Introduction
Agkistrodon acutus is one of the most common
venomous snakes in China and Vietnam (1). A. acutus venom contains
metalloproteinases, phospholipase C-type lectin-like proteins and
serine proteases (2,3). Snakebites of A. acutus can cause
acute reactions, including tissue inflammation, edema, necrosis and
hemorrhage, as well as clotting abnormalities, which can induce
multiple organ failure (4,5).
The treatment of A. acutus bites remains a
controversial topic. Conventionally, animal-derived antivenom is
administered to patients, but the clinical use of animal-derived
antivenom has been limited due to frequent allergic reactions and
serum sickness (6). In the last
decade, antigen-specific monoclonal antibodies have been applied
therapeutically as antivenoms (7).
However, the clinical application of monoclonal antibodies derived
from animal sources is limited by immunogenicity and short
half-life (8). Genetically
engineered antibodies have been constructed to reduce
immunogenicity and enhance performance (9). A basic functional unit of the antibody,
the single-chain variable fragment (scFv) region, confers antigen
specificity. Expressed scFv proteins possess advantages such as
lower molecular weight, improved tissue penetration, and the
potential to be prepared in vitro (10). Phage libraries of scFv gene
repertoires are powerful tools for the isolation and identification
of scFv molecules targeting specific antigens (11).
In the present study, to identify a potential
treatment strategy for A. acutus bites, lymphocytes of two
patients bitten by A. acutus were used to generate an scFv
library, constructed in the T7 phage display system. The affinity
of selected scFv proteins for A. acutus venom was probed by
ELISA. The high-affinity scFv genes were introduced to a
prokaryotic expression vector and functionally expressed in
vitro.
Materials and methods
Ethics approval
The present study was approved by the Ethics
Committee of Southwest Hospital of the Third Military Medical
University (Chongqing, China). All patients provided written
informed consent for their inclusion in the study.
RNA extraction and cDNA synthesis
Two female patients (Patient 1: Female, 21 years
old, admitted to Southwest Hospital Emergency Department on August
4, 2011; Patient 2: Female, 33 years old, admitted to Southwest
Hospital Emergency Department on August 14, 2011) who were
hospitalized within 24 h of being bitten by A. acutus were
recruited to the current study. The patients were then treated with
10 tablets (0.4 g/tablet) every 6 h of oral Jidesheng medicine
(Jinghua Pharmaceutical Group Co., Ltd., Nantong city, China), a
traditional Chinese snakebite medicine, together with rabies virus
vaccine injection (Shanghai Serum Bio-technology Co., Ltd.,
Shanghai city, China). Blood samples (5 ml) taken respectively at
48 and 96 h after treatments were pooled, and lymphocytes were
isolated using Lymphocyte Cell Separation Medium (Sangon Biotech
Co., Ltd., Shanghai, China), according to the manufacturer's
protocol. Total RNA was extracted from isolated lymphocytes using
TRNzol-A+ Reagent (Tiangen Biotech Co., Ltd., Beijing, China),
according to the manufacturer's protocol, then mRNA was purified
from extracted total RNA using Oligotex mRNA Mini kits (Qiagen
GmbH, Hilden, Germany). Different purify cDNA was prepared
respectively using reverse transcription using M-MLV Reverse
Transcriptase (Invitrogen; Thermo Fisher Scientific, Inc., Waltham,
MA, USA) with random hexamers starting with 1–5 µg of total RNA or
mRNA to a final volume of 20 µl.
Generation of human scFv gene
repertoire
Primers for PCR amplification of human variable
regions of light (VL) and heavy (VH) chain genes were designed
according to the degenerate primers described by Sblattero et
al (12). The 5′ ends of the
human VL forward primers and VH reverse primers were modified
include EcoRI and HindIII sites, respectively, for
ligation into T7Select 10–3b DNA vector (EMD Millipore, Billerica,
MA, USA). In order to splice the VL and VH amplicons, a
complementary overlapping sequence (Linker-F and Linker-R) encoding
a flexible linker of 12 amino acids was added to the 5′ ends of the
human VL reverse primers and VH forward primers (13). Primers were designed to generate scFv
genes using splicing by overlap extension-polymerase chain reaction
(SOE-PCR) (Table I). PCR
amplification of the human VL and VH genes was performed in a 50 µl
mixture containing 10 nM of each forward and reverse primer, 4 µl
cDNA, 0.25 mM each dNTP mixture, 10 µl 5×PrimeSTAR®
Buffer (Mg2+ plus) and 0.5 units PrimeSTAR®
polymerase (Takara Biotechnology Co., Ltd., Dalian, China). The
amplification conditions comprised an initial denaturation at 95°C
for 5 min, followed by 35 PCR cycles of 95°C for 20 sec, 53°C for
20 sec and 72°C for 30 sec, and a final extension step of 72°C for
5 min. All amplified VL and VH genes were purified using the
Qiaquick Gel Extraction kit (Qiagen GmbH) in accordance with the
manufacturer's protocol. The human scFv gene was generated by
SOE-PCR. Briefly, the SOE-PCR reaction contained 100 ng of purified
VL products and 100 ng of purified VH products, 10 nM of VH-scFv-F
and VL1/VL2-scFv-R primers, 0.25 mM each dNTP mixture,
1×PrimeSTAR® Buffer (Mg2+ plus) and 0.5 units
PrimeSTAR® polymerase. The cycling conditions for
SOE-PCR were identical to those aforementioned. Total PCR products
were subjected to 1.5% agarose gel electrophoresis and then
visualized with ethidium bromide.
 | Table 1.Primer sequences for PCR and splicing
by overlap extension-PCR. |
Table 1.
