Introduction
Polycystic liver disease (PLD) is a group of genetic
disorders, which may either only be present in the liver (isolated
PLD), involve the kidney [autosomal-dominant polycystic kidney
disease (ADPKD)], or, as a rare occurrence, be complicated with
congenital hepatic fibrosis (CHF) (1). Regardless of the distinct genetic
mutation, the natural progression is mostly similar between
different types. Most patients with PLD are asymptomatic and only a
minority of patients is symptomatic with persistently normal liver
functions (2). The expansion of
liver cysts causes extensive hepatomegaly and serious abdominal
symptoms. Current medical treatments have indicated potential
therapeutic effects, but further research is still required
(3,4). Liver transplantation remains the only
definitive curative procedure. Other surgical interventions may
relieve symptoms by reducing liver cysts. These palliative
disease-directed interventions are associated with high rates of
recurrence, morbidity and mortality, which may eventually
complicate liver transplantation (5). However, liver transplantation remains
controversial due to the shortage of donors, as well as high
morbidity and mortality. The present study revealed that
appropriately selected patients with extensive hepatic involvement
or HBV-associated decompensated cirrhosis complicated with PLD
treated by liver transplantation had an excellent long-term
prognosis compared with those subjects receiving other surgical
treatments. The present study aimed to explore the indications for
and safety of liver transplantation for PLD.
Materials and methods
Patients
A retrospective analysis of 11 patients with PLD who
were diagnosed at the Third Affiliated Hospital of Sun Yat-Sen
University (Guangzhou, China) between May 2004 and September 2013
was performed. The study was supervised and approved by the Ethics
Committee of the Third Affiliated Hospital, Sun Yat-Sen University
[Guangzhou, China; no. (2018)02-006-011]. Written informed consent
was obtained from all of the patients or the patients' first-degree
relatives.
Examination
The diagnosis of PLD was defined as the presence of
>20 liver cysts revealed by ultrasonography, computed tomography
(CT) scan or magnetic resonance imaging (MRI) scan (6). Furthermore, if PLD presented as the
extrarenal progression in ADPKD, ≥4 liver cysts were regarded as
sufficient for diagnosis (7). The
severity and classification of PLD were defined based on the Gigot
criteria (8). The routine
examinations, including electrocardiography, chest radiograph or
chest CT and blood test, were performed for pre-operative
assessment. The Child-Pugh scores (9,10), as
well as the score of the Short-Form (36) Health Survey (SF-36) questionnaire,
the Eastern Cooperative Oncology Group (ECOG) performance status
(11) and the Clavien-Dindo
classification (12) were determined
as the risk factors of orthotopic liver transplantation (OLT) and
the short- and long-term prognosis.
Treatment and follow-up
The 11 patients underwent modified piggyback OLT or
combined liver-kidney transplantation (CLKTx) according to the
degree of renal dysfunction and the regional anatomical structure
during surgery. A total of six patients (Patients 2, 5, 7 and 9–11)
were indicated to have a severely decreased quality of life (ECOG
≥3). End-stage liver disease progressed from isolated PLD and
hepatitis B virus (HBV)-associated cirrhosis were indications in
the other five patients. Of note, Patients 10 and 11 underwent
CLKTx due to complication with renal dysfunction. The data of the
prior surgical history, warm ischemia time, cold ischemia time,
time of anhepatic phase, volume of ascites, type of surgery,
biochemical or radiologic examinations, post-operative
complications and immunosuppressive treatment were recorded.
Patients were followed up until January 2018.
Statistical analysis
The survival of patients was assessed with a
Kaplan-Meier analysis using GraphPad Prism 6.0 software (version
6.01; GraphPad Software, Inc. San Diego, CA, USA). Measurement data
were expressed as mean ± standard deviation or median
[interquartile range (IQR), Q1-Q3].
