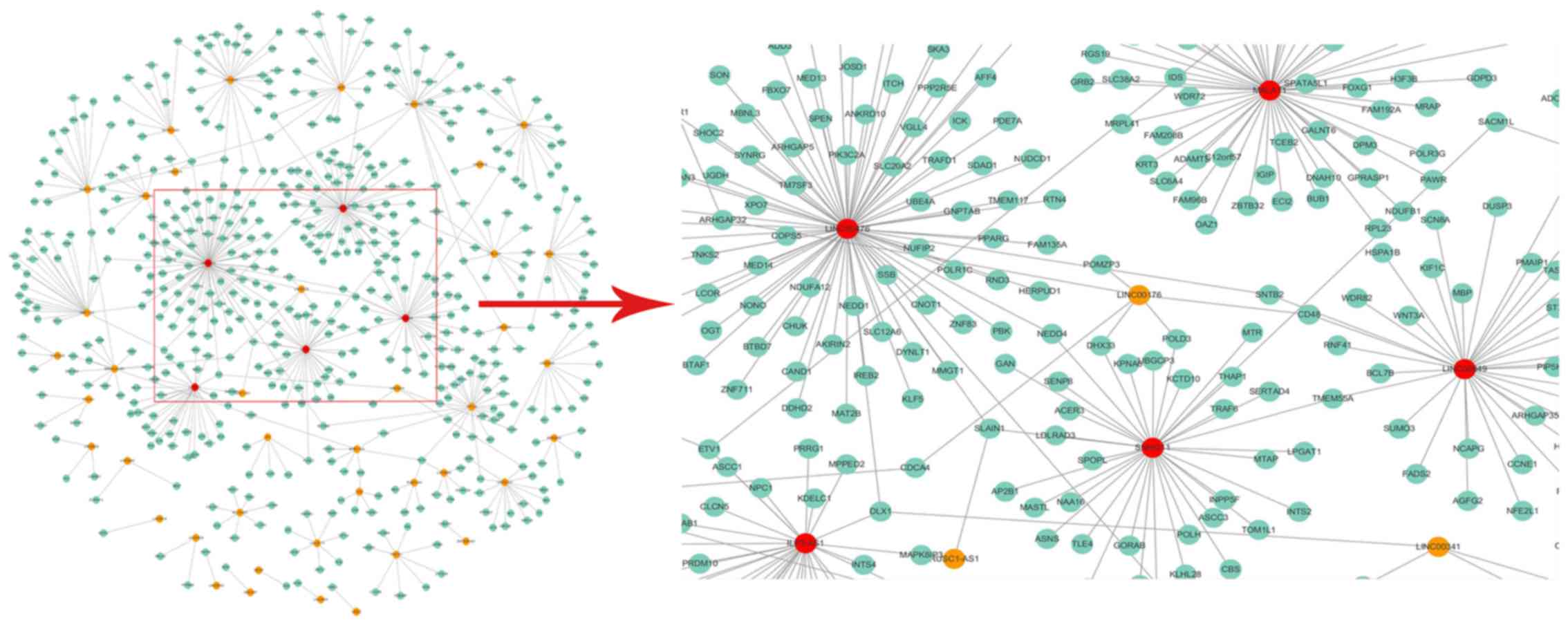
coexpression ceRNA interactions. After mapping the network graph,
it was found that there were 46 lncRNAs, 522 mRNAs and 569
interactions (Fig. 4). In addition,
the results showed that the lncRNA nodes were always in the central
region of the network, whereas the mRNA nodes were always in the
outer layer. Therefore, in the LMCN of PCa, the lncRNAs dominated
in the central regulatory function. Using the cytoscape software to
analyze the ceRNA interactions, it was found that LINC00476,
MALAT1, SNHG11, LINC00649, and ILF3-AS1 had more nodes and higher
BC compared to other lncRNAs. It suggests that these 5 lncRNAs are
likely to be prognostic markers in PCa.
Prediction of GO analysis and
pathway
In order to further verify the potential function of
lncRNAs, the functional enrichment of mRNAs was analyzed, which
were significantly associated with lncRNA and the function of
lncRNA can be predicted by the mature function of mRNA. The mRNAs
which are closely related to the central lncRNAs of PCa have an
obvious enrichment in a function whose ID is GO: 0030522, and the
involved genes are DAB2, ASXL1, CTNNB1, TRIM16, SAFB, DDRGK1,
VDR, EZH2, TWIST1, CCNE1, HSPA1B, PUM1, NOTCH2, TRAF6, NEDD4,
MED13, ITCH, PPARG, MED14, CHUK, CNOT1, BRD8, and SRC.