Primer sequences for PCR and splicing
by overlap extension-PCR.
| Primer | Sequence
(5′-3′) |
|---|
| VH-01-F |
TCGAGCGAATTCTCAGGTGCAGCTGCAGGAGTCSG |
| VH-02-F |
TCGAGCGAATTCTGAGGTGCAGCTGKTGGAGWCY |
| VH-03-F |
TCGAGCGAATTCTCAGGTGCAGCTGGTGSARTCTGG |
| VH-01-R |
GCCTCCACCTGATGAGGAGACRGTGACCAGGGT |
| VH-02-R |
GCCTCCACCTGACGATGGGCCCTTGGTGGARGC |
| VH-03-R |
GCCTCCACCTGAGGTTGGGGCGGATGCACTCC |
| VL1-01-F |
GGTGGAGGCTCGGATATTGTGMTGACBCAGWCTCC |
| VL1-02-F |
GGTGGAGGCTCGCAGTCTGTSBTGACGCAGCCGCC |
| VL1-01-R |
ATGGTCAAGCTTTTTGATYTCCASCTTGGTCC |
| VL1-02-R |
ATGGTCAAGCTTTTTAATCTCCAGTCGTGTCC |
| VL2-01-F |
GGTGGAGGCTCGCAGCCTGTGCTGACTCARYC |
| VL2-02-F |
GGTGGAGGCTCGCAGDCTGTGGTGACYCAGGAGCC |
| VL2-03-F |
GGTGGAGGCTCGTCCTATGAGCTGAYRCAGCYACC |
| VL2-01-R |
ATGGTCAAGCTTTAGGACGGTSASCTTGGTCC |
| VL2-02-R |
ATGGTCAAGCTTGAGGACGGTCAGCTGGGTGC |
| VH-scFv-F |
ACGTTATCCTCGAGCGAATTCTCAGGTG |
| V1-scFv-R |
ACGGAAGTTATGGTCAAGCTTTTT |
| V2-scFv-R |
ACGGAAGTTATGGTCAAGCTTTAGGAC |
| Linker-F |
TCAGGTGGAGGCGGTTCTGGCGGAGGTGGCTCAGGCGGTGGAGGCTCG |
| Linker-R |
CGAGCCTCCACCGCCTGAGCCACCTCCGCCAGAACCGCCTCCACCTGA |
Cloning of human scFv gene repertoire
into T7Select10-3b vector
The human scFv gene products were digested with
restriction enzymes EcoRI and HindIII (New England
BioLabs, Inc., Ipswich, MA, USA). After purification of the
digested products, cohesive ligation of the scFv gene products
(0.12 pM) with T7Select 10-3b EcoRI/HindIII Vector
Arms (0.04 pM) (Merck, USA) was performed using T4 DNA Ligase (New
England BioLabs, Inc.) at 16°C for 12 h. The ligation products were
added directly to 25 µl T7 Packaging Extracts (EMD Millipore) and
incubated for 2 h at 22°C for in vitro packaging. Sterile
lysogeny broth (270 µl) was added to stop the reaction. The primary
scFv library was amplified by liquid lysate amplification according
to the T7Select system manual. The titers of the primary and
amplified library were determined by plaque assay, as described by
the system manual.
ScFv library screening
To screen for antigen-specific scFv that bound to
A. acutus venom expressed in T7 phages, biopanning was
performed to enrich venom-specific scFv phages according to the
T7Select system manual. A. acutus venom protein was
purchased from Guduo Biotechnology Inc. (Shanghai, China). Venom
protein was dissolved in PBS to yield a stock solution of 10 mg/ml,
and diluted in PBS to 100 ng/ml for biopanning. A total of 12 phage
clones were selected from output phages of the fourth round of
biopanning, and their reactivity with venom protein was analyzed by
phage ELISA (14).
Phage ELISA
Briefly, ELISA plates were coated with venom protein
at a concentration of 10 µg/ml in 100 µl Coating buffer (15 mM
Na2CO3, 35 mM NaHCO3, pH 9.6) per
well overnight at 4°C. After soaking each well of the plate three
times for 2 min with 200 µl PBS containing 0.05% Tween-20 (PBST),
the plate was blocked with PBST containing 5% bovine serum albumin
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) at room temperature
for 4 h. Each well was then soaked with PBST three times in the
conditions as aforementioned. Selected phages (1×108
pfu) were added to each well and incubated at 37°C for 1 h. Bound
phages were detected by horseradish peroxidase (HRP)-conjugated
anti-T7 tag antibody (cat no. 69084; Merck KGaA), followed by a
coloration reaction using the substrate
3,3′5,5′-tetramethylbenzidine (TMB; Tiangen Biotech Co., Ltd.).
Absorbance at 450 nm was measured using a Synergy HT spectrometer
(BioTek Instruments, Inc., Winooski, VT, USA). The phages generated
by the T7Select control insert were used as negative controls. The
sample OD450 value/negative control OD450 value was determined, and
values >2 were considered positive.
Sequencing, expression and
purification of scFv
According to T7Select® System Manual, the
phages with positive signal were amplified by PCR, which was
performed in a 50 µl mixture containing: 10 nM sequencing primers
T7selectUP and T7selectDOWN (Novagen, Merck, Germany) 2 µl phage
(>1×108 pfu), 0.25 mM of each dNTP mixture, 10 µl 5
×PrimeSTAR® Buffer (Mg2+ plus) and 0.5 units
PrimeSTAR® polymerase (Takara Biotechnology Co., Ltd.).
The amplification conditions comprised an initial denaturation at
95°C for 5 min, followed by 35 PCR cycles at 95°C for 15 sec, 50°C
for 15 sec and 72°C for 30 sec, and a final extension step at 72°C
for 5 min. Products of PCR were sent to BGI Corporation (Shenzhen,
China) for DNA sequencing. Positive PCR products and the pET28a (+)
vector (Novagen) were digested with restriction enzymes EcoR
I and Hind III. Then the digested vector and PCR products
were ligated with T4 DNA ligase in a ratio of 1:3 overnight at 16°C
The Escherichia coli BL21(DE3) competent cells were
transformed with the ligation product and cultured on an LB medium
plate (10 g/l Tryptone, 5 g/l Yeast Extract, 10 g/l NaCl, 1.5%
Agar; Sangon Biotech Co., Ltd.) containing 50 µg/ml kanamycin for
12 h at 37°C to grow a positive colony that included the
pET28a(+)-scFv recombinant plasmid. Positive colonies were selected
and cultured on 200 ml auto-induction medium (12 g/l tryptone, 24
g/l yeast extract, 0.8% glycerol, 5 g/l lactose, 0.15 g/l glucose,
2 mM MgSO4, 0.38% aspartic acid, 17 mM
KH2PO4, 72 mM K2HPO4)
at 25°C. At 24 h after auto-induction culture, the cells were
harvested by centrifugation (4,200 × g, 4°C, 10 min), suspended in
ice-cold lysis buffer (50 mM NaH2PO4, 300 mM
NaCl and 5 mM imidazole, 1 mM phenylmethylsulfonyl fluoride, pH
8.0) and lysed by sonication (10 times, 4°C, 10 sec interval).