Results
Patients characteristics
A total of 11 recipients (6 males and 5 females)
underwent modified piggyback OLT (n=9) or CLKTx (n=2) for PLD
between May 2004 and September 2013 at the Transplantation Center
of the Third Affiliated Hospital of Sun Yat-Sen University
(Guangzhou, China), accounting for 1.09% of all OLTs performed at
our center. Recipient characteristics are summarized in Table I. The recipients' median age was 56
years (IQR, 52–57 years). All recipients underwent pre-operative CT
scan or MRI scan to confirm the diagnosis (6 cases of isolated PLD
and 5 of ADPKD). In fact, among the PLD subgroup, only 1 patient
(Patient 7) was diagnosed with isolated PLD, while the other 5
patients all had a history of hepatitis B, which progressed to
hepatic failure. The hepatitis B of all of those 5 patients was
under effective control by anti-HBV drugs (HBs antigen was negative
and HBV-DNA quantification <100 copies/ml). Abdominal CT scan
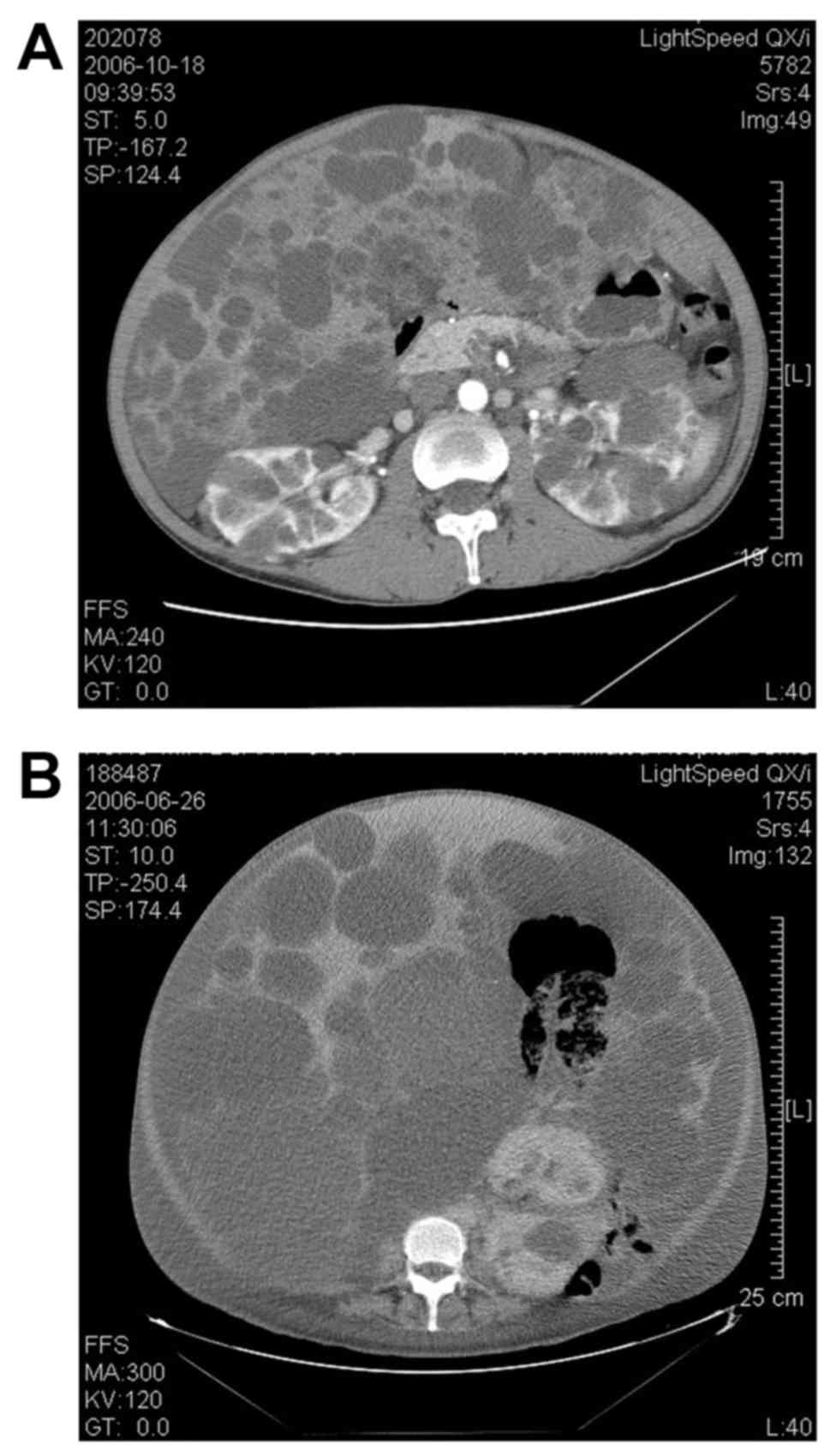
revealed that 7 patients (63.6%) had Gigot type II PLD and 4
(36.4%) had Gigot type III PLD; representative CT scans are
presented in Fig. 1. A total of
eight patients suffered from massive hepatomegaly with abdominal
pain, discomfort, shortness of breath and limited daily activities
(8 had an ECOG score of ≥3 and 3 had an ECOG score of 2). There
were 8 cases (72.7%) of Child-Pugh Class B, and 3 cases (27.3%) of
Child-Pugh Class C. A history of abdominal surgery was present in 5
patients. A total of 4 patients underwent palliative surgical
intervention for PLD. Specifically, Patient 4 underwent segmental
hepatectomy, Patient 7 underwent aspiration-sclerotherapy combined
with fenestration, Patient 9 underwent aspiration-sclerotherapy and
Patient 10 underwent aspiration. Patient 1 underwent splenectomy
due to hypersplenism associated with chronic hepatitis B.
 | Table I.Patient characteristics. |
Table I.
Patient characteristics.
| Patient no. | Age (years) | Sex | Diagnosis | Child-Pugh
score | ECOG score | Prior
procedures |
|---|
| 1 | 64 | Male | Isolated PLD,
hepatitis B | C | 3 | Splenectomy |
| 2 | 49 | Female | ADPKD | C | 4 |
|
| 3 | 52 | Male | Isolated PLD,
hepatitis B | B | 2 |
|
| 4 | 57 | Male | Isolated PLD,
hepatitis B | B | 2 | Segmental
hepatectomy |
| 5 | 47 | Male | ADPKD | B | 3 |
|
| 6 | 54 | Male | Isolated PLD,
hepatitis B | B | 2 |
|
| 7 | 57 | Male | Isolated PLD | B | 3 |
Aspiration-sclerotherapy;
fenestration |
| 8 | 68 | Female | Isolated PLD,
hepatitis B | B | 3 |
|
| 9 | 56 | Female | ADPKD | C | 3 |
Aspiration-sclerotherapy |
| 10 | 56 | Female | ADPKD | B | 4 | Aspiration |
| 11 | 57 | Female | ADPKD | B | 3 |
|
Peri-operative period
During the liver transplantation period, the median
ascites volume was 1,000 ml (IQR, 600–4,000 ml), the mean warm
ischemia time was 4.36±1.74 min, the mean cold ischemia time was
7.73±2.53 h, and the anhepatic phase time was 43.91±12.53 min;
detailed data are presented in Tables
II and III. In Patient 2, the
difficult explantation of the recipient liver resulted in massive
hemorrhage and increased operation time during the separation
procedure of the hepatic ligaments. The mean post-operative
hospitalization duration was 45.4±15.3 days and the mean length of
stay at the intensive care unit was 4.1±1.9 days.
Methylprednisolone combined with Tacrolimus was used for basic
immunosuppressive management. Patient 4 exhibited an improved
therapeutic response upon addition of Rapamycin. Mycophenolate
mofetil was added due to the high level of serum creatinine in
Patients 6 and 7. According to the Clavien-Dindo classification
system, six patients were classified as grade II based on total
parenteral nutrition post-transplantation. In particular, Patient 9
experienced early acute rejection after OLT, which was effectively
managed by increasing the Tacrolimus dose and providing steroid
hormone aggressive therapy. A total of 2 patients were classified
to grade IIIa for intrahepatic or extrahepatic bile duct stricture.
The condition in Patient 1 was temporarily relieved by endoscopic
retrograde cholangiopancreatography and Patient 6 was cured by
percutaneous transhepatic cholangiodrainage combined with balloon
dilatation. Patient 5, who was classified as grade IVa, was
subjected to re-transplantation due to hepatic artery stricture at
45 days after the first OLT. Patients 10 and 11 were classified as
grade V due to death within 1 month post-CLKTx. Patient 10 died of
multiple organ dysfunction syndrome (MODS) induced by fungal
pneumonia and Patient 11 died of primary non-function (PNF)
progressed from acute kidney rejection.
| Table II.Peri-operative characteristics. |
Table II.
Peri-operative characteristics.
| Patient no. | Procedure | Ascites (ml) | WIT (min) | CIT (h) | Anhepatic phase
(min) | Clavien-Dindo
classification | Immunosuppressive
regimen |
|---|
| 1 | Modified piggyback
OLT | 3,000 | 4 | 7 | 45 | IIIa | MP +
Tacrolimus |
| 2 | Modified piggyback
OLT | 6,000 | 4 | 12 | 48 | II | MP +
Tacrolimus |
| 3 | Modified piggyback
OLT | 2,000 | 3 | 5 | 45 | II | MP +
Tacrolimus |
| 4 | Modified piggyback
OLT | 700 | 3 | 9 | 50 | II | MP + Tacrolimus +
Rapamycin |
| 5 | Modified piggyback
OLT | 1,000 | 3 | 10 | 30 | IVa | MP +
Tacrolimus |
| 6 | Modified piggyback
OLT | 600 | 5 | 11 | 30 | IIIa | MP + Tacrolimus +
MMF |
| 7 | Modified piggyback
OLT | 900 | 5 | 7 | 70 | II | MP + Tacrolimus +
MMF |
| 8 | Modified piggyback
OLT | 500 | 3 | 8 | 27 | II | MP +
Tacrolimus |
| 9 | Modified piggyback
OLT | 7,000 | 9 | 4 | 55 | II | MP +
Tacrolimus |
| 10 | CLKTx | 4,000 | 4 | 6 | 38 | V | MP +
Tacrolimus |
| 11 | CLKTx | 300 | 5 | 6 | 45 | V | MP +
Tacrolimus |
| Table III.Post-operative morbidity and
mortality. |
Table III.
Post-operative morbidity and
mortality.
| Patient no. | Duration of
hospitalization (days) | ICU stay
(days) | Complications | Intervention | Follow-up | Cause of death |
|---|
| 1 | 48 | 3 | Intrahepatic bile
duct stenosis | ERCP | Dead (122
months) | Ischemia
cholangitis |
| 2 | 42 | 3 |
|
| Alive (137
months) |
|
| 3 | 43 | 3 |
|
| Alive (132
months) |
|
| 4 | 37 | 5 |
|
| Alive (132
months) |
|
| 5 | 62 | 6 | Hepatic artery
strictures | Re-LTx | Alive (33
months) |
|
| 6 | 50 | 3 | Common bile duct
stenosis | PTCD combined
balloon dilatation |
| Alive (123
months) |
| 7 | 44 | 4 |
|
| Alive (111
months) |
|
| 8 | 59 | 3 |
|
| Alive (103
months) |
|
| 9 | 68 | 9 | Acute
rejection | Steroid hormone
aggressive therapy | Alive (50
months) |
|
| 10 | 12 | 3 | Fungal
pneumonia | Anti-infective
therapy | Dead (12 days) | MODS |
| 11 | 34 | 3 | Acute kidney
rejection | Steroid hormone
aggressive therapy | Dead (34 days) | PNF |
Survival following liver
transplantation
All patients received ABO blood type-compatible and
whole-cadaver graft transplantation. The mortality rate was 18.2%
(n=2) and the morbidity rate was 54.5% (n=6) during the
hospitalization period. These included 2 cases of biliary
complications, 1 case of hepatic artery strictures that received
re-transplantation at 45 days post-OLT and 1 case of acute liver
rejection. Of note, there was 1 case of fungal pneumonia (Patient
10), who died due to MODS at 12 days post-CLKTx, while another case
(Patient 11) died of PNF at 34 days post-CLKTx. The median
follow-up period was 111 months (IQR, 33–132 months). Only Patient
1 died due to ischemia cholangitis at 124 months post-OLT.
According to the SF-36 questionnaire, the mean physical component
summary (PCS) score was 87.1±6.9 and the mean mental component
summary (MCS) score was 81.5±6.4, which indicated that the
surviving patients were capable of normal activity, with only a few
symptoms or signs of disease (Table
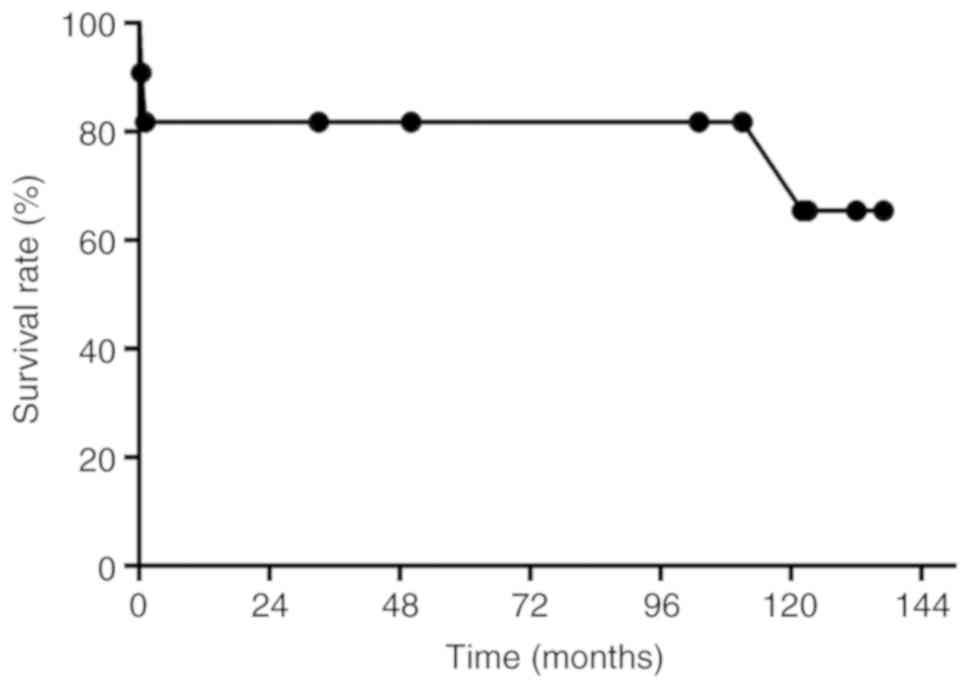
III). The actuarial 1-, 5- and 10-year patient survival rates
during the follow-up period were 81.8, 81.8 and 65.5%, respectively
(Fig. 2).
Discussion
PLD is a rare congenital disorder, which is
pathologically developed from isolated hepatic cysts (isolated
PLD), advanced secondary to kidney cysts (ADPKD), Caroli's disease
or CHF. Regarding the genetic diagnosis of primary PLD, mutations
in three genes, protein kinase substrate 80K-H (PRKCSH) (13,14),
SEC63 (15) and low-density
lipoprotein receptor-related protein 5 (16), are known to be associated with the
pathology, with PRKCSH being the most frequent (~15%) (17). As pathogenic genes for ADPKD, PKD1
(~80–85% of cases) and PKD2 (~10–15% of cases) have been identified
(18). In addition, mutations in
PKHD1 are causative for Caroli's disease as well as CHF (19,20). The
diagnostic criterion for primary PLD is usually defined as the
presence of >20 cysts by ultrasonography, CT or MRI scan,
particularly CT or MRI scan. For the diagnosis of ADPKD, >4
liver cysts are sufficient if the patient's family background
includes inherited autosomal dominant isolated polycystic liver
(6,7).
Although isolated PLD and ADPKD are genetically
distinct, the clinical progression is largely similar. Most
patients are initially asymptomatic (21). However, with the continuous expansion
of liver cysts, a small number of patients develops massive
hepatomegaly-associated abdominal symptoms, including pain,
anorexia, dyspnea and decreased mobility (22). Age (35–46 years), sex (female),
exogenous estrogens and multiple pregnancies are high-risk factors
for liver cyst development and progression (23–25). In
general, the levels of γ-glutamyltransferase, alkaline phosphatase
and carbohydrate antigen 19-9 may rise in severe PLD, while the
levels of liver enzymes are normal in the majority of patients.
Other complications induced by massive cysts or chronic liver
disease include cyst infection, ascites, spontaneous peritonitis,
hepatic venous outflow obstruction, inferior vena cava syndrome and
portal vein compression, which may result in irreversible hepatic
failure (26–29).
Treatments for PLD aim to reduce the volume or
numbers of liver cyst-associated complications in symptomatic
patients. The pharmaceutical management may be the primary
intervention applied to slowly progressing patients without severe
complications or to those refusing surgical procedures. Given that
oral estrogens increase liver cyst progression, any estrogen
therapy should be terminated as early as possible (30). Somatostatin analogue inhibits cystic
fluid secretion and proliferation by reducing intracellular levels
of cyclic adenosine monophosphate. According to a randomized
controlled trial reported by van Hogan et al (31) and van Keimpema et al (32), 120 mg lanreotide autogel for 6 months
every 28 days or 40 mg octreotide for 1 year every 28 days achieved
an absolute decrease in liver cysts in patients with isolated PLD,
while somatostatin analogue appeared to have no beneficial effect
in ADPKD patients. Furthermore, certain clinical trials including
mammalian target of rapamycin inhibitor or ursodeoxycholic acid
management also demonstrated positive effects in reducing cyst
volume in PLD (33,34). Although the medical approach provides
a considerable benefit in PLD patients, discontinuation of therapy
would result in immediate recurrence of cyst growth (35).
At present, surgical approaches, including
aspiration and sclerotherapy, fenestration and segmental
hepatectomy are adapted to candidates with isolated large
symptomatic cysts or Gigot type I and II PLD. These palliative
disease-directed interventions are able to effectively relieve
hepatomegaly-associated symptoms, but are linked to a high rate of
recurrence, morbidity and mortality. Of note, the invasive
procedures fail to change the natural course of the disease because
symptoms usually recur due to growth of new cysts or re-growth of
already treated cysts, leading to re-operation. The potential
drawback of the open-abdominal procedure is the increased risk of
intra-abdominal adhesions, which may complicate future liver
transplantation (36).
Liver transplantation is the only curative option in
patients with severe PLD (37). The
indication for transplantation is extremely disabling symptoms that
lead to a severely decreased quality of life. In addition,
untreatable complications are indications for liver
transplantation, including hepatic venous outflow obstruction,
inferior vena cava syndrome, and portal vein compression or Gigot
type III PLD. Following the 2008 meeting of The American Society of
Transplantation, guidelines for simultaneous liver-kidney
transplantation were published by Eason et al (38). These guidelines recommend that CLKTx
should be performed in patients with chronic kidney disease
complicated with portal hypertension or hepatorenal syndrome that
have been on dialysis for >8 weeks and patients with a
glomerular filtration rate of ≤30 ml/min when the patients have
end-stage liver disease. Recently, Coquillard et al
(39) reported that CLKTx for ADPKD
was associated with better outcomes compared with patients who
received CLKTx for other indications. Additionally, patients with
ADPKD in the CLKTx group also had significantly better outcomes
compared with patients with ADPKD undergoing liver transplant alone
(39). Transplantation for PLD
remains controversial due to the shortage of donors, high morbidity
and mortality. However, since the first liver transplantation
performed for isolated PLD by Kwok and Lewin (40) in 1988 and the first CLKTx for PLD by
Starzl et al (41) in 1990,
with the accumulation of surgical experience, the increased use of
marginal donors and the improvement of immunosuppressant agents,
transplantation has become a viable and safe treatment option for
PLD and its implementation is increasing (36,42–44). As
most patients have normal Model for End-stage Liver Disease (MELD)
scores due to preserved liver function, the MELD exception criteria
is advocated (45).
In the present study, a total of six patients with
PLD (1 case of isolated PLD and 5 of ADPKD) received LTx or CLKTx
due to a severely decreased quality of life (ECOG score ≥3).
Concurrent hepatic decompensation was indicated for the operation
in Patients 2 and 9. Furthermore, complicated renal dysfunction was
an indication for the CLKTx in Patients 10 and 11. End-stage liver
disease progressed from isolated PLD and HBV-associated cirrhosis
were indications in the other five patients. Although hepatitis B
was under effective control by routine treatment with anti-HBV
drugs, the HBV-associated cirrhosis accelerated the progression and
increased the severity of PLD to a certain extent. Based on our
experience regarding OLT for end-stage liver disease associated
with hepatitis B, it is recommended that PLD with hepatitis B that
progressed to decompensated cirrhosis is the primary indication for
transplantation; however, further clinical evidence is required to
support this. The difficult and hazardous procedure was mainly
attributed to the explantation of the polycystic liver, including
massive enlarged liver (Patient 2), hepatic decompensation (Patient
3) and volume depletion during the reperfusion phase (Patients 10
and 11). In the present study, prior intervention did not appear to
be a risk factor for transplantation. During the
peri-transplantation period, the mortality and morbidity was 18.2
and 54.5%, respectively. The two mortalities occurred within 1
month post-CLKTx and were due to hepatic and renal dysfunction,
which tremendously increased the risk of the operation. Except for
the two CLKTx cases, the morbidity and mortality in the present
study was similar to that of patients with other indications for
liver transplantation at our center (data not shown). At the time
of writing of the manuscript, only 1 patient (Patient 1) died due
to ischemia cholangitis at 122 months post-OLT. The mean PCS score
was 87.1±6.9 and the mean MCS score was 81.5±6.4. A total of 8
patients were alive with good graft function and high quality of
life. The actuarial 1-, 5-, and 10-year patient survival rates
during the follow-up period were 81.8, 81.8 and 65.5%,
respectively.
In conclusion, PLD is a rare congenital disorder,
which is mainly diagnosed by radiological examinations. The
pharmaceutical intervention or palliative disease-directed
interventions may temporarily relieve hepatomegaly-associated
symptoms. Due to the high recurrence rate, almost all of the cases
require re-treatment. Although segmental hepatectomy is an
effective procedure for patients with cysts in at least one hepatic
segment, it may potentially complicate OLT in the future. Due to
limiting factors, including the shortage of donors, as well as the
higher mortality and morbidity compared with those of other
surgical procedures, transplantation for a benign disease is an
ethical issue. Liver transplantation is the only curative procedure
for PLD, and the present study demonstrated that the intervention
is relatively safe and leads to good long-term prognosis and high
quality of life. Liver transplantation may be the primary
therapeutic option in cases of progressive or advanced PLD where
other forms of therapy to palliate symptoms were insufficient. Of
note, PLD is a rare congenital disorder, only accounting for 1.09%
of cases receiving OLT at our center. The small sample size was a
limitation of the present single-center study and a multi-center
study should be a future endeavor.
Acknowledgements
The authors would like to thank Dr Qiao-Lan Zheng
from the Institute of Journal Publishing Center of the Third
Affiliated Hospital, Sun Yat-Sen University (Guangzhou, China) for
reviewing the statistical method of this study.
Funding
No funding received.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Author contributions
CX and WC designed the study. GC, YY and CX
performed the liver transplantations. FD, HT and XF collected the
data. FD, HZ and WC analyzed the data and wrote the manuscript.
Ethics approval and written informed
consent
The study was supervised and approved by the Ethics
Committee of the Third Affiliated Hospital, Sun Yat-Sen University
[Guangzhou, China; no. (2018)02-006-011]. Written informed consent
was obtained from all of the patients or the patients' first-degree
relatives.
Patients' consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Gevers TJ and Drenth JP: Diagnosis and
management of polycystic liver disease. Nat Rev Gastroenterol
Hepatol. 10:101–108. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Que F, Nagorney DM, Gross JB Jr and Torres
VE: Liver resection and cyst fenestration in the treatment of
severe polycystic liver disease. Gastroenterology. 108:487–494.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hoshino J, Ubara Y, Suwabe T, Sumida K,
Hayami N, Mise K, Hiramatsu R, Hasegawa E, Yamanouchi M, Sawa N, et
al: Intravascular embolization therapy in patients with enlarged
polycystic liver. Am J Kidney Dis. 63:937–944. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Temmerman F, Ho TA, Vanslembrouck R,
Coudyzer W, Billen J, Dobbels F, van Pelt J, Bammens B, Pirson Y
and Nevens F: Lanreotide reduces liver volume, but might not
improve muscle wasting or weight loss, in patients with symptomatic
polycystic liver disease. Clin Gastroenterol Hepatol.
13:2353–2359.e2351. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Aussilhou B, Douflé G, Hubert C, Francoz
C, Paugam C, Paradis V, Farges O, Vilgrain V, Durand F and Belghiti
J: Extended liver resection for polycystic liver disease can
challenge liver transplantation. Ann Surg. 252:735–743. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Drenth JP, Chrispijn M, Nagorney DM,
Kamath PS and Torres VE: Medical and surgical treatment options for
polycystic liver disease. Hepatology. 52:2223–2230. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Qian Q: Isolated polycystic liver disease.
Adv Chronic kidney Dis. 17:181–189. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gigot JF, Jadoul P, Que F, Van Beers BE,
Etienne J, Horsmans Y, Collard A, Geubel A, Pringot J and Kestens
PJ: Adult polycystic liver disease: Is fenestration the most
adequate operation for long-term management? Ann Surg. 225:286–294.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Pugh RN, Murray-Lyon IM, Dawson JL,
Pietroni MC and Williams R: Transection of the oesophagus for
bleeding oesophageal varices. Br J Surg. 60:646–649. 1973.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Child CG and Turcotte JG: Surgery and
portal hypertension. Major Probl Clin Surg. 1:1–85. 1964.PubMed/NCBI
|
|
11
|
Oken MM, Creech RH, Tormey DC, Horton J,
Davis TE, McFadden ET and Carbone PP: Toxicity and response
criteria of the eastern cooperative oncology group. Am J Clin
Oncol. 5:649–655. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Clavien PA, Barkun J, de Oliveira ML,
Vauthey JN, Dindo D, Schulick RD, de Santibañes E, Pekolj J,
Slankamenac K, Bassi C, et al: The Clavien-Dindo classification of
surgical complications: Five-year experience. Ann Surg.
250:187–196. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li A, Davila S, Furu L, Qian Q, Tian X,
Kamath PS, King BF, Torres VE and Somlo S: Mutations in PRKCSH
cause isolated autosomal dominant polycystic liver disease. Am J
Hum Genet. 72:691–703. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Drenth JP, te Morsche RH, Smink R,
Bonifacino JS and Jansen JB: Germline mutations in PRKCSH are
associated with autosomal dominant polycystic liver disease. Nat
Genet. 33:345–347. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Davila S, Furu L, Gharavi AG, Tian X, Onoe
T, Qian Q, Li A, Cai Y, Kamath PS, King BF, et al: Mutations in
SEC63 cause autosomal dominant polycystic liver disease. Nat Genet.
36:575–577. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cnossen WR, te Morsche RH, Hoischen A,
Gilissen C, Chrispijn M, Venselaar H, Mehdi S, Bergmann C, Veltman
JA and Drenth JP: Whole-exome sequencing reveals LRP5 mutations and
canonical Wnt signaling associated with hepatic cystogenesis. Proc
Natl Acad Sci USA. 111:5343–5348. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Perugorria MJ, Masyuk TV, Marin JJ,
Marzioni M, Bujanda L, LaRusso NF and Banales JM: Polycystic liver
diseases: Advanced insights into the molecular mechanisms. Nat Rev
Gastroenterol Hepatol. 11:750–761. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Rossetti S, Consugar MB, Chapman AB,
Torres VE, Guay-Woodford LM, Grantham JJ, Bennett WM, Meyers CM,
Walker DL, Bae K, et al: Comprehensive molecular diagnostics in
autosomal dominant polycystic kidney disease. J Am Soc Nephrol.
18:2143–2160. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ward CJ, Hogan MC, Rossetti S, Walker D,
Sneddon T, Wang X, Kubly V, Cunningham JM, Bacallao R, Ishibashi M,
et al: The gene mutated in autosomal recessive polycystic kidney
disease encodes a large, receptor-like protein. Nat Genet.
30:259–269. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wills ES, Roepman R and Drenth JP:
Polycystic liver disease: Ductal plate malformation and the primary
cilium. Trends Mol Med. 20:261–270. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Everson GT, Taylor MR and Doctor RB:
Polycystic disease of the liver. Hepatology. 40:774–782. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Swenson K, Seu P, Kinkhabwala M, Maggard
M, Martin P, Goss J and Busuttil R: Liver transplantation for adult
polycystic liver disease. Hepatology. 28:412–415. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Alvaro D, Barbaro B, Franchitto A, Onori
P, Glaser SS, Alpini G, Francis H, Marucci L, Sterpetti P,
Ginanni-Corradini S, et al: Estrogens and insulin-like growth
factor 1 modulate neoplastic cell growth in human
cholangiocarcinoma. Am J Pathol. 169:877–888. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chapman AB: Cystic disease in women:
Clinical characteristics and medical management. Adv Ren Replace
Ther. 10:24–30. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bae KT, Zhu F, Chapman AB, Torres VE,
Grantham JJ, Guay-Woodford LM, Baumgarten DA, King BF Jr, Wetzel
LH, Kenney PJ, et al: Magnetic resonance imaging evaluation of
hepatic cysts in early autosomal-dominant polycystic kidney
disease: The consortium for radiologic imaging studies of
polycystic kidney disease cohort. Clin J Am Soc Nephrol. 1:64–69.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Rajoriya N, Tripathi D, Leithead JA,
Gunson BK, Lord S, Ferguson JW and Hirschfield GM: Portal
hypertension in polycystic liver disease patients does not affect
wait-list or immediate post-liver transplantation outcomes. World J
Gastroenterol. 22:9966–9973. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Barbier L, Ronot M, Aussilhou B, Cauchy F,
Francoz C, Vilgrain V, Soubrane O, Paradis V and Belghiti J:
Polycystic liver disease: Hepatic venous outflow obstruction
lesions of the non-cystic parenchyma have major consequences.
Hepatology. 68:652–662. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Dumont PN, Rode A, Mabrut JY, Ducerf C and
Golse N: Polycystic liver disease complicated by obstructive
jaundice. J Visc Surg. 153:149–151. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Temmerman F, Dobbels F, Ho TA, Pirson Y,
Vanslembrouck R, Coudyzer W, Bammens B, van Pelt J, Pirenne J and
Nevens F: Development and validation of a polycystic liver disease
complaint-specific assessment (POLCA). J Hepatol. 61:1143–1150.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sherstha R, McKinley C, Russ P,
Scherzinger A, Bronner T, Showalter R and Everson GT:
Postmenopausal estrogen therapy selectively stimulates hepatic
enlargement in women with autosomal dominant polycystic kidney
disease. Hepatology. 26:1282–1286. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hogan MC, Masyuk TV, Page LJ, Kubly VJ,
Bergstralh EJ, Li X, Kim B, King BF, Glockner J, Holmes DR, et al:
Randomized clinical trial of long-acting somatostatin for autosomal
dominant polycystic kidney and liver disease. J Am Soc Nephrol.
21:1052–1061. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
van Keimpema L, Nevens F, Vanslembrouck R,
van Oijen MG, Hoffmann AL, Dekker HM, de Man RA and Drenth JP:
Lanreotide reduces the volume of polycystic liver: A randomized,
double-blind, placebo-controlled trial. Gastroenterology.
137:1661–1668.e1661-1662. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
D'Agnolo HM, Kievit W, Takkenberg RB,
Riaño I, Bujanda L, Neijenhuis MK, Brunenberg EJ, Beuers U, Banales
JM and Drenth JP: Ursodeoxycholic acid in advanced polycystic liver
disease: A phase 2 multicenter randomized controlled trial. J
Hepatol. 65:601–607. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Qian Q, Du H, King BF, Kumar S, Dean PG,
Cosio FG and Torres VE: Sirolimus reduces polycystic liver volume
in ADPKD patients. J Am Soc Nephrol. 19:631–638. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chrispijn M, Nevens F, Gevers TJ,
Vanslembrouck R, van Oijen MG, Coudyzer W, Hoffmann AL, Dekker HM,
de Man RA, van Keimpema L and Drenth JP: The long-term outcome of
patients with polycystic liver disease treated with lanreotide.
Aliment Pharmacol Ther. 35:266–274. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Baber JT, Hiatt JR, Busuttil RW and
Agopian VG: A 20-year experience with liver transplantation for
polycystic liver disease: Does previous palliative surgical
intervention affect outcomes? J Am Coll Surg. 219:695–703. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Temmerman F, Missiaen L, Bammens B,
Laleman W, Cassiman D, Verslype C, van Pelt J and Nevens F:
Systematic review: The pathophysiology and management of polycystic
liver disease. Aliment Pharmacol Ther. 34:702–713. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Eason JD, Gonwa TA, Davis CL, Sung RS,
Gerber D and Bloom RD: Proceedings of consensus conference on
simultaneous liver kidney transplantation (SLK). Am J Transplant.
8:2243–2251. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Coquillard C, Berger J, Daily M, Shah M,
Mei X, Marti F and Gedaly R: Combined liver-kidney transplantation
for polycystic liver and kidney disease: Analysis from the United
Network for Organ Sharing dataset. Liver Int. 36:1018–1025. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kwok MK and Lewin KJ: Massive hepatomegaly
in adult polycystic liver disease. Am J Surg Pathol. 12:321–324.
1988. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Starzl TE, Reyes J, Tzakis A, Mieles L,
Todo S and Gordon R: Liver transplantation for polycystic liver
disease. Arch Surg. 125:575–577. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Gedaly R, Guidry P, Davenport D, Daily M,
Ronsenau J, Shah M, Cooper MA and Hundley J: Peri-operative
challenges and long-term outcomes in liver transplantation for
polycystic liver disease. HPB (Oxford). 15:302–306. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zamora-Valdés D, Contreras AG and Mercado
MA: Liver transplantation for polycystic liver disease. J Am Coll
Surg. 219:11922014. View Article : Google Scholar
|
|
44
|
van Keimpema L, Nevens F, Adam R, Porte
RJ, Fikatas P, Becker T, Kirkegaard P, Metselaar HJ, Drenth JP,
European Liver and Intestine Transplant Association: Excellent
survival after liver transplantation for isolated polycystic liver
disease: An European Liver Transplant Registry study. Transpl Int.
24:1239–1245. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Arrazola L, Moonka D, Gish RG and Everson
GT: Model for end-stage liver disease (MELD) exception for
polycystic liver disease. Liver Transpl 12 (12 Suppl). S110–S111.
2006. View Article : Google Scholar
|