Introduction
Chronic granulomatous disease (CGD) is a rare
primary immunodeficiency disease caused by reduced nicotinamide
adenine dinucleotide phosphate oxidase defects, which may impair
the ability of phagocytes to kill peroxidase-positive bacteria and
fungi, resulting in repeated and severe infections (1). The most common infection sites include
the lungs, skin, lymph nodes and liver (1). For patients with CGD, an excessive
inflammatory reaction gradually forms a granuloma; however, the
clinical manifestations are varied, which makes early diagnosis
difficult (2,3). US and European statistics have reported
incidence rates of CGD of 1/200,000-1/250,000 (4,5). At
present, there is no precise data regarding the incidence of CGD in
China; however, as the attention paid to primary immunodeficiency
diseases increases and the assessment strategies improve, an
increasing number of CGD diagnoses are made (6,7). As the
prognosis of this disease is poor, it is important to perform
pedigree analyses and pre-natal diagnoses in affected families. In
the present study, the clinical manifestations, pathogenic gene
mutations and patterns of inheritance were assessed in a family
with CGD to demonstrate the importance of next-generation
sequencing (NGS) for the pre-natal diagnosis of this disease.
Materials and methods
Patient information
The proband, a 36-day-old male, was born to
non-consanguineous Chinese parents. He was admitted to the hospital
due to discovery of a neck lump 10 days previously. In the
neo-natal period, the proband had been hospitalized at the
neo-natal department of a local hospital due to neo-natal
hyperbilirubinemia, neo-natal pneumonia and congenital
hypothyroidism. He received oral levothyroxine tablets following
discharge from the hospital. The mother was ‘gravida 3, para 3’,
and the patient was the third child. The first child of the
proband's parents, a male, died when he was 7 months old due to
severe pneumonia, severe sepsis, septicemia (due to Burkholderia
cepacia), intracranial infection and multiple organ failure.
The second child of the proband's parents was a healthy 1-year-old
female. The proband was born at week 39 of gestation with a birth
weight of 3.7 kg. No CGD-associated mutation within the family was
known. Approximately 10 months after the birth of the proband, the
mother became pregnant again.
Accessory examination
Routine laboratory examinations on blood, urine and
stool samples were conducted. Blood tests were conducted using the
Mindray blood cell analyzer (model no. BC-5000; Shenzhen Mindray
Bio-Medical Electronics Co., Ltd., Shenzhen, China). Urinalysis was
tested by manual standard methods (8) combined with dry chemistry test paper.
Following the addition of isotonic saline to slides containing
stool samples, they were view in several random fields for direct
smear light microscopy using a magnification of ×10 or ×40 as
appropriate. Erythrocyte sedimentation rate [ESR; the Westergren
method (9)], C-reactive protein
(CRP) and T cells with interferon-γ release assays were performed.
CRP content was measured with the Full C-Reactive Protein
Quantification Test kit (Shanghai Upper Bio-Tech Pharma Co., Ltd.,
Shanghai, China) according to the manufacturer's protocol. T cells
with interferon-γ release assays were conducted using the TB-IGRA
kit (cat. no. TB-0296; WANTAI BioPharm Co., Ltd., Beijing, China).
A chest radiograph was also performed. Immune globulin detection
was measured by immuno-nephelometry using the BNII instrument (Dade
Bering Inc., Deerfield, IL, USA), and a biopsy of the cervical
lymph nodes was conducted. A biopsy of the cervical lymph nodes and
immune globulin detection were conducted.
Measurement of the respiratory burst
activity
In brief, 1 ml peripheral blood was collected in a
heparin anti-coagulant tube and was sent for detection within 2 h.
Phorbol ester (cat. no. P8139) and dihydrorhodamine (DHR; cat. no.
D1054) were purchased from Sigma-Aldrich (Merck KGaA, Darmstadt,
Germany). Ammonium chloride, EDTA and potassium carbonate were
provided by Gold Wall Reagent Co. Ltd. (Kunshan, China). A working
solution of hemolysin was composed of 8.29 g ammonium chloride,
0.037 g EDTA, 1.0 g potassium carbonate and 1 l deionized water. As
for flow cytometry, the analysis was performed using CELLQuest
software (version 5.1; BD Biosciences, San Jose, CA, USA). The
detection was performed as described by Richardson et al
(10). Briefly, three samples were
taken were placed in separate polypropylene tubes and were used as
stimulated, resting and reagent blank tests. The tubes were
successively incubated with working DHR solution (30 µg/ml) at 37°C
for 5 min and FACS lysing solution (BD Biosciences) at room
temperature for 10 min. Following the centrifugation of the samples
at 150 × g at room temperature for 5 min, they were washed with PBS
and then fixed with 1% paraformaldehyde at 4°C for 30 min to
stabilize the cells. To measurement the activity, the instrument
was set up with a reagent blank sample and neutrophils were
selected by gating. The negative contrast was adjusted to
101 within the histogram using FL1 as the abscissa. Data
were collected from all tubes, with 3,000 granulocytes obtained for
each sample. During analysis, granulocytes were selected and
displayed in a specific area using the forward angle and the
lateral angle. A DHR fluorescence histogram was obtained in the
gate and the average number of fluorescent signals was
recorded.
Genetic sequencing
To determine the molecular cause of the disease in
the proband, genetic sequencing, including NGS and Sanger
sequencing, was performed. NGS was performed as previously
described (11), with the
modification that only 310 genes were captured. A gene library was
prepared using peripheral blood, detected with the Agilent
Bioanalyzer 2100 (Agilent Technologies, Inc., Santa Clara, CA,
USA), and sequenced using the Illumina Hiseq2500 platform
(Illumina, Inc., San Diego, CA, USA). The sequencing data were then
processed using BclToFastq software (version 2; Illumina, Inc.) and
aligned with the National Center for Biotechnology Information
human reference genome (HG-19) using the Burrows-Wheeler aligner
(12,13). Genome Analysis Toolkit software
(GATK-3.7 software; Broad Institute, Cambridge, MA, USA) was used
for identifying single nucleotide polymorphisms and
insertion-deletions in the sequences. Protein biological function
was predicted by using MutationAssessor (14), Provean (15) and PolyPhen-2 (16).
For Sanger sequencing, genomic DNA was extracted
with Qiagen FlexiGene DNA kit (cat. no. 512206; Qiagen GmbH,
Hilden, Germany) according the protocol provided by the
manufacturer. The DNA was used for amplification was performed
according with the method described previously (11) using an annealing temperature of 58°C.
The primers used were 5′-AGGGTGCCTTGGTTAGAATAGC-3′ (forward) and
5′-TCTGTTGGGCATGAATTCAATC-3′ (reverse) to generate a product 362 bp
in length. The polymerase chain reaction (PCR) products were
sequenced using an ABI 3730XL (Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) and analyzed with DNASTAR 5.0 software (DNASTAR,
Inc., Madison, WI, USA).
Pre-natal diagnosis of the fetus
For pre-natal diagnosis, 30 ml amniotic fluid was
extracted at week 18 of pregnancy and was sent to the laboratory in
a sterile culture tube. Following centrifugation, amniotic fluid
DNA was extracted using the Qiagen FlexiGene DNA kit and PCR
amplification was performed as mentioned above. In addition, the
chromosomes were analyzed to identify the sex of the fetus.
Briefly, 25 ml amniotic fluid was centrifuged for 10 min at 295 × g
and room temperature. After the supernatant was discarded, amniotic
fluid culture medium (CHANG Amnio®; Irvine Scientific
Sales Company, Inc., Santa Ana, CA, USA) was added to prepare cell
suspension. The cell suspension was inoculated in culture flask and
cultured in 37°C, 5% CO2 incubator. The medium was
changed for 7–8 days and the cells were harvested after 10 days.
The cells were stained with Geimsa (Beijing Solarbio Science &
Technology Co., Ltd., Beijing, China) at room temperature for 3–5
min and karyotype analysis was performed under a phase contrast
microscope at a magnification of ×100. These slides were examined
systematically using an image analyzer system and Cyto-Vision
software (version 3.93; Applied Imaging Corp., San Jose, CA, USA).
DNA amplification products were sequenced using the ABI 3730XL
(Thermo Fisher Scientific, Inc.) and analyzed using DNASTAR 5.0
software (DNASTAR, Inc.). In order to avoid experimental errors,
PCR and sequencing experiments were repeated two times for the six
samples.
Results
Clinical characteristics
To gain insight into the general condition of the
proband, a physical examination was performed on admission. The
infant had a normal development and was well nourished. The skin
around his ears was red and skin damage was noted. Swollen lymph
nodes (the size of peas to peanuts) were present at the back of the
ears and the neck. The nodes were active and smooth, with no
fusion, no pain reaction and no adhesion to the surrounding
tissue.
Accessory examination results
In order to provide an accurate diagnosis, an
additional examination was undertaken. Routine blood results were
as follows: White blood cells, 21.48×109/l (normal
range, 4–10×109/l); neutrophil ratio (N%), 44.7% (normal
range, 50–70%); hemoglobin, 92 g/l (normal range, 110–140 g/l); and
platelets, 416×109/l (normal range,
100–300×109/l), indicating infection and mild anemia.
Stool and urine were normal on routine examination. Chest
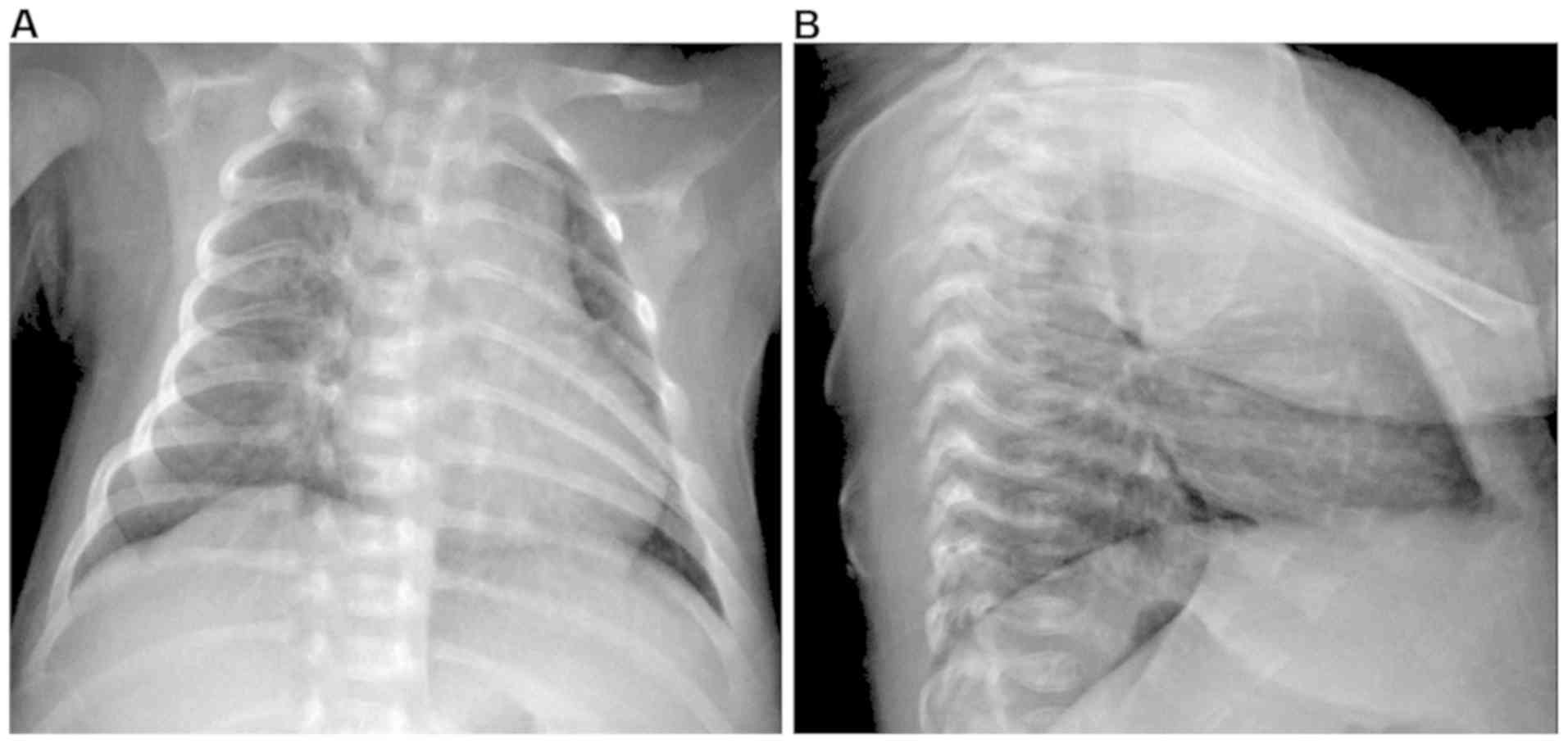
radiography demonstrated thickened and blurred markings, as well as
scattered spots in the bilateral lung tissues (Fig. 1), indicating bronchopneumonia.
C-reactive protein was 43.4 mg/l (normal range, <10 mg/l) and
the erythrocyte sedimentation rate was 67 mm/h (normal range, 0–20
mm/h), suggesting the presence of bacterial infection. The
detection of Mycobacterium tuberculosis in T cells with
interferon-γ release assay demonstrated that the level of
Mycobacterium tuberculosis was 238.5 pg/ml (normal range,
0–14 pg/ml). Biopsy of a left cervical lymph node revealed lymph
node granulomatous inflammation, with central necrosis, flakey
debris and rapid coagulation necrosis, indicating tuberculosis. The
biopsy staining was periodic acid-Schiff-negative and acid-fast
stain-negative. Complete immunity detection revealed immunoglobulin
(Ig)A, 0.52 g/l (normal range, 0.03–0.82 g/l), IgM, 1.10 g/l
(normal range, 0.15–1.09 g/l) and IgG, 6.74 g/l (normal range,
7–14.8 g/l), which was considered as normal. No other abnormalities
were noted.
Respiratory burst activity
Neutrophil samples were analyzed to determine any
defects in the phagocytosis and oxidation function of neutrophils.
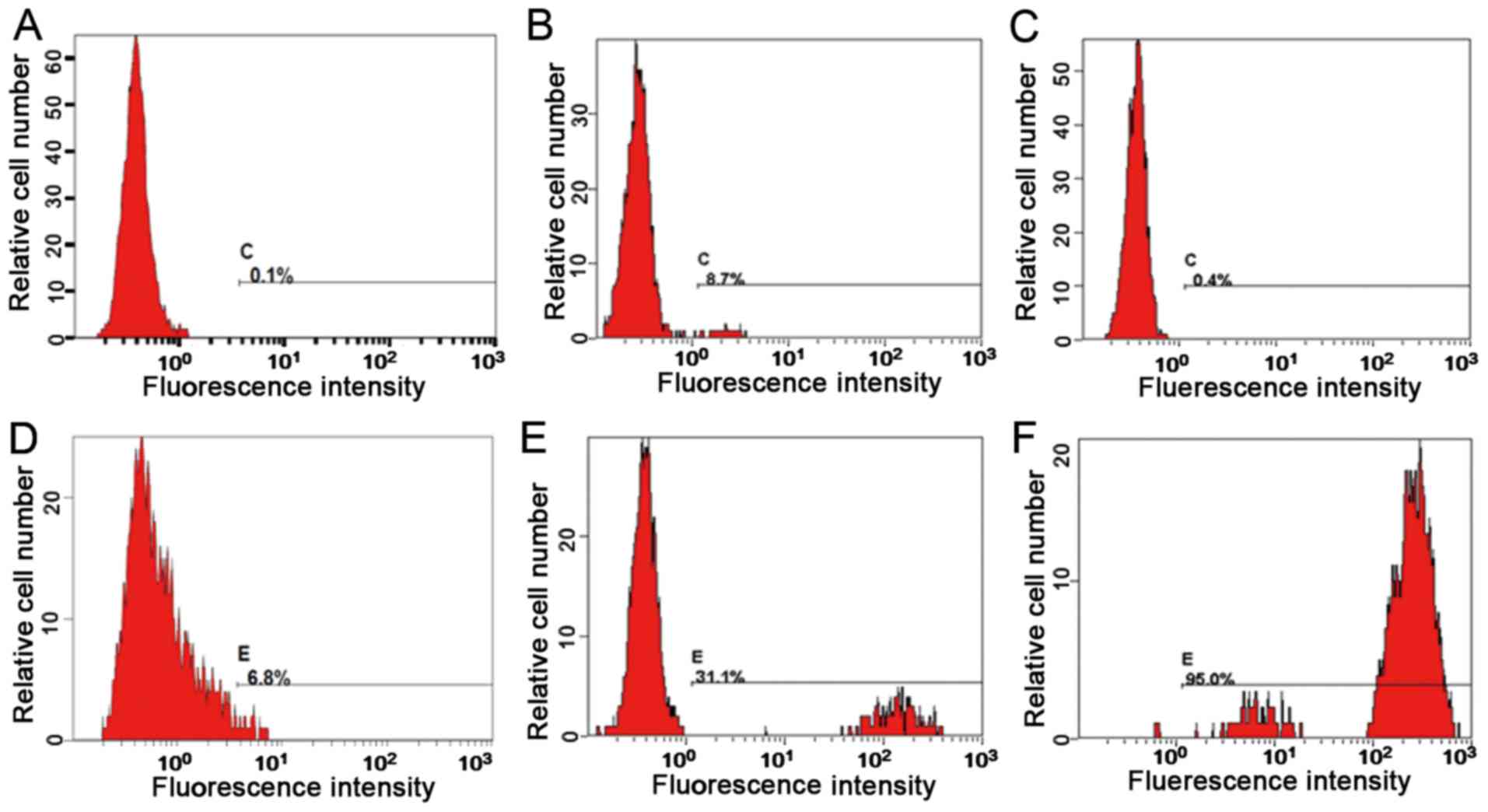
Representative flow cytometry fluorescence intensity histograms are
provided in Fig. 2 and the
quantitative results are presented in Table I. The neutrophil activation rate and
the stimulation index of the proband, the proband's father and the
proband's mother were 6.8% and 0.4, 95% and 696.1, and 31.1% and
85.1, respectively. It is assumed that the proband exhibited a
neutrophil oxygen eruption deficiency.
 | Table I.Measurement of neutrophil function in
the family (mean fluorescence intensity). |
Table I.
Measurement of neutrophil function in
the family (mean fluorescence intensity).
| Subject | Control | Phorbol 12-myristate
13-acetate | Neutrophil activation
rate (%) | Stimulation
index |
|---|
| Proband | 0.452 | 1.2 | 6.8 | 0.4 |
| Proband's mother | 0.51 | 43.4 | 31.1 | 85.1 |
| Proband's father | 0.412 | 287 | 95.0 | 696.1 |
Genetic analysis
In order to investigate the molecular etiology,
genetic analysis, including NGS and Sanger sequencing, was
performed. For the panel of 310 genes included, a total of 361
variants were identified. Among these variants, 262 were benign, 35
were likely benign, 59 were of uncertain significance and four were
likely pathogenetic. The four variants were c.1447_1448del
(NM_030917) in FIP1L1, p.Arg487Glyfs*3 (NM_000431) in MVK,
c.863C>T, p.Pro288Leu (NM_001002235) in SERPINA1, as well as
c.880G>A, p.Asp294Asn and c.1520_1521del, p.Lys508Aspfs*10
(NM_000397) in cytochrome b-245 β chain (CYBB), with a depth of 90,
118, 102 and 133×, respectively. The variants in the first three
genes are known to cause diseases different from the clinical
features of the proband, therefore, they were excluded as they were
unlikely to be pathogenic genes. CYBB is known to induce a disease
similar to that of the patient (17,18).
Following filtering, the c.1520_1521del, p.Lys508Aspfs”10
(NM_000397) variant of the CYBB gene in the proband was considered
as the etiological factor. The proband's mother was a carrier and
the father was wild-type. CYBB is located on the X sex chromosome
and CYBB mutations have been previously reported to be the cause of
the X-linked recessive CGD (19). A
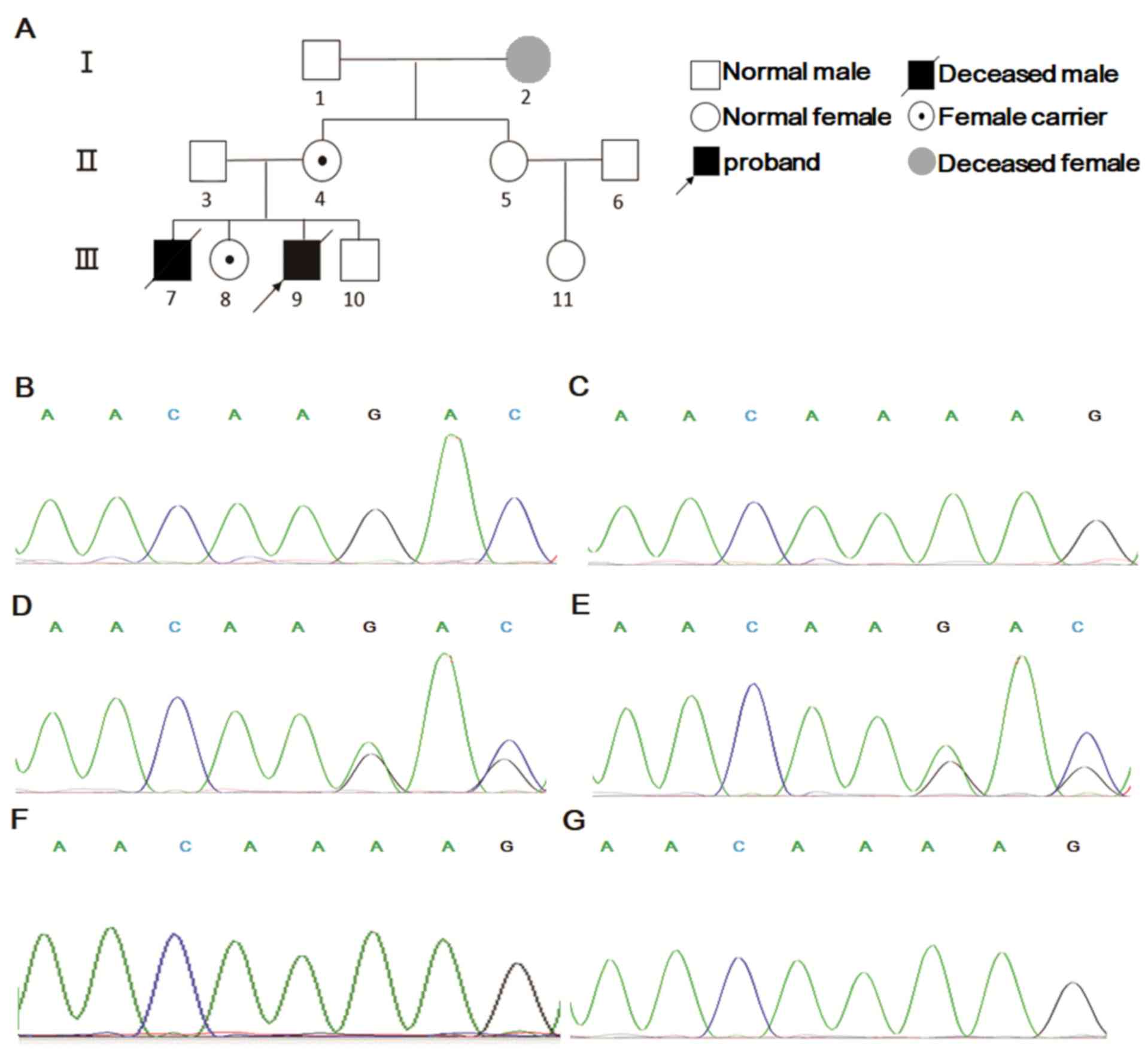
pedigree diagram of the family was presented in Fig. 3A. Sanger sequencing further validated
the results of NGS (Fig. 3B-E). In
addition, Sanger sequencing was applied to the elder sister and the
aunt (the mother's sister) of the proband, which revealed that the
sister was a carrier and the aunt possessed a wild-type genotype
(Fig. 3F and G). In summary, the
proband and his sister possessed a c.1520_1521del, p.Lys508Aspfs*10
(NM_000397) variant in the CYBB gene passed on from their
mother.
Clinical outcome
Fever was noted on the day of admission, and thus,
cephathiamidin, meropenem and vancomycin were administered to treat
potential infection. Ig was used for symptomatic treatment.
However, the fever did not subside, the swelling of the cervical
lymph nodes was not reduced and the patient experienced seizures.
The parents refused further treatment and discharged the child.
Pre-natal diagnosis
Molecular genetic diagnosis was performed for the
proband; however, follow-up revealed that he had died 21 days after
discharge. To prevent the mother from giving birth to another
infant with the same disease, amniotic cells were collected and
cultured for further analysis during a subsequent pregnancy. As
demonstrated in Fig. 4, karyotype
analysis indicated that the fetus was male. Sanger sequencing of
amniotic cell DNA indicated no variant in the CYBB gene (Fig. 3G). Considering the pre-natal
diagnosis result, the mother continued the pregnancy and gave birth
to a healthy infant.
Discussion
CGD has two genetic types: Autosomal recessive CGD
and X-linked recessive CGD. X-linked recessive CGD accounts for
65–70% of all CGD cases (5). In the
present study, the proband had a healthy elder sister and an elder
brother that exhibited repeated infection at the age of 7 months
and died due to severe pneumonia and Klebsiella sepsis. Considering
the clinical information, it is probable the elder brother also had
CGD. The clinical manifestations of this disease are variable;
infection of the respiratory tract, skin, lymph nodes and digestive
tract, and granuloma are the most common features (5). The proband of the present study
initially presented with neck lymphadenopathy but without any other
obvious symptoms. He had been vaccinated with Bacillus
Calmette-Guérin against tuberculosis, but had no scar. On the basis
of T-cell detection and lymph node biopsy results, he was highly
suspected to have a tuberculosis infection. The family history
suggested that a primary immunodeficiency disease was involved.
Within this pedigree, children that present with lymph node
enlargement, pathogen infection and potential immunodeficiency, CGD
should be considered as a possible cause in addition to blood
system disorders and connective tissue diseases.
Laboratory analysis demonstrated that the number of
leukocytes was increased in the proband, suggesting the presence of
infection. Ig was also was slightly increased. An increase of
γ-globulin is a common phenomenon in children and it has
implications for further clinical symptoms (20). The diagnosis of CGD is based on
direct determination of the production of superoxide.
Dihydrorhodamine flow cytometry has gradually replaced nitroblue
tetrazolium analysis due to its relatively simple operation, and
its ability to distinguish the X linkage and autosomal patterns by
flow cytometry. In addition, to a certain extent, the prognosis of
pediatric patients with CGD may be estimated using this method. It
was previously demonstrated that the activity of residual
neutrophils is positively correlated with survival rate (21). The activity of neutrophils in this
case was significantly decreased, the onset was early, and the
prognosis was poor, which is consistent with previous report
(21). Flow cytometry is sensitive
to even a small change in the number of functional neutrophils
(21). In the present study, the
proband was diagnosed using this method, and his mother and sister
were screened and identified as carriers. Pre-natal diagnosis for
primary immunodeficiency disease by gene detection has been applied
for numerous years (22).
CGD may be caused by mutations in the CYBB gene,
which is 30 kb in length and contains 13 exons. The mutations are
widespread, without hot-spot mutation. No associations between
genotype and phenotype have been identified thus far. Genetic
analysis confirmed that the proband of the present study possessed
the CYBB gene mutation c.1520_1521del, (NM_000397). This is a novel
variant and may cause a frameshift from Lys at the 508 amino acid
site. This variant was identified as the molecular cause of the
disease and abnormal neutrophil respiratory burst function was also
observed. In recent years, flow cytometry has been used for the
pre-natal diagnosis of CGD (23).
Flow cytometry is a fast and sensitive method for detecting CGD;
however, the accuracy of this method requires further evaluation
(24). In the present case, the
mother desired to have more children; therefore, genetic testing
was performed using amniotic fluid cell culture during her
subsequent pregnancy. Chromosome karyotype analysis and genetic
testing suggested that the mother was a carrier and the fetus was a
normal male. Peripheral blood was collected from the infant
following birth and the genetic test results were also normal.
Testing was also performed on other members of the pedigree. The
sister of the proband was also a mutation carrier, while the
proband's aunt had a normal genotype.
A method of CGD treatment that may cure the disease
is immune reconstruction, including hematopoietic stem cell
transplantation (HSCT) and gene therapy. Although HSCT has various
difficulties, including the retrieval of a suitable match and
graft-versus-host disease following transplantation, HSCT has been
used to eradicate CGD in numerous cases (25,26).
Gene therapy is still in the experimental stages, and it may take a
number of years until it is commonly used as a treatment for CGD
worldwide (27).
Acknowledgements
Not applicable.
Funding
No funding received.
Availability of data and materials
The data for present study are available from the
corresponding author on reasonable request.
Authors' contributions
LZ and XH designed the study. FP collected the
clinical data, performed the experiments, and drafted and wrote the
manuscript. RZ participated in clinical data collection. All
authors read and approved the final version of the manuscript.
Ethics approval and consent to
participate
The present study was approved by the ethics
committee of Hunan Provincial People's Hospital (Changsha, China)
and informed consent was obtained from the aunt and the parents of
the proband.
Patient consent for publication
Not applicable.
Competing interests
All authors declare that they have no competing
interests.
References
|
1
|
Segal BH, Leto TL, Gallin JI, Malech HL
and Holland SM: Genetic, biochemical, and clinical features of
chronic granulomatous disease. Medicine. 79:170–200. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Roos D and de Boer M: Molecular diagnosis
of chronic granulomatous disease. Clin Exp Immunol. 175:139–149.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rawat A, Vignesh P, Sharma A, Shandilya
JK, Sharma M, Suri D, Gupta A, Gautam V, Ray P, Rudramurthy SM, et
al: Infection profile in chronic granulomatous disease: A 23-year
experience from a tertiary care center in north India. J Clin
Immunol. 37:319–328. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Winkelstein JA, Marino MC, Johnston RB Jr,
Boyle J, Curnutte J, Gallin JI, Malech HL, Holland SM, Ochs H, Quie
P, et al: Chronic granulomatous disease. Report on a national
registry of 368 patients. Medicine (Baltimore). 79:155–169. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
van den Berg JM, van Koppen E, Ahlin A,
Belohradsky BH, Bernatowska E, Corbeel L, Español T, Fischer A,
Kurenko-Deptuch M, Mouy R, et al: Chronic granulomatous disease:
The European experience. PLoS One. 4:e52342009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Arnold DE and Heimall JR: A review of
chronic granulomatous disease. Adv Ther. 34:2543–2557. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Beghin A, Comini M, Soresina A, Imberti L,
Zucchi M, Plebani A, Montanelli A, Porta F and Lanfranchi A:
Chronic granulomatous disease in children: A single center
experience. Clin Immunol. 188:12–19. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Grinstead GF, Scott RE, Stevens BS, Ward
VL and Wilson DM: The Ames Clinitek 200/Multistix 9 urinalysis
method compared with manual and microscopic methods. Clin Chem.
33:1660–1662. 1987.PubMed/NCBI
|
|
9
|
Shelat SG, Chacosky D and Shibutani S:
Differences in erythrocyte sedimentation rates using the Westergren
method and a centrifugation method. Am J Clin Pathol. 130:127–130.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Richardson MP, Ayliffe MJ, Helbert M and
Davies EG: A simple flow cytometry assay using dihydrorhodamine for
the measurement of the neutrophil respiratory burst in whole blood:
Comparison with the quantitative nitrobluetetrazolium test. J
Immunol Methods. 219:187–193. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jin D, Yu T, Zhang L, Wang T, Hu J, Wang Y
and Yang XA: Novel NFU1 variants induced MMDS behaved as special
leukodystrophy in Chinese sufferers. J Mol Neurosci. 62:255–261.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li H and Durbin R: Fast and accurate
long-read alignment with Burrows-Wheeler transform. Bioinformatics.
26:589–595. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li H and Durbin R: Fast and accurate short
read alignment with Burrows-Wheeler transform. Bioinformatics.
25:1754–1760. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Reva B, Antipin Y and Sander C: Predicting
the functional impact of protein mutations: Application to cancer
genomics. Nucleic Acids Res. 39:e1182011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Choi Y and Chan AP: PROVEAN web server: A
tool to predict the functional effect of amino acid substitutions
and indels. Bioinformatics. 31:2745–2747. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Adzhubei IA, Schmidt S, Peshkin L,
Ramensky VE, Gerasimova A, Bork P, Kondrashov AS and Sunyaev SR: A
method and server for predicting damaging missense mutations. Nat
Methods. 7:248–249. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Iwasaki J, Kondo T, Darmanin S, Ibata M,
Onozawa M, Hashimoto D, Sakamoto N and Teshima T: FIP1L1 presence
in FIP1L1-RARA or FIP1L1-PDGFRA differentially contributes to the
pathogenesis of distinct types of leukemia. Ann Hematol.
93:1473–1481. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Silva D, Oliveira MJ, Guimaraes M, Lima R,
Gomes S and Seixas S: Alpha-1-antitrypsin (SERPINA1) mutation
spectrum: Three novel variants and haplotype characterization of
rare deficiency alleles identified in Portugal. Respir Med.
116:8–18. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Roos D: Chronic granulomatous disease. Br
Med Bull. 118:50–63. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wolach B, Gavrieli R, de Boer M, van
Leeuwen K, Berger-Achituv S, Stauber T, Ben Ari J, Rottem M,
Schlesinger Y, Grisaru-Soen G, et al: Chronic granulomatous
disease: Clinical, functional, molecular, and genetic studies. The
Israeli experience with 84 patients. Am J Hematol. 92:28–36. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kuhns DB, Alvord WG, Heller T, Feld JJ,
Pike KM, Marciano BE, Uzel G, DeRavin SS, Priel DA, Soule BP, et
al: Residual NADPH oxidase and survival in chronic granulomatous
disease. N Engl J Med. 363:2600–2610. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Newburger PE, Cohen HJ, Rothchild SB,
Hobbins JC, Malawista SE and Mahoney MJ: Prenatal diagnosis of
chronic granulomatous disease. N Engl J Med. 300:178–181. 1979.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kulkarni M, Gupta M and Madkaikar M:
Phenotypic prenatal diagnosis of chronic granulomatous disease: A
useful tool in the absence of molecular diagnosis. Scand J Immunol.
86:486–490. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Newburger PE: Comment on: Phenotypic
prenatal diagnosis of chronic granulomatous disease: A useful tool
in the absence of molecular diagnosis. Scand J Immunol. 87:572018.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ahlin A and Fasth A: Chronic granulomatous
disease-conventional treatment vs. hematopoietic stem cell
transplantation: An update. Curr Opin Hematol. 22:41–45. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Norman M, David C, Wainstein B, Ziegler
JB, Cohn R, Mitchell R, O'Brien T, Russell S, Trahair T, Trickett
A, et al: Haematopoietic stem cell transplantation for primary
immunodeficiency syndromes: A 5-year single-centre experience. J
Paediatr Child Health. 53:988–994. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
De Ravin SS, Li L, Wu X, Choi U, Allen C,
Koontz S, Lee J, Theobald-Whiting N, Chu J, Garofalo M, et al:
CRISPR-Cas9 gene repair of hematopoietic stem cells from patients
with X-linked chronic granulomatous disease. Sci Transl Med.
9(pii): eaah34802017. View Article : Google Scholar : PubMed/NCBI
|