After centrifugation at 10,000 × g for 20 min at 4°C, the scFv
protein in soluble fraction was purified using Ni-nitrilotriacetic
acid (NTA) agarose beads (Qiagen GmbH) at 4°C. Briefly, 1 ml of
Ni-NTA agarose was loaded into a QIAGEN-tip 500 column (Qiagen
GmbH) and equilibrated with 10 ml of lysis buffer [50 mM
NaH2PO4, 300 mM NaCl and 5 mM imidazole (pH
8.0)]. The soluble fraction was loaded onto the equilibrated
columns. After loading, the column was washed with 50 ml of washing
buffer (50 mM NaH2PO4, 300 mM NaCl and 20 mM
imidazole, pH 8.0). The bound scFv protein was eluted with 5 ml of
elution buffer (50 mM NaH2PO4, 300 mM NaCl
and 250 mM imidazole, pH 8.0) and 1 ml fractions were collected.
The fractions were then dialyzed against PBS for 3 h. The protein
concentration was determined using a BCA protein assay kit
(Beyotime Institute of Biotechnology, Haimen, China).
SDS-PAGE and western blot
analysis
Purity of the Ni-NTA-purified scFv was examined by
SDS-PAGE and western blot analysis. 20 µg purified proteins were
separated on a 12% polyacrylamide gel at a constant electric
current of 15 mA for 1.5 h and visualized by Coomassie blue
staining. For western blot analysis, the proteins were transferred
onto nitrocellulose membranes using the wet transfer method at a
constant voltage of 80 V for 2 h. The membrane was blocked with
PBST containing 5% non-fat milk at room temperature for 1 h, then
the blot was probed overnight at 4°C with a mouse anti-6×His tag
antibody (cat no. CW0082A; CWBio, Beijing, China) at 1:2,000
dilution. Following incubation with an HRP-conjugated goat
anti-mouse IgG antibody (cat no. A0216; Beyotime Institute of
Biotechnology) at 1:10,000 dilution for 2 h at room temperature,
immune reactive bands were detected with an enhanced
chemiluminescence kit (EMD Millipore).
ELISA analysis of the activity of
anti-venom scFv
ELISA plates were coated with venom protein and
utilized for the assay as described above. ScFv protein (2 µg) was
added to each well and incubated at 37°C for 2 h. After washing
with PBST three times in conditions as aforementioned, Horseradish
peroxidase-labeled goat anti-human IgG (H+L; cat. no. A0201;
Beyotime, China) was added to the plate and incubated at 37°C for 1
h. After washing with PBST three times in conditions as
aforementioned, bound scFv protein was detected by a colorimetric
assay with TMB as the substrate. Absorbance at 450 nm was measured.
Horseradish peroxidase-labeled goat anti-human IgG (H+L) was used
as the negative control and PBS solution was used as the blank
control. The sample OD450 value/negative control OD450 value was
determined, and values >2 were considered positive.
Surface plasmon resonance
analysis
The binding kinetics of soluble scFv and venom were
analyzed using BiacoreX (GE healthcare Life Sciences, Little
Chalfont, UK). The Kd value of each purified scFv
was calculated.
Results
Amplification of the VL and VH
genes
Total mRNA, extracted and purified from the
lymphocytes of patients, in addition to total RNA, was used as a
template for reverse transcription of a cDNA substrate, then the VL
and VH genes were amplified by PCR. As described previously, mRNA
was a preferable substrate for specific and efficient
amplification, in comparison with total RNA (14). Therefore the PCR products amplified
from mRNA were used for library cloning. The major VL and VH PCR
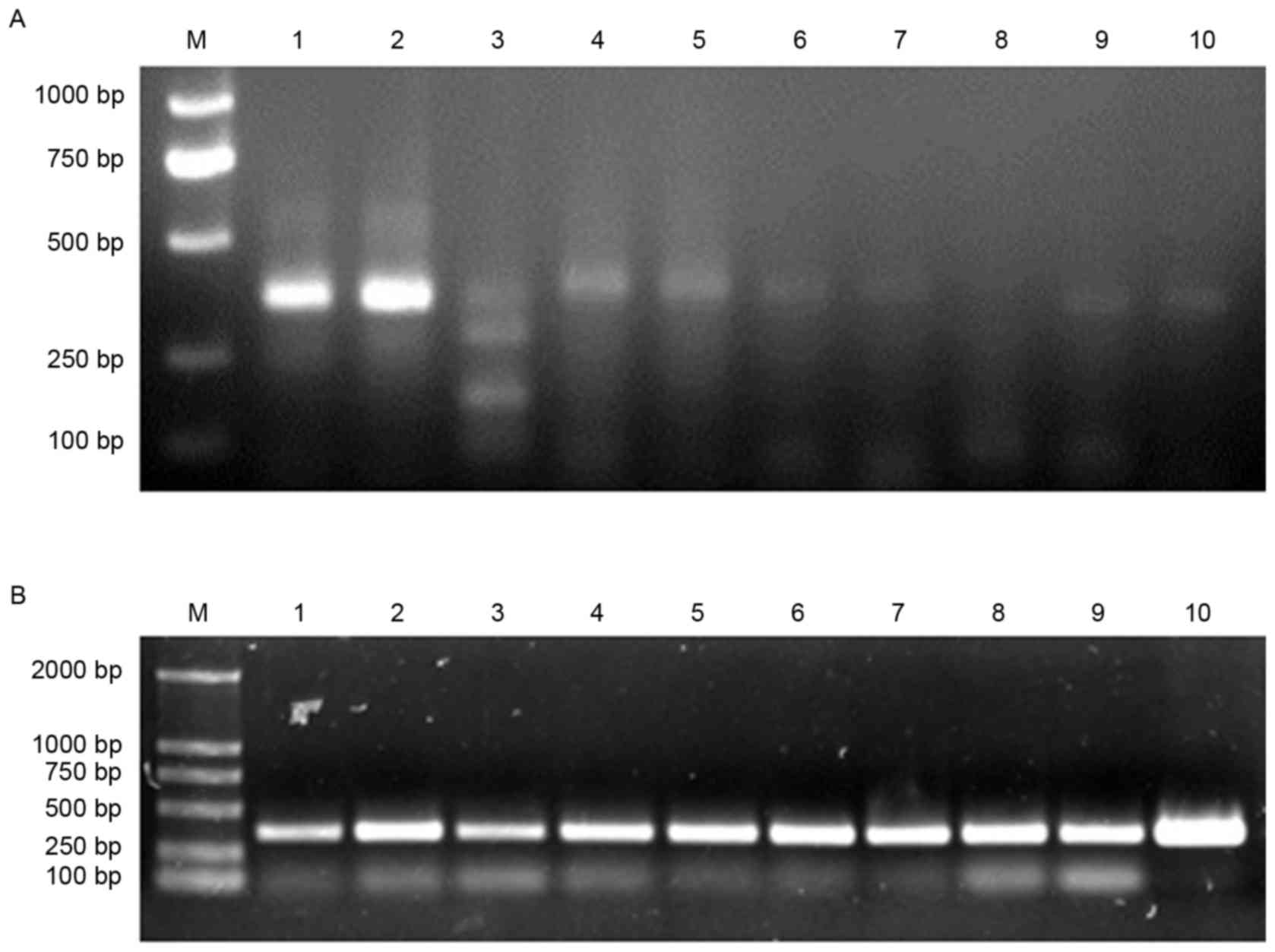
product sizes were approximately 360 bp in length (Fig. 1). All VL and VH genes amplified by
reverse transcription PCR were mixed and purified by gel filtration
(Fig. 2A). Then, SOE-PCR was
performed to generate full human scFv genes. The resulting PCR
products were approximately 750 bp in length (Fig. 2B).