The pathway of the lncRNA enrichment was identified by the Fisher
Test based on the Kyoto Encyclopedia of Genes and Genomes (KEGG)
database. After correction, the pathway with P-value <0.05 was
the pathway for enrichment of mRNAs that was significantly
associated with lncRNA, indicating they may be the pathway
regulated by the lncRNAs (Table
I).
 | Table I.The prediction of GO analysis and
pathways. |
Table I.
The prediction of GO analysis and
pathways.
| Pathway | P-value (FDR) | Number of mRNA |
|---|
| 4144 Endocytosis
[PATH:hsa04144] |
0.0178827965638394 | 18 |
| 4510 Focal adhesion
[PATH:hsa04510] |
0.020522962511304 | 16 |
| 4740 Olfactory
transduction [PATH:hsa04740] |
0.020522962511304 | 2 |
| 5200 Pathways in
cancer [PATH:hsa05200] |
0.0196461610273246 | 26 |
| 5222 Small cell lung
cancer [PATH:hsa05222] |
0.0196461610273246 | 10 |
| 5169 Epstein-Barr
virus infection [PATH:hsa05169] |
0.020522962511304 | 16 |
Discussion
The early treatments of PCa include surgical
treatment, androgen blockade and radiotherapy, but most patients
still suffered from recurred tumors which will destroy the bone
marrow and lead to death (11).
Therefore, it is urgent to identify the molecular mechanisms to
confirm potential effective therapeutic targets for improving
patient survival. Bioinformatics is one of the interdisciplinary
disciplines involved in many fields, and one of its core elements
was to study the internal expression regulatory mechanisms of genes
and to reveal essential laws of human diseases (12). lncRNAs are longer than 200
nucleotides but the functional open reading frames are lackig.
lncRNAs have important clinical value of diagnosis, prognosis and
treatment because of their biological properties, so that it has
been extensively studied. The studies indicated that lncRNAs
participate in a variety of cell biological processes such as
cellular proliferation, differentiation, and DNA damage responses
(13). At present, the aberrant
expression of lncRNAs occurs in many human diseases. Aiello et
al using RNA-ChIP and ChIRP found that in PCa patients HOTAIR
and MALAT1 were relevant to both ERα/ERβ detectable at chromatin
level (14). The results showed that
MALAT1 silencing was relevant for PCa, and disclosing potential
perspective manipulations in terms of transcription regulation of
prognostic genes.
It has been identified that lncRNA overexpression is
concerned with the progression and pathology of PCa (15). The PCa-specific LncRNAs affected PCa
cell self-renewal, proliferation, survival, metastasis, and
apoptosis by either transcriptional or post-transcriptional
regulation. Therefore some of them are involved in distinct
subtypes of PCa (16). To make clear
the intermodulation relationship between lncRNA and mRNA, a
functional LMCN was constructed. First the gene expression profile
collected in GEO database was preprocessed, resulting in obtaining
14,451 genes. Then the miRNA-mRNA interactions and lncRNA-miRNA
pinsections from starBase v2.0, were studied and new gene
expression profile and interaction pairs though overlapping
miRNA-mRNA and lncRNA-miRNA interaction were obtained. The new gene
expression profile included 8,522 genes, and the new interaction
pairs included 265,782 pairs of miRNA-mRNA interactions and 598
pairs of lncRNA-miRNA. Finally, in order to assess the significance
of miRNAs shared between each mRNA and lncRNA, competitive
lncRNA-mRNA interactions were identified using the hypergeometric
test, and the prediction of lncRNA function used the mature mRNA
function.
The lncRNAs are implicated in regulation of PCa
generation and development, and their functions are accomplished by
adjusting the expression level of protein-coding genes. In this
study, the interaction between lncRNA and mRNA was analyzed using
software, and most of the central points were lncRNA. The power law
distributions shown as R2=0.999 indicated the LMCN
network has a high degree of confidence. Compared the number of
nodes and BC values among different central lncRNAs, LINC00476,
MALAT1, SNHG11, LINC00649 and ILF3-AS1 had the deeper interaction
with more mRNAs, therefore these 5 lncRNAs were more likely to
serve as the prognostic markers of PCa. MALAT1 is also named NEAT2,
and was a highly conserved lncRNA. Studies have shown that it was
overexpressed in multifarious human tumors and linked to enhanced
cell migration, invasion and proliferation of cancers. The human
tumors affected by MALAT1 included glioma (17), pancreatic (18), prostate (19) and lung cancer (20). Sebastian et al thought that
the cancer metastasis was a complex process in which the cells
deviated from the cancer primary site then traveled through the
lymphatic or circulatory systems to become secondary tumors
(21). They used PCa-osteoblast
interaction microarrays to recognize the new factors which promote
PCa metastasis to bone. In the result, they found that MALAT1 was
not only one of the differentially expressed genes, but also
regulated by Sost. It meant that the proliferation, migration, or
invasion of PCa cells may be regulated by the change of MALAT1
expression. Unlike MALAT1, the other four lncRNAs had no verified
experiments or studies in connection with PCa. However, it was
discovered that the expression of ILF3-AS1 is negatively correlated
with that of miR-200b/a/429 in melanoma tissues (22). ILF3-AS1 is a melanoma-upregulated
lncRNA induces cell invasion, migration and proliferation through
negatively regulating miR-200b/a/429. Chaudhry investigateded the
miRNA modulation mechanism in human cells treated by radiotherapy
(23). SNHG11 was induced in TK6 and
WTK1 cells, and its expression level was followed by a decline.
SNHG11 has been well-characterized in cancer. Although the direct
study between these four lncRNAs and PCa was lacking, their effects
on various cancers were unquestionable. Therefore, the potential
lncRNA biomarkers (e.g. LINC00476, MALAT1, SNHG11, LINC00649 and
ILF3-AS1) have been inferred, which can be used for diagnosis,
evaluation and gene-targeted therapy of PCa. Further studies need
to be carried out for verifying this inference. The GO analysis
suggested that the occurrence and progression of PCa were
associated with intracellular receptor signaling pathway. The
Fisher test indicated that lncRNAs may be involved in Endocytosis
and Focal adhesion.
In conclusion, this study structured an LMCN
landscape of 147 PCa samples with a new method. The LMCN landscape
was used to observe the specific topological properties and
synergistic, competitive effects of lncRNAs, and it revealed
regulatory interactions with coding mRNAs in PCa.
Acknowledgements
Not applicable.
Funding
This study is supported by the Natural Science
Foundation of Shandong (grant no. ZR201709290020).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
GY and LG conceived the study and drafted the
manuscript. YX acquired the data. WS and MY analyzed the data and
revised the manuscript. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Kalsbeek AMF, Chan EKF, Corcoran NM,
Hovens CM and Hayes VM: Mitochondrial genome variation and prostate
cancer: a review of the mutational landscape and application to
clinical management. Oncotarget. 8:71342–71357. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lee SU and Cho KH: Multimodal therapy for
locally advanced prostate cancer: The roles of radiotherapy,
androgen deprivation therapy, and their combination. Radiat Oncol
J. 35:189–197. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Puche-Sanz I, Alvarez-Cubero MJ,
Pascual-Geler M, Rodriguez-Martinez A, Delgado-Rodriguez M,
Garcia-Puche JL, Expósito J, Robles-Fernández I, Entrala-Bernal C,
Lorente JA, et al: A comprehensive study of circulating tumour
cells at the moment of prostate cancer diagnosis: Biological and
clinical implications of EGFR, AR and SNPs. Oncotarget.
8:70472–70480. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Terracciano D, Terreri S, de Nigris F,
Costa V, Calin GA and Cimmino A: The role of a new class of long
noncoding RNAs transcribed from ultraconserved regions in cancer.
Biochim Biophys Acta. 1868:449–455. 2017.
|
|
5
|
Sun Y, Zou X, He J and Mao Y:
Identification of long non-coding RNAs biomarkers associated with
progression of endometrial carcinoma and patient outcomes.
Oncotarget. 8:52604–52613. 2017.PubMed/NCBI
|
|
6
|
Su W, Xu M, Chen X, Chen N, Gong J, Nie L,
Li L, Li X, Zhang M and Zhou Q: Long noncoding RNA ZEB1-AS1
epigenetically regulates the expressions of ZEB1 and downstream
molecules in prostate cancer. Mol Cancer. 16:1422017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jia J, Li F, Tang XS, Xu S, Gao Y, Shi Q,
Guo W, Wang X, He D and Guo P: Long noncoding RNA DANCR promotes
invasion of prostate cancer through epigenetically silencing
expression of TIMP2/3. Oncotarget. 7:37868–37881. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Misawa A, Takayama K, Urano T and Inoue S:
Androgen-induced long noncoding RNA (lncRNA) SOCS2-AS1 promotes
cell growth and inhibits apoptosis in prostate cancer cells. J Biol
Chem. 291:17861–17880. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wu H, Wu R, Chen M, Li D, Dai J, Zhang Y,
Gao K, Yu J, Hu G, Guo Y, et al: Comprehensive analysis of
differentially expressed profiles of lncRNAs and construction of
miR-133b mediated ceRNA network in colorectal cancer. Oncotarget.
8:21095–21105. 2017.PubMed/NCBI
|
|
10
|
Zhou M, Diao Z, Yue X, Chen Y, Zhao H,
Cheng L and Sun J: Construction and analysis of dysregulated
lncRNA-associated ceRNA network identified novel lncRNA biomarkers
for early diagnosis of human pancreatic cancer. Oncotarget.
7:56383–56394. 2016.PubMed/NCBI
|
|
11
|
Makishima H, Ishikawa H, Tanaka K, Mori Y,
Mizumoto M, Ohnishi K, Aihara T, Fukumitsu N, Okumura T and Sakurai
H: A retrospective study of late adverse events in proton beam
therapy for prostate cancer. Mol Clin Oncol. 7:547–552. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ready D, Yagiz K, Amin P, Yildiz Y, Funari
V, Bozdag S and Cinar B: Mapping the STK4/Hippo signaling network
in prostate cancer cell. PLoS One. 12:e01845902017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wu Y, Liang S, Xu B, Zhang R, Zhu M, Zhou
W, Zhang S, Guo J, Xu L and Zhu H: Long noncoding RNA eosinophil
granule ontogeny transcript inhibits cell proliferation and
migration and promotes cell apoptosis in human glioma. Exp Ther
Med. 14:3817–3823. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Aiello A, Bacci L, Re A, Ripoli C,
Pierconti F, Pinto F, Masetti R, Grassi C, Gaetano C, Bassi PF, et
al: MALAT1 and HOTAIR long non-coding RNAs play opposite role in
estrogen-mediated transcriptional regulation in prostate cancer
cells. Sci Rep. 6:384142016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ahadi A, Brennan S, Kennedy PJ, Hutvagner
G and Tran N: Long non-coding RNAs harboring miRNA seed regions are
enriched in prostate cancer exosomes. Sci Rep. 6:249332016.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mehra R, Udager AM, Ahearn TU, Cao X, Feng
FY, Loda M, Petimar JS, Kantoff P, Mucci LA and Chinnaiyan AM:
Overexpression of the long non-coding RNA SChLAP1 independently
predicts lethal prostate cancer. Eur Urol. 70:549–552. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cao S, Wang Y, Li J, Lv M, Niu H and Tian
Y: Tumor-suppressive function of long noncoding RNA MALAT1 in
glioma cells by suppressing miR-155 expression and activating FBXW7
function. Am J Cancer Res. 6:2561–2574. 2016.PubMed/NCBI
|
|
18
|
Jiao F, Hu H, Yuan C and Wang L, Jiang W,
Jin Z, Guo Z and Wang L: Elevated expression level of long
noncoding RNA MALAT-1 facilitates cell growth, migration and
invasion in pancreatic cancer. Oncol Rep. 32:2485–2492. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ren S, Liu Y, Xu W, Sun Y, Lu J, Wang F,
Wei M, Shen J, Hou J, Gao X, et al: Long noncoding RNA MALAT-1 is a
new potential therapeutic target for castration resistant prostate
cancer. J Urol. 190:2278–2287. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tano K, Mizuno R, Okada T, Rakwal R,
Shibato J, Masuo Y, Ijiri K and Akimitsu N: MALAT-1 enhances cell
motility of lung adenocarcinoma cells by influencing the expression
of motility-related genes. FEBS Lett. 584:4575–4580. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sebastian A, Hum NR, Hudson BD and Loots
GG: Cancer-osteoblast interaction reduces sost expression in
osteoblasts and up-regulates lncRNA MALAT1 in prostate cancer.
Microarrays (Basel). 4:503–519. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chen X, Liu S, Zhao X, Ma X, Gao G, Yu L,
Yan D, Dong H and Sun W: Long noncoding RNA ILF3-AS1 promotes cell
proliferation, migration, and invasion via negatively regulating
miR-200b/a/429 in melanoma. Biosci Rep. 37(pii): BSR201710312017.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chaudhry MA: Expression pattern of small
nucleolar RNA host genes and long non-coding RNA in X-rays-treated
lymphoblastoid cells. Int J Mol Sci. 14:9099–9110. 2013. View Article : Google Scholar : PubMed/NCBI
|