Introduction
Breast cancer remains a leading cause of fatality in
women worldwide (1). According to
the expression of estrogen receptor α (ERα), progesterone receptor
(PR) and human epidermal growth factor receptor-2 (Her2), breast
cancer can be classified into three different types, including
ERα+, Her2+ and triple negative breast cancer (TNBC) (2). Due to the lack of ERα, PR and Her2
expression in patients with TNBC, gemcitabine-based chemotherapy is
one of the major procedures for the treatment of TNBC (3). However, the survival of patients with
TNBC receiving chemotherapy is compromised due to intrinsic or
acquired resistance to gemcitabine (4). The molecular mechanism for gemcitabine
resistance is common among various cancer types, including breast
cancer, which is complicated and still poorly understood (5).
MicroRNAs (miRs) are small non-coding RNAs that were
identified decades ago (6).
Deregulation of miRs has been reported to contribute to various
diseases, including cancer (7,8). During
cancer progression, miRs regulate their target genes to promote or
inhibit cancer cell proliferation, metastasis, apoptosis and drug
resistance (9–11). In breast cancer, dysregulation of miR
patterns is also associated with chemoresistance (12). Several miRs, including miR-34a,
miR-21 and miR-489, have been identified to promote chemoresistance
via targeting their specific target genes (13–15).
In breast cancer, a set of genes have been proved to
contribute to gemcitabine resistance via deoxycytidine
monophosphate deaminase (DCTD) (16). DCTD catalyzes the deamination of dCMP
to dUMP, and its underexpression can induce dNTP pool imbalance,
which affects DNA amplification (17). Thus, decreased DCTD would reduce
gemcitabine self-potentiation so as to cause drug resistance
(18). However, to the best of our
knowledge the regulatory mechanism of DCTD in gemcitabine-resistant
breast cancer has not been studied yet.
In the current study, miR-620 levels were assessed
in gemcitabine-resistant TNBC cells. Luciferase assays, reverse
transcription-quantitative polymerase chain reaction (RT-qPCR) and
western blot analysis were performed to assess if DCTD, a gene
associated with gemcitabine resistance, was directly regulated by
miR-620 in TNBC cells. Furthermore, TNBC cells were assessed
following the inhibition of miR-620 or silencing of DCTD in TNBC
cells. The present study further extended current understandings on
chemoresistance of TNBC and implied miR-620 as a promising
prognostic and therapeutic target for patients with
gemcitabine-resistant TNBC.
Materials and methods
Patients
Patients who received radiotherapy, chemotherapy or
surgery were excluded from the current study. Between December 2011
and December 2014, 36 TNBC samples were obtained from patients with
TNBC in Liaocheng People's Hospital (Liaocheng, China). The
adjacent normal tissues were extracted ≥5 cm away from the tumor
tissues. Patients (20 males and 16 females) were aged from 43 to 65
years old. A total of 21 gemcitabine-sensitive patients with TNBC
[who had reached pCR (pathologic complete response) following
gemcitabine-based neoadjuvant chemotherapy] and 15 patients with
gemcitabine-resistant TNBC (tumor size was the same or larger at
the time of surgical removal) were enrolled in the research. The
detailed clinicopathologic features are indicated in Table I. All patients provided written
consent prior to surgery, and the present study was approved by the
Ethics Committee of Liaocheng People's Hospital. Tissues were
immediately frozen at liquid nitrogen with the temperature of
−196°C for RNA extraction.
 | Table I.The clinicopathologic features of
patients with triple negative breast cancer. |
Table I.
The clinicopathologic features of
patients with triple negative breast cancer.
| Clinicopathologic
features | Case (n) | Percent (%) |
|---|
| Age (years) |
|
|
| ≥50 | 25 | 69.44 |
|
<50 | 11 | 30.56 |
| Response to
emcitabine |
|
|
|
Sensitive | 21 | 58.33 |
|
Resistant | 15 | 41.67 |
| Tumor size (3
cm) |
|
|
| ≥3 | 12 | 33.33 |
|
<3 | 24 | 66.67 |
| TNM stage |
|
|
| I–II | 26 | 72.22 |
|
III–IV | 10 | 27.78 |
| Menopause state |
|
|
| Yes | 13 | 36.11 |
| No | 23 | 63.89 |
Cell culture
Human TNBC MDA-MB-231 and BT549 cell lines were
obtained from American Type Culture Collection (Manassas, VA, USA).
Three gemcitabine-resistant MDA-MB-231 cell lines (MDA-MB-231rGEM1,
MDA-MB-231rGEM2 and MDA-MB-231rGEM3) were generated by continuous
exposure of MDA-MB-231 cells to increasing concentrations of
gemcitabine for >12 months as described previously study
(19). All cell lines were cultured
in Dulbecco's Minimum Essential Medium (Gibco; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) containing 10% fetal bovine
serum (Hyclone, Logan, UT, USA) in an incubator containing 5%
CO2. For analysis of the gemcitabine response, increased
concentrations of gemcitabine were added into the culture medium of
MDA-MB-231 cells and gemcitabine-resistant MDA-MB-231 cells for 0,
24, 48 and 72 h. Subsequently, cells were subjected to further
experiments.
Inhibition and overexpression of
miR-620
miR-NC inhibitor, miR-620 inhibitor, miR-NC mimic
and miR-620 mimic were purchased from Shanghai GenePharma Co., Ltd.
(Shanghai, China). The sequences were as follows: miR-NC inhibitor,
5′-UUCUCCGAACGUGUCACGUUU-3′; miR-620 inhibitor,
5′-AUUUCUAUAUCUCCAUUU-3′; miR-NC mimic, 5′-UCGCUUGGUGCAGGUCGGG-3′;
miR-620 mimic, 5′-AUGGAGAUAGAUAUAGAAAUUU-3′. The control group was
treated with Lipofectamine 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.). A total of 30 nM miR-NC inhibitor, miR-620
inhibitor, miR-NC mimic and miR-620 mimic were transfected with
Lipofectamine 2000 into cells at 37°C 24 h prior to subsequent
experiments.
RNA extraction and RT-qPCR
Total RNA of tissues and cells was isolated using
the miRNeasy mini kit (Qiagen, Inc., Valencia, CA, USA) according
to the manufacturer's instructions. cDNA was synthesized using the
M-MLV kit (Thermo Fisher Scientific, Inc.). Following this, qPCR
was performed to detect the expression levels of specific genes
using SYBR Premix Ex Taq (Takara Bio, Inc., Otsu, Japan). GAPDH and
U6 served as internal controls for mRNA and miR, respectively. The
primer sequences were as follows: Stem loop primer,
5′-CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGATTTCTA-3′; miR-620-forward
(F): 5′-GCCGAGATGGAGATAGATAT-3′; miR-620-reverse (R),
5′-CTCAACTGGTGTCGTGGA-3′; DCTD-F, 5′-TGCAAGAAACGGGACGACTAT-3′;
DCTD-R, 5′-ATCACTGCACCCATTTGGCAT-3′; U6-F, 5′-CTCGCTTCGGCAGCACA-3′,
and U6-R, 5′-AACGCTTCACGAATTTGCGT-3′; GAPDH-F,
5′-GAAATCCCATCACCATCTTCCAGG-3′ and GAPDH-R,
5′-GAGCCCCAGCCTTCTCCATG-3′. The PCR conditions were as follows:
95°C for 5 min followed by 40 cycles of amplification at 95°C for
30 sec, 57°C for 30 sec and 72°C for 30 sec. The relative
expression levels of indicated genes were calculated using the
2−ΔΔCq method (20).
Western blot analysis
GAPDH antibody was purchased from Zhejiang Kangchen
Biotech Co., Ltd. (Wuhan, China). DCTD antibody was bought from
Santa Cruz Biotechnology, Inc. (Dallas, TX, USA). Secondary
antibodies against rabbit and mouse were bought from Abcam
(Cambridge, MA, USA). Cell lysates were prepared using
radioimmunoprecipitation assay lysis buffer (Beyotime Institute of
Biotechnology, Haimen, China) with protease inhibitor (Sigma
Aldrich; Merck KGaA, Darmstadt, Germany). Protein concentration was
determined using the BCA method. Proteins (15 µg/lane) were
separated by SDS-PAGE using an 8% gel and transferred to a
polyvinylidene difluoride membrane (EMD Millipore, Billerica, MA,
USA). Following blocking in 5% non-fat milk at room temperature for
2 h, the membranes were washed with Tris-buffered saline with
Tween-20 and incubated with the primary antibodies against DCTD
(cat. no. ab183607) and GAPDH (cat. no. ab181602; both 1:1,000;
Abcam) overnight at 4°C. On the following day, the membranes were
incubated in horseradish peroxidase-conjugated goat anti-rabbit
secondary antibodies (cat. no. ab6721; 1:2,000; Abcam) for 1 h at
room temperature. The bands were developed using an enhanced
chemiluminescence detection agent (Thermo Fisher Scientific, Inc.)
and the images were obtained with a densitometer (GS-700; Bio-Rad
Laboratories, Inc., Hercules, CA, USA). GAPDH was used as an
internal control. Densitometry was achieved by Image J version
1.8.0 (National Institutes of Health, Bethesda, MD, USA).
Cell proliferation assay and
calculation of IC50
The cell proliferation assay was performed with Cell
Counting Kit-8 (CCK8; Dojindo Molecular Technologies, Inc.,
Kumamoto, Japan). MDA-MB-231, MDA-MB-231rGEM1 and MDA-MB-231rGEM2
cells (1×103/well) were seeded into 96-well plates with
DMEM with 10% FBS (Invitrogen; Thermo Fisher Scientific, Inc.)
containing vehicle (0.9% NaCl in water) or various concentrations
of gemcitabine (0, 0.1, 1, 10, 100 and 100 nM; Selleck Chemicals,
Houston, TX, USA) and sustained at 37°C for 72 h. Following this,
10 µl CCK8 solution was added into each well and incubated at 37°C
for 1 h. For each well, the absorbance at 450 nm was measured using
a microplate reader (Bio-Rad Laboratories, Inc.). The
IC50 (the cell proliferation that was inhibited by 50%
compared with cells in control group) was calculated using CompuSyn
software (version 1.0; ComboSyn, Inc., Paramus, NJ, USA).
Cell apoptosis assay
Cell apoptosis was detected using the Annexin V/Dead
Cell Apoptosis Kit (Invitrogen; Thermo Fisher Scientific, Inc.)
according to the manufacturer's protocol. Cells were trypsinized
and suspended in annexin binding buffer. Following this, propidium
iodide (PI) and annexin v-fluorescein isothiocyanate (FITC) were
added into the cell suspension and incubated for 15 min. The
stained cells were analyzed on a BD FACSCalibur flow cytometer (BD
Biosciences, San Jose, CA, USA). Cells that were positive for
annexin V-FITC staining and negative staining for PI were
considered as early apoptotic cells, whereas cells that were
positive for Annexin V-FITC staining and PI staining were
considered as late apoptotic cells. The results were analyzed using
FlowJo software (version 10.3; FlowJo LLC, Ashland, OR, USA).
Construction and transfection of
plasmid
The full length of DCTD was amplified from the cDNA
of 293 cells and ligated into pcDNA3 (YouBio, Changsha, China). For
the overexpression of DCTD, 2 µg pcDNA3-DCTD was mixed with
Lipofectamine 2000 in Opti-MEM (Invitrogen; Thermo Fisher
Scientific, Inc.) for 15 min and then added into the culture medium
in each well of the 6-well plates. The control group was
transfected with 2 µg pcDNA3 plasmid with the same method.
Following 24 h of incubation, the cells were subjected to further
experiments.
Luciferase reporter assay
The binding site between DCTD and miR-620 was first
predicted by TargetScan (http://www.targetscan.org/vert_71/). The
3′-untranslated region (UTR) of DCTD was amplified from cDNA of 293
and inserted into pGL-3 (Promega Corporation, Madison, WI, USA).
The mutated DCTD 3′-UTR was generated using site-directed
mutagenesis kit (Agilent Technologies, Inc., Santa Clara, CA, USA)
according to the manufacturer's protocol. 293 cells were
cotransfected with pGL3-DCTD 3′-UTR wild-type (WT) or pGL3-DCTD
3′-UTR mutant (Mut), miR-620 mimics or miR-NC mimics and internal
control Renilla plasmid using Lipofectamine 2000
(Invitrogen; Thermo Fisher Scientific, Inc.). Following a total of
24 h, the activity of luciferase and Renilla activity was
detected using a Dual Luciferase Reporter Assay Kit (Promega
Corporation) according to the manufacturer's protocol.
Statistical analysis
All data were analyzed using GraphPad Prism 5.0
software (GraphPad Software, Inc., La Jolla, CA, USA), and results
were presented as the mean ± standard deviation. Differences
between two groups were compared using a Student's t-test.
Differences among three or more groups were analyzed with one-way
analysis of variance followed by Newman-Keuls analysis. The
correlation between miR-620 and DCTD mRNA expression was analyzed
by a Spearman's correlation coefficient test. P<0.05 was
considered to indicate a statistically significant difference.
Results
Generation of gemcitabine-resistant
MDA-MB-231 cell lines
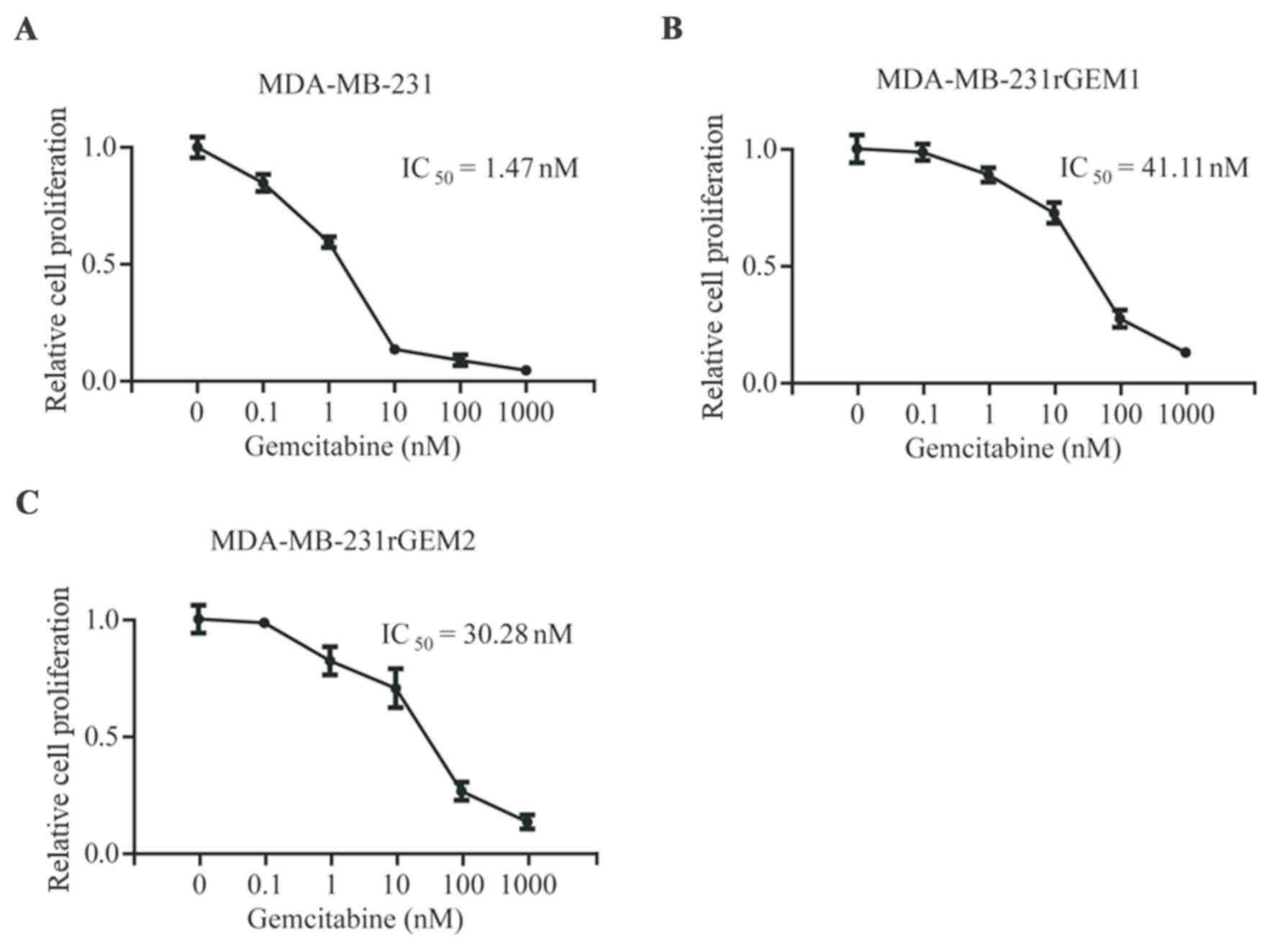
To explore the molecular mechanism of gemcitabine
resistance in TNBC, gemcitabine-resistant MDA-MB-231 cell lines
were developed by continuous exposure of MDA-MB-231 cells to
gemcitabine. As indicated in Fig. 1,
MDA-MB-231 cells were sensitive towards gemcitabine
(IC50=1.47 nM), gemcitabine-resistant MDA-MB-231 cells
(MDA-MB-231rGEM1 and MDA-MB-231rGEM2) were relatively insensitive
towards gemcitabine (IC50=41.11 nM and
IC50=30.28 nM, respectively). These results indicated
that MDA-MB-231rGEM1 and MDA-MB-231rGEM2 were useful
gemcitabine-resistant TNBC models.
DCTD is upregulated in
gemcitabine-resistant MDA-MB-231 cells
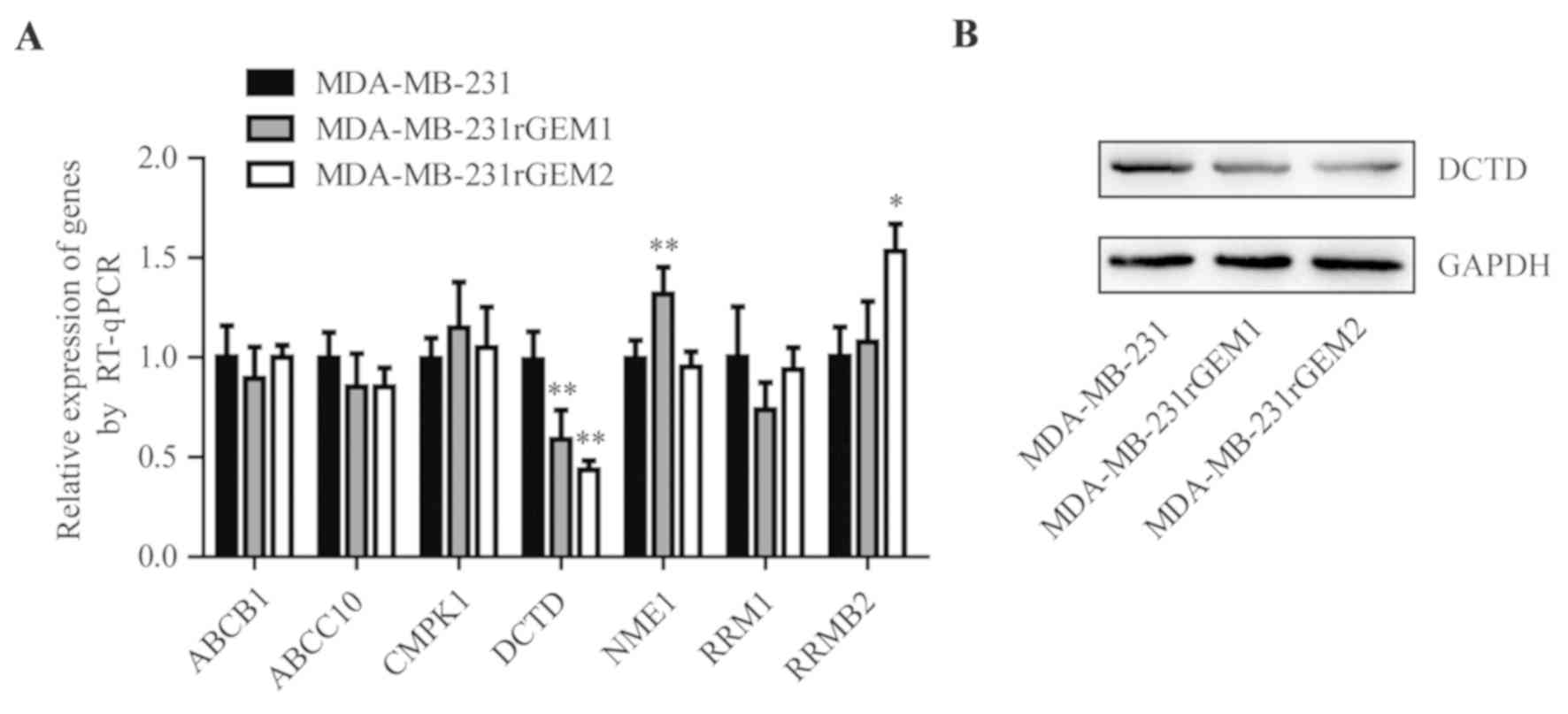
Previous research identified that deregulation of 7
genes (ABCB1, ABCC10, CMPK1, DCTD, NME1, RRM1 and RRMB1) was
involved in the development of gemcitabine resistance (16). Consequently, the mRNA levels of these
7 genes were detected in MDA-MB-231, MDA-MB-231rGEM1 and
MDA-MB-231rGEM2 cells. RT-qPCR data revealed that DCTD mRNA
expression levels were significantly decreased in MDA-MB-231rGEM1
and MDA-MB-231rGEM2 cells compared with that in MDA-MB-231 cells
(Fig. 2A). In addition, DCTD protein
expression levels were also downregulated in MDA-MB-231rGEM1 and
MDA-MB-231rGEM2 cells when compared with MDA-MB-231 cells (Fig. 2B).
Upregulation of miR-620 in
gemcitabine-resistant MDA-MB-231 cells negatively regulates
DCTD
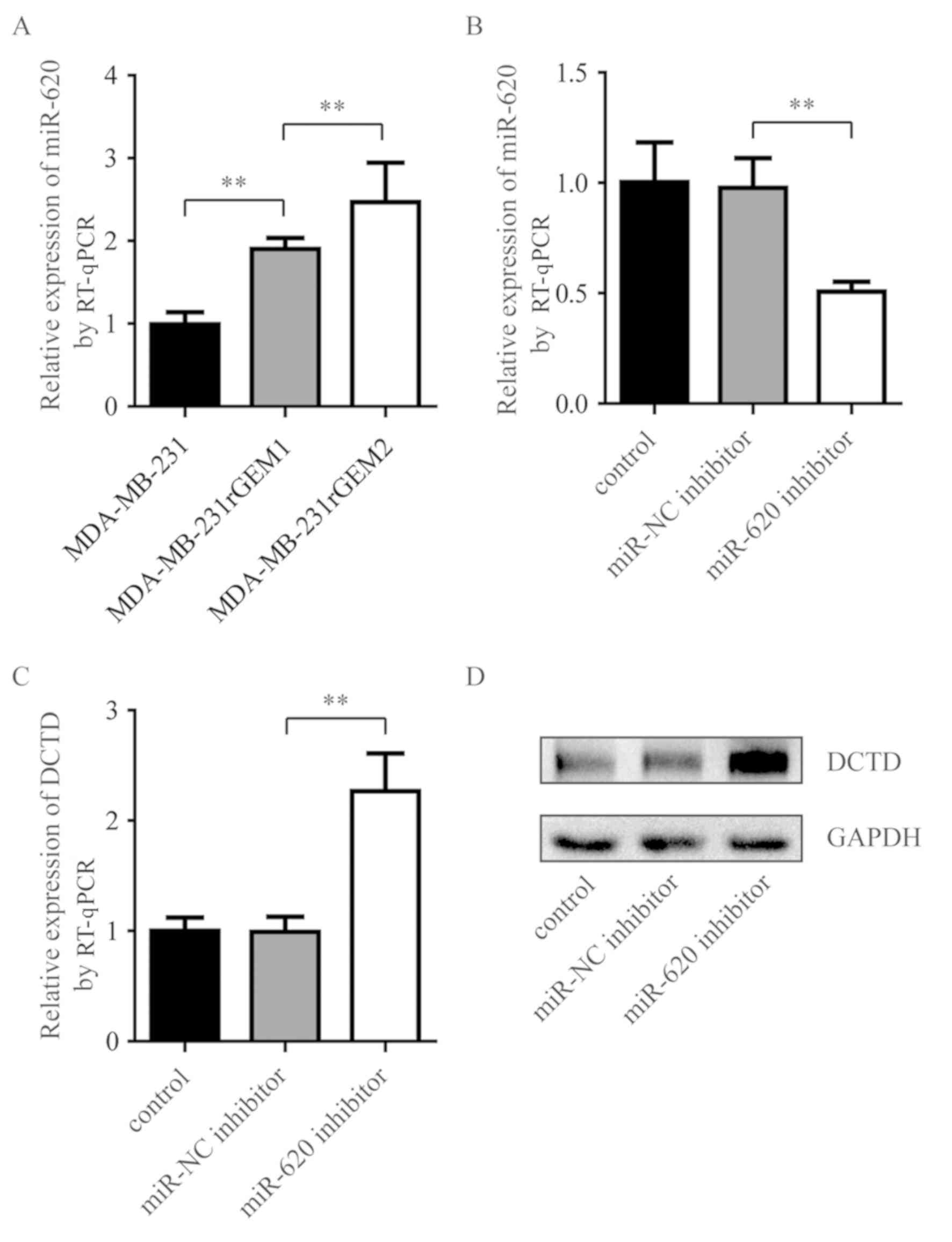
miR-620 expression levels were measured in
MDA-MB-231rGEM1, MDA-MB-231rGEM2 cells and their parental
MDA-MB-231 cells. miR-620 expression levels were significantly
upregulated in MDA-MB-231rGEM1 compared with parental MDA-MB-231
cells. In addition, miR-620 expression levels were significantly
upregulated in MDA-MB-231rGEM2 cells compared with MDA-MB-231rGEM1
cells (Fig. 3A). To investigate
whether elevation of miR-620 contributed to the deregulation of
DCTD in gemcitabine-resistant MDA-MB-231 cells, an miR-620
inhibitor was used to explore the association between miR-620 and
DCTD. Compared with MDA-MB-231rGEM1 cells transfected with miR-NC
inhibitor, transfection with miR-620 inhibitor significantly
decreased miR-620 expression levels and increased DCTD mRNA
expression levels (Fig. 3B and C).
Additionally, inhibition of miR-620 upregulated DCTD protein
expression levels in MDA-MB-231rGEM1 cells. These data suggested
that miR-620 may contribute to gemcitabine resistance via
regulation of DCTD.
DCTD is directly regulated by
miR-620
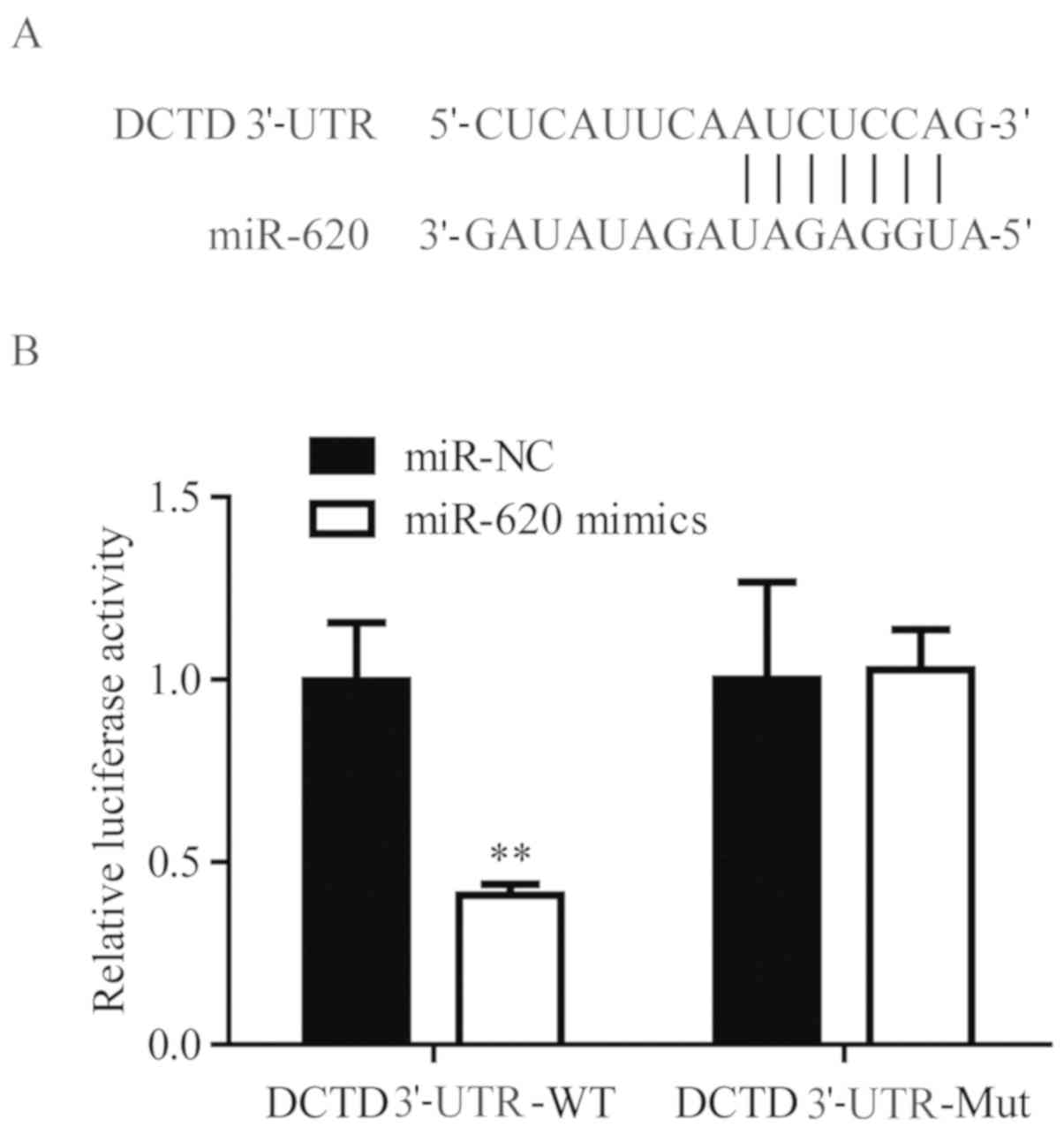
Using TargetScan, it was predicted DCTD was a target
gene of miR-620 (Fig. 4A). To
validate the regulatory association between miR-620 and DCTD, dual
luciferase activity was performed. In 293, miR-620 mimics
significantly suppressed the luciferase activity of cells
transfected with DCTD 3′-UTR-WT, but not DCTD 3′-UTR-Mut, when
compared with miR-NC mimics (Fig.
4B). These results confirmed that miR-620 directly regulated
DCTD mRNA expression levels via binding to its 3′-UTR.
miR-620 contributes to gemcitabine
resistance through DCTD
To verify whether miR-620 and DCTD were involved in
the development of gemcitabine resistance, DCTD was overexpressed
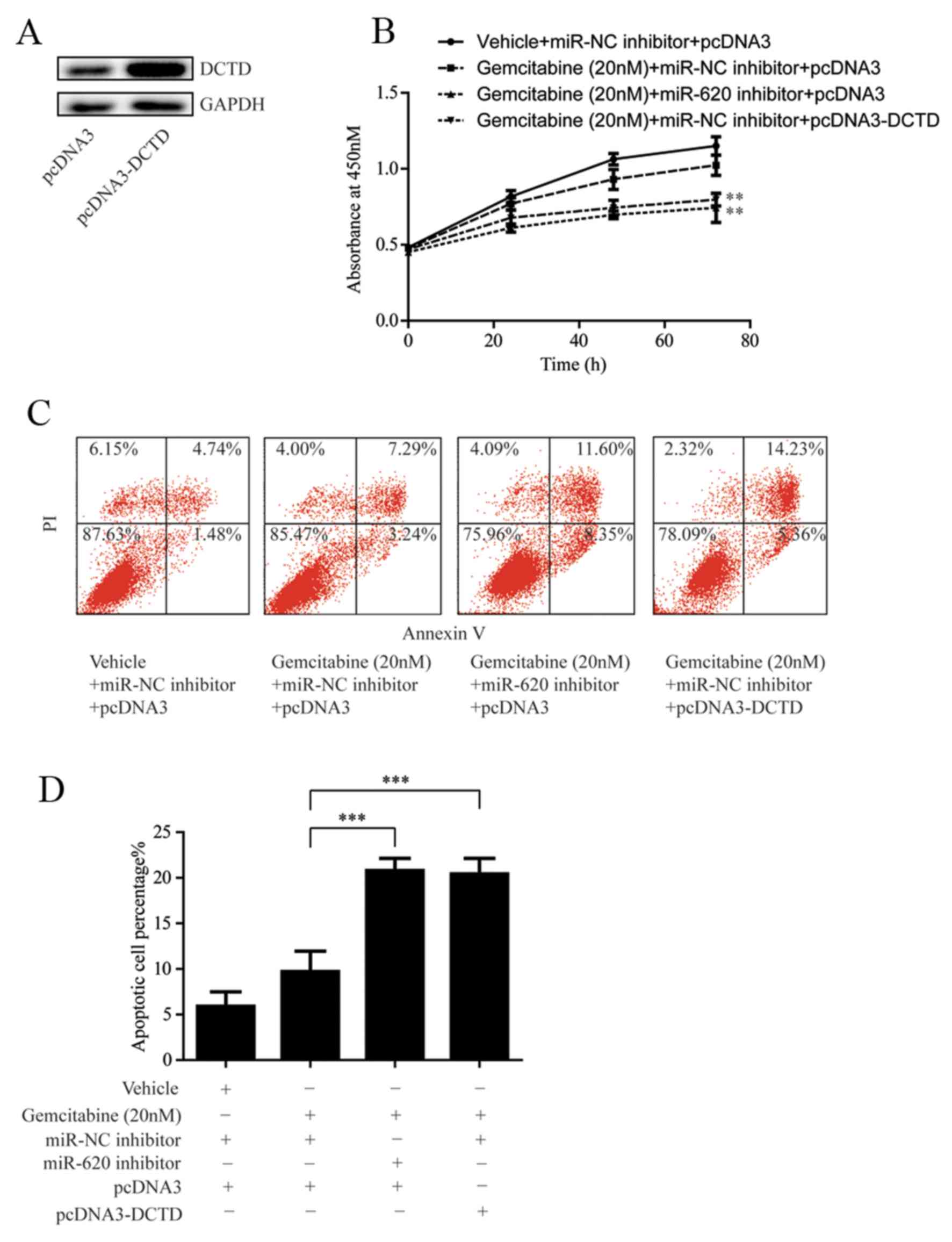
in MDA-MB-231rGEM1 cells via transfection of pcDNA3-DCTD (Fig. 5A). Subsequently, cell proliferation
of MDA-MB-231rGEM1 cells was determined following 20 nM gemcitabine
treatment with or without transfection of miR-620 inhibitor or
pcDNA3-DCTD. As indicated in Fig.
5B, miR-620 inhibition or overexpression of DCTD significantly
enhanced the sensitivity of MDA-MB-231rGEM1 cells to gemcitabine
(Fig. 5B). Consistently,
transfection with miR-620 inhibitor or pcDNA3-DCTD significantly
induced cell apoptosis in MDA-MB-231rGEM1 cells in response to 20
nM gemcitabine treatment (Fig. 5C and
D). These results confirmed the hypothesis that upregulation of
miR-620 conferred gemcitabine resistance in TNBC cells via
regulation of DCTD.
Altered expression of miR-620 and DCTD
contributes to gemcitabine resistance in patients with TNBC