Generation and screening of the scFv
library
The primary scFv library was generated through
cloning the scFv gene repertoire into the T7Select10-3b vector and
in vitro packaging. The primary library was amplified by
liquid lysate method. The titer of the primary library was
4.6×109 pfu/ml. The A. acutus venom-specific
clones were enriched by a biopanning procedure, which was performed
four times, and the resultant titer of enriched phages was
5.2×1014 pfu/ml.
Identification of positive scFv
genes
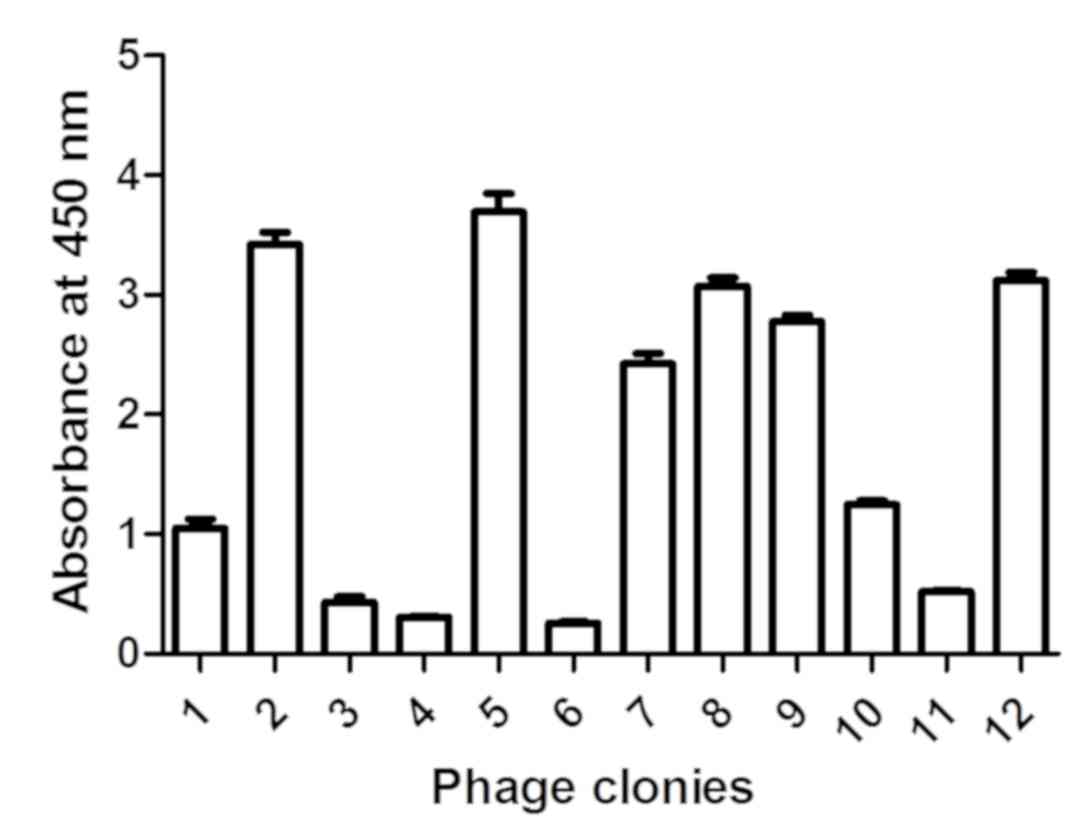
A total of 12 clones were selected from the fourth
generation phage library, and the affinity of these clones for
venom was assessed by phage ELISA. Of these phages, 50% exhibited
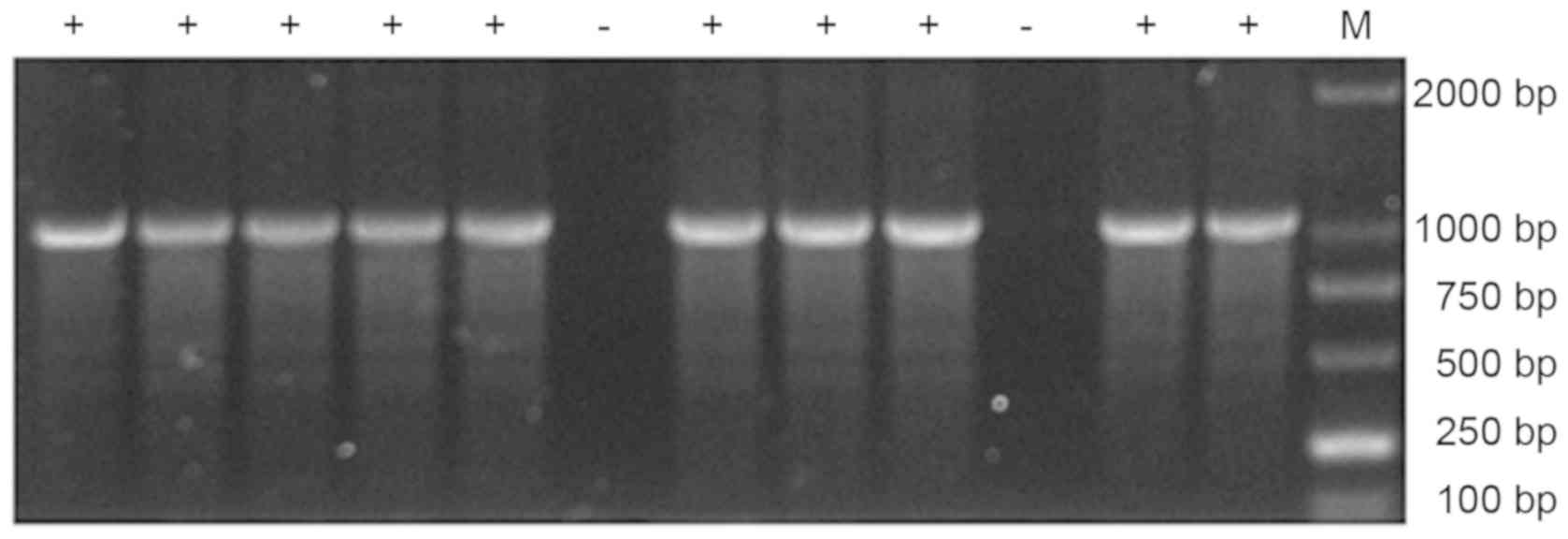
affinity for venom (Fig. 3). PCR
products of these phage clones were analyzed by agarose gel
electrophoresis (Fig. 4) and
positive products were sequenced. DNA sequence alignment
demonstrated that positive scFv genes were heterozygote forms of
the VL-Linker (12 animo acids)-VH (data not shown).
Soluble expression of scFv and
evaluation of affinity to venom
Four scFv genes (B1-05, E1-03, E1-08 and
G1-09) were cloned in multiple cloning sites of the pET-28a (+)
vector and four scFv proteins were expressed in E. coli BL21
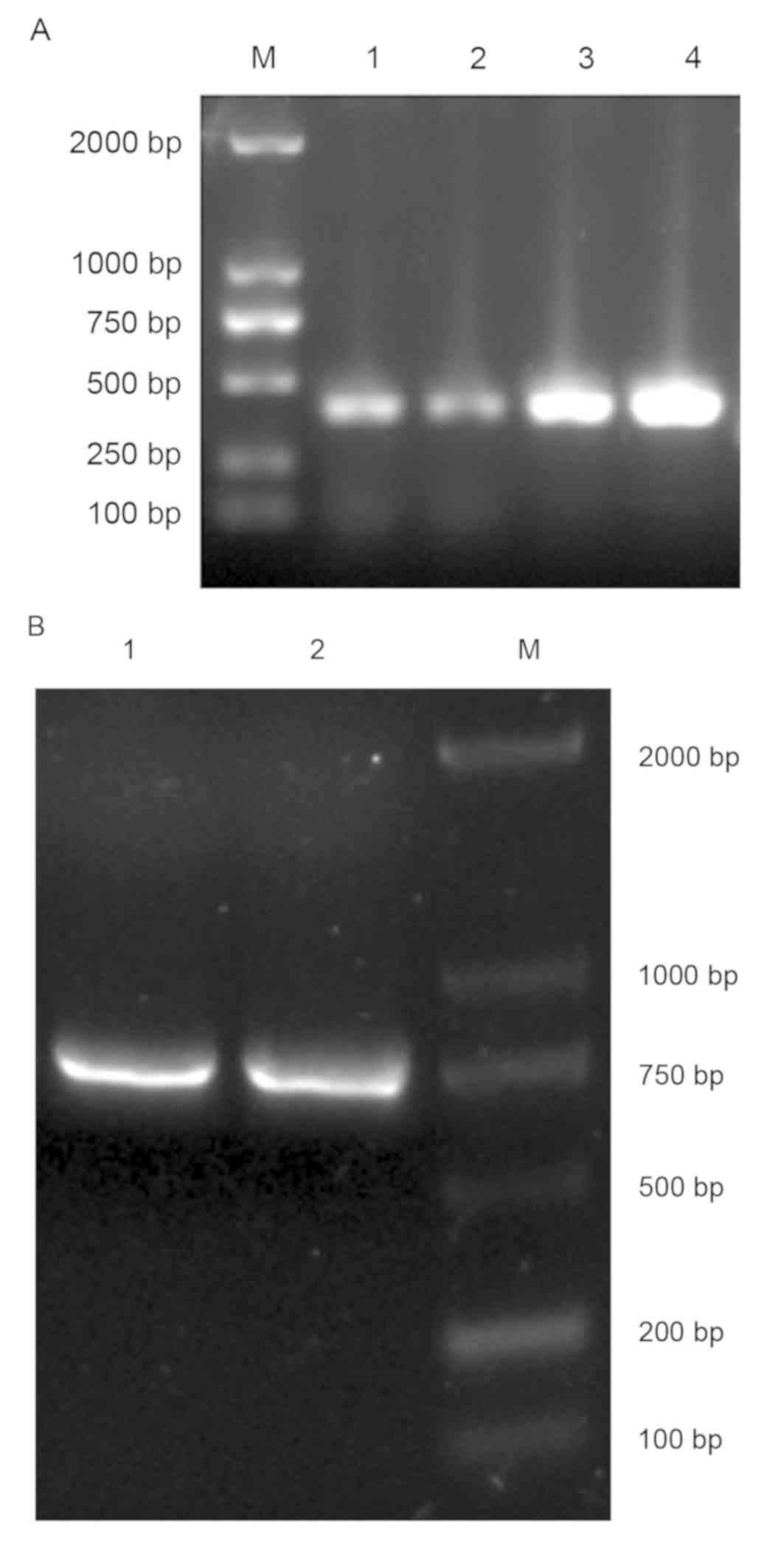
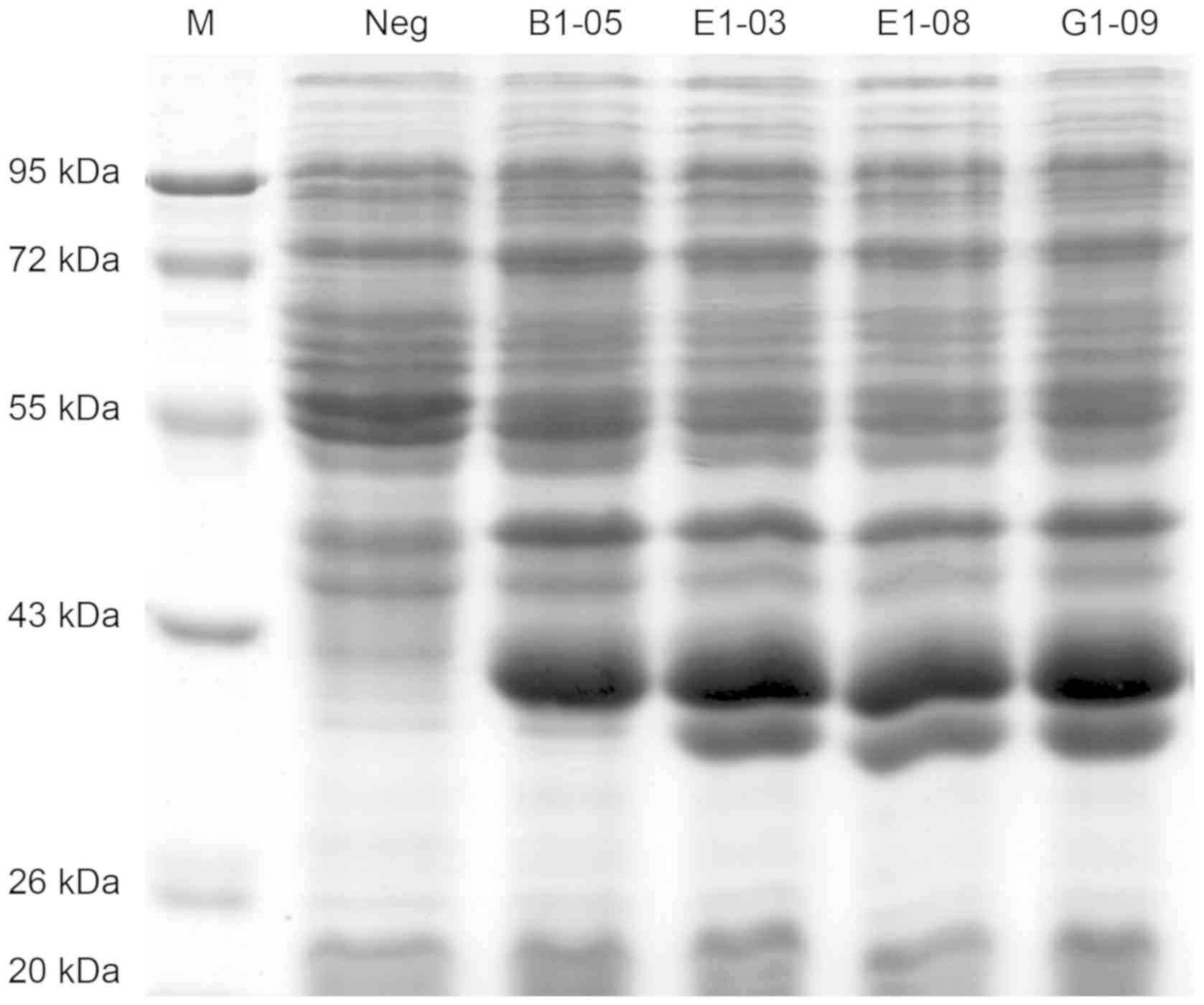
(DE3) by auto-induction (Fig. 5).
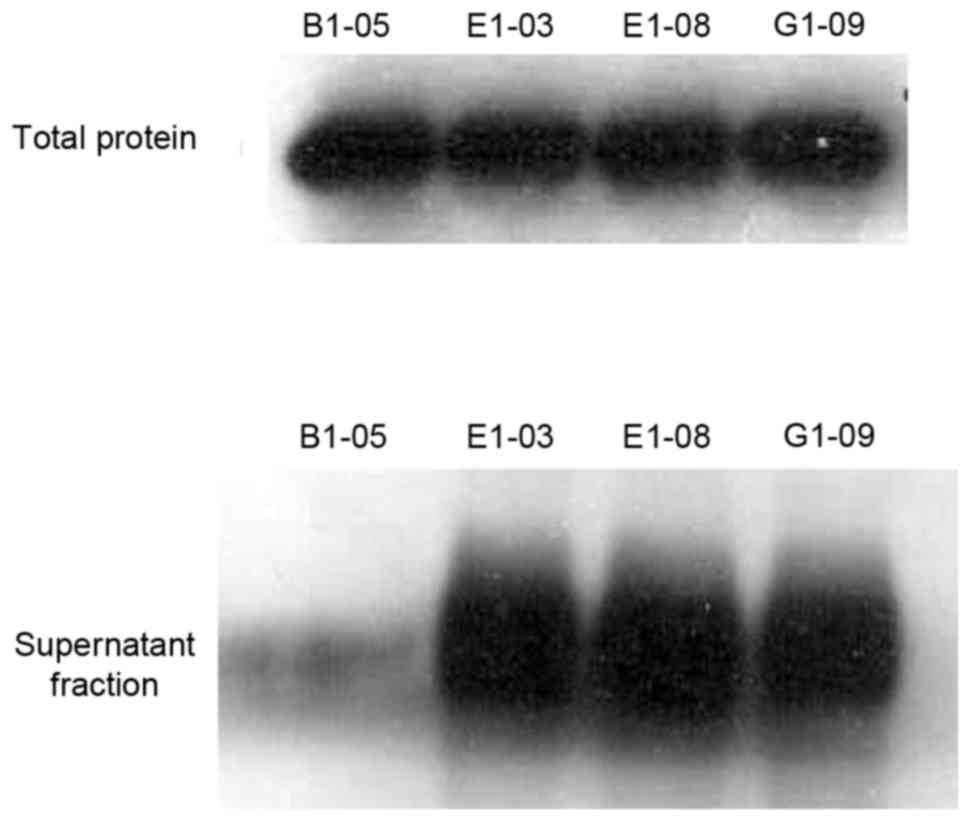
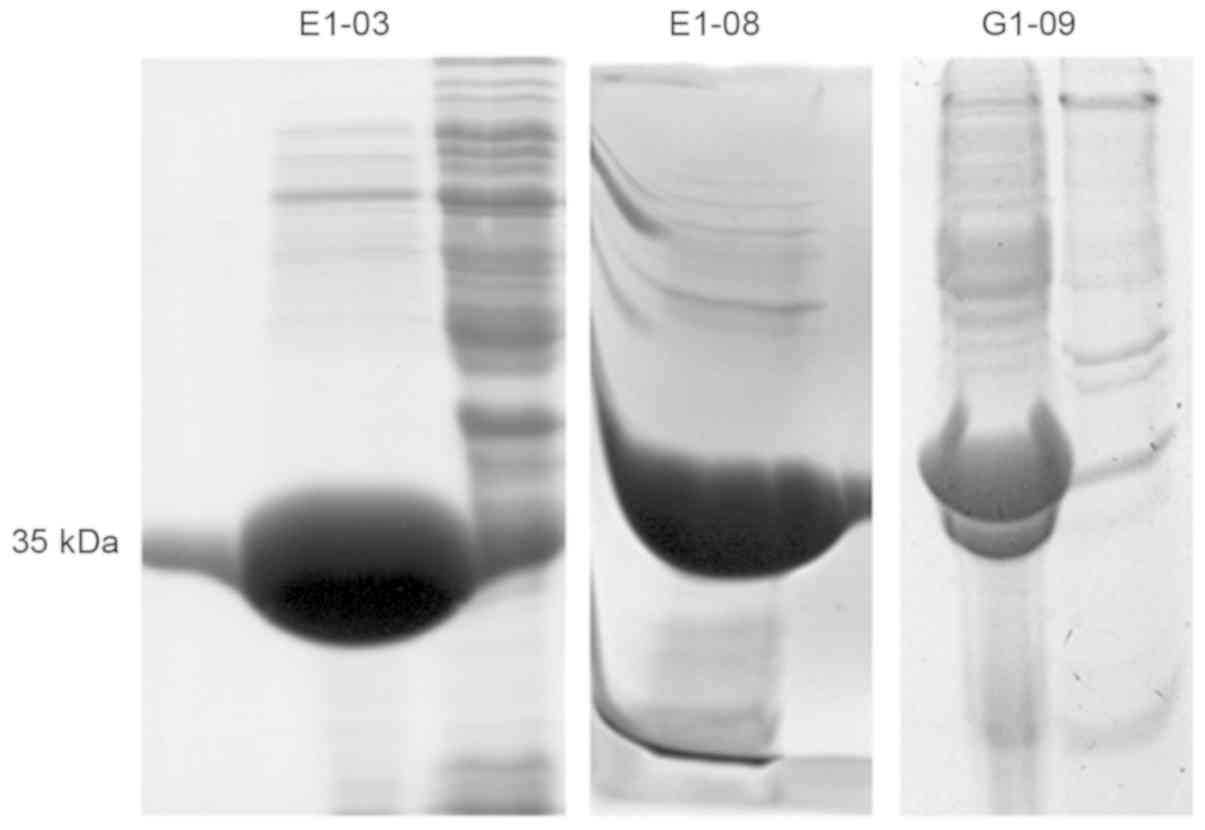
Three of the four proteins exhibited solubility (Fig. 6) and could be purified and
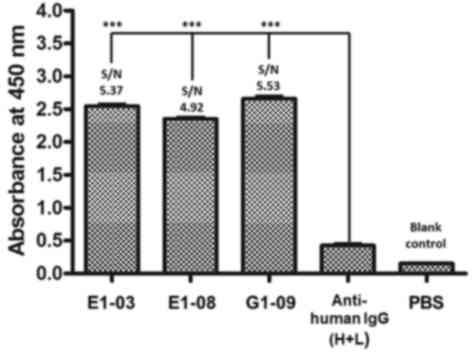
concentrated using a Ni-NTA agarose column (Fig. 7). ELISA results as shown in Fig. 8, PBS is blank control, anti-human IgG
(H+L) is negative control, three monomeric scFv showed positive
reactions (S/N value > 2), demonstrated that scFv peptides
possessed specific affinity for A. acutus venom.
Affinity analysis of anti-venom scFv
by surface plasmon resonance
The affinity of the purified monomeric scFv and
venom was analyzed using BiacoreX. As presented in Table II, the Kon,
Koff, and Kd values of
different scFvs were examined. The Kd value
varied between 27.06 and 39.11 nmol/l, indicating that the selected
scFv exhibited a higher affinity activity.
| Table II.Affinity analysis of anti-venom scFv
by surface plasmon resonance. |
Table II.
Affinity analysis of anti-venom scFv
by surface plasmon resonance.
| scFv | Kon
(1/ms) | Koff
(1/s) | Kd
(nmol/l) |
|---|
| E1-03 |
3.38×105 |
4.62×10−3 | 30.58 |
| E1-08 |
3.22×105 |
1.84×10−2 | 39.11 |
| G1-09 |
2.74×106 |
6.39×10−2 | 27.06 |
Discussion
Natural A. acutus venom contains complex
antigenic components (5), so
therapeutic antibodies have thus far proven difficult to isolate
using monoclonal antibody technology. ScFv cloning is an
alternative method by which antigen-specific fusion immunoglobulin
proteins can be generated. Cloning of scFv libraries has been
widely used to generate specific scFv, which exhibit improved
pharmacokinetic properties compared with an intact antibody,
including better tissue penetration and rapid blood clearance
(15–17). The present study aimed to identify
A. acutus venom-specific antibodies. For this purpose, mRNAs
were purified from the lymphocytes of patients who had been bitten
by A. acutus. The immune response to the venom was likely to
have amplified the repertoire of toxin-specific immunoglobulin
genes. The amplicons of the VL and VH domains were connected by a
flexible linker of 12 amino acids by SOE-PCR, generating an scFv
gene library, which was then packaged in the T7Select Phage Display
system. Unlike other phage display systems, the T7Select system is
capable of displaying non-membrane proteins. In addition, due to
the small size of scFv PCR products, non-membrane proteins can be
well presented in the T7Select system (18–21).
After four rounds of biopanning, specific anti-venom scFv
presenting phages were enriched. Of the 12 randomly selected scFv
clones, 50% exhibited affinity for venom in ELISA. DNA sequencing
demonstrated that the sequences of these scFv clones varied (data
not shown), further demonstrating the complexity of A.
acutus venom.
To confirm that the encoded proteins of these scFv
clones exhibited specificity to venom, four scFv sequences were
selected from sequencing data to perform prokaryotic expression in
E. coli BL21(DE3). In previous reports, recombinant scFv
protein is often present in the form of an inclusion body protein
during prokaryotic expression in E. coli; however, this
original structure and its activity are lost following inclusion
body renaturation (22–25). In the present study, due to high
expression levels and a high probability of solubility for foreign
proteins, the method of auto-induction was substituted for
isopropyl β-D-1-thiogalactopyranoside (IPTG)-induction for protein
expression (26). Indeed, the four
scFv proteins selected exhibited insoluble expression or no
expression in a preliminary experiment with IPTG-induction (data
not shown). By contrast, three scFv proteins were purified using
Ni-NTA chromatography for soluble expression by auto-induction
culture. Each of these three scFv proteins exhibited a specific
function to detect venom as antibodies in ELISA.
In the present study, an anti-A. acutus venom
scFv phage library was constructed to select specific scFvs against
venom. The recombinant proteins of these scFv genes were also
screened and were determined to be soluble and expressed in E.
coli. These results provide a foundation for the preparation of
humanized therapeutic antibody in vitro to treat A.
acutus bites.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant no. 81071537), the
Tackling Program of Science and Technology of Chongqing (grant no.
CSTC2010AC5026) and the Clinical Innovative Foundation of The Third
Military Medical University (grant no. 2009XLC15).
Availability of data and materials
All data generated or analyzed during the present
study are included in this published article.
Authors' contributions
LZ and YC performed the experiments and prepared the
manuscript, under the supervision of ML. ML designed the current
study and gave technical advice. JT, XC and QX acquired the data
and provided critical advice during manuscript preparation. JT and
ML revised the final text. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Informed consent was obtained in all cases, and
protocols were approved by the ethical committee of Third Military
Medical University (Chongqing, China).
Patient consent for publication
The study was performed with the patients' informed
consent.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Kong Y, Huo JL, Xu W, Xiong J, Li YM and
Wu WT: A novel anti-platelet aggregation tripeptide from
Agkistrodon acutus venom: Isolation and characterization. Toxicon.
54:103–109. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Feng J, Chen T, Zhou M and Shaw C: Cloning
of cDNAs and molecular characterisation of C-type lectin-like
proteins from snake venoms. Toxicon. 60:1363–1369. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jiang W, Ma T, Su X, Qiu P and Yan G:
Enzymatic activities and functional characterization of a novel
recombinant snake venom proteinase from Agkistrodon acutus.
Biochimie. 91:277–287. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Luo S, Wang R, Jiang W, Lin X, Qiu P and
Yan G: A novel recombinant snake venom metalloproteinase from
Agkistrodon acutus protects against taurocholate induced severe
acute pancreatitis in rats. Biochimie. 92:1354–1361. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Villalta M, Pla D, Yang SL, Sanz L, Segura
A, Vargas M, Chen PY, Herrera M, Estrada R, Cheng YF, et al: Snake
venomics and antivenomics of Protobothrops mucrosquamatus and
Viridovipera stejnegeri from Taiwan: Keys to understand the
variable immune response in horses. J Proteomics. 75:5628–5645.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Markland FS and Swenson S: Snake venom
metalloproteinases. Toxicon. 62:3–18. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Azofeifa-Cordero G, Arce-Estrada V,
Flores-Díaz M and Alape-Girón A: Immunization with cDNA of a novel
P-III type metalloproteinase from the rattlesnake Crotalus durissus
durissus elicits antibodies which neutralize 69% of the hemorrhage
induced by the whole venom. Toxicon. 52:302–308. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Stas P and Lasters I: Immunogenicity of
therapeutic antibodies. Med Sci (Paris). 25:1070–1077. 2009.(In
French). View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chames P and Baty D: The future of
antibody fragments, made of a single immunoglobulin domain. Med Sci
(Paris). 25:1159–1162. 2009.(In French). View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cabezas S, Rojas G, Pavon A, Alvarez M,
Pupo M, Guillen G and Guzman MG: Selection of phage-displayed human
antibody fragments on Dengue virus particles captured by a
monoclonal antibody: Application to the four serotypes. J Virol
Methods. 147:235–243. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tang KH, Yusoff K and Tan WS: Display of
hepatitis B virus PreS1 peptide on bacteriophage T7 and its
potential in gene delivery into HepG2 cells. J Virol Methods.
159:194–199. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sblattero D and Bradbury A: A definitive
set of oligonucleotide primers for amplifying human V regions.
Immunotechnology. 3:271–278. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Haidaris CG, Malone J, Sherrill LA, Bliss
JM, Gaspari AA, Insel RA and Sullivan MA: Recombinant human
antibody single chain variable fragments reactive with Candida
albicans surface antigens. J Immunol Methods. 257:185–202. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang S, Zheng C, Liu Y, Zheng H and Wang
Z: Construction of multiform scFv antibodies using linker peptide.
Genet J Genomics. 35:313–316. 2008. View Article : Google Scholar
|
|
15
|
Bhatia S, Gangil R, Gupta DS, Sood R,
Pradhan HK and Dubey SC: Single-chain fragment variable antibody
against the capsid protein of bovine immunodeficiency virus and its
use in ELISA. J Virol Methods. 167:68–73. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Studier WF: Protein production by
auto-induction in high-density shaking cultures. Protein Expr
Purif. 41:207–234. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Monnier PP, Vigouroux RJ and Tassew NG: In
vivo applications of single chain Fv (Variable Domain) (scFv)
fragments. Antibodies. 2:193–208. 2013. View Article : Google Scholar
|
|
18
|
Sun D, Shi H, Chen J, Shi D, Zhu Q, Zhang
H, Liu S, Wang Y, Qiu H and Feng L: Generation of a mouse scFv
library specific for porcine aminopeptidase N using the T7 phage
display system. J Virol Methods. 182:99–103. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sheets MD, Amersdorfer P, Finnern R,
Sargent P, Lindqvist E, Schier R, Hemingsen G, Wong C, Gerhart JC
and Marks JD: Efficient construction of a large nonimmune phage
antibody library: The production of high-affinity human
single-chain antibodies to protein antigens. Proc Natl Acad Sci
USA. 95:6157–6162. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lamberski JA, Thompson NE and Burgess RR:
Expression and puriWcation of a single-chain variable fragment
antibody derived from a polyol-responsive monoclonal antibody.
Protein Expr Purif. 47:82–92. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wan L, Zeng L, Chen L, Huang Q, Li S, Lu
Y, Li Y, Cheng J and Lu X: Expression, purification, and refolding
of a novel immunotoxin containing humanized single-chain fragment
variable antibody against CTLA4 and the N-terminal fragment of
human perforin. Protein Expr Purif. 48:307–313. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Heo MA, Kim SH, Kim SY, Kim YJ, Chung J,
Oh MK and Lee SG: Functional expression of single-chain variable
fragment antibody against c-Met in the cytoplasm of Escherichia
coli. Protein Expr Purif. 47:203–209. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhao Q, Chan YW, Lee SS and Cheung WT:
One-step expression and purification of single-chain variable
antibody fragment using an improved hexahistidine tag phagemid
vector. Protein Expr Purif. 68:190–195. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yang T, Yang L, Chai W, Li R, Xie J and
Niu B: A strategy for high-level expression of a single-chain
variable fragment against TNFα by subcloning antibody variable
regions from the phage display vector pCANTAB 5E into pBV220.
Protein Expr Purif. 76:109–114. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bu D, Zhou Y, Tang J, Jing F and Zhang W:
Expression and purification of a novel therapeutic single-chain
variable fragment antibody against BNP from inclusion bodies of
Escherichia coli. Protein Expr Purif. 92:203–207. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Deckert PM: Current constructs and targets
in clinical development for antibody-based cancer therapy. Curr
Drug Targets. 10:158–175. 2009. View Article : Google Scholar : PubMed/NCBI
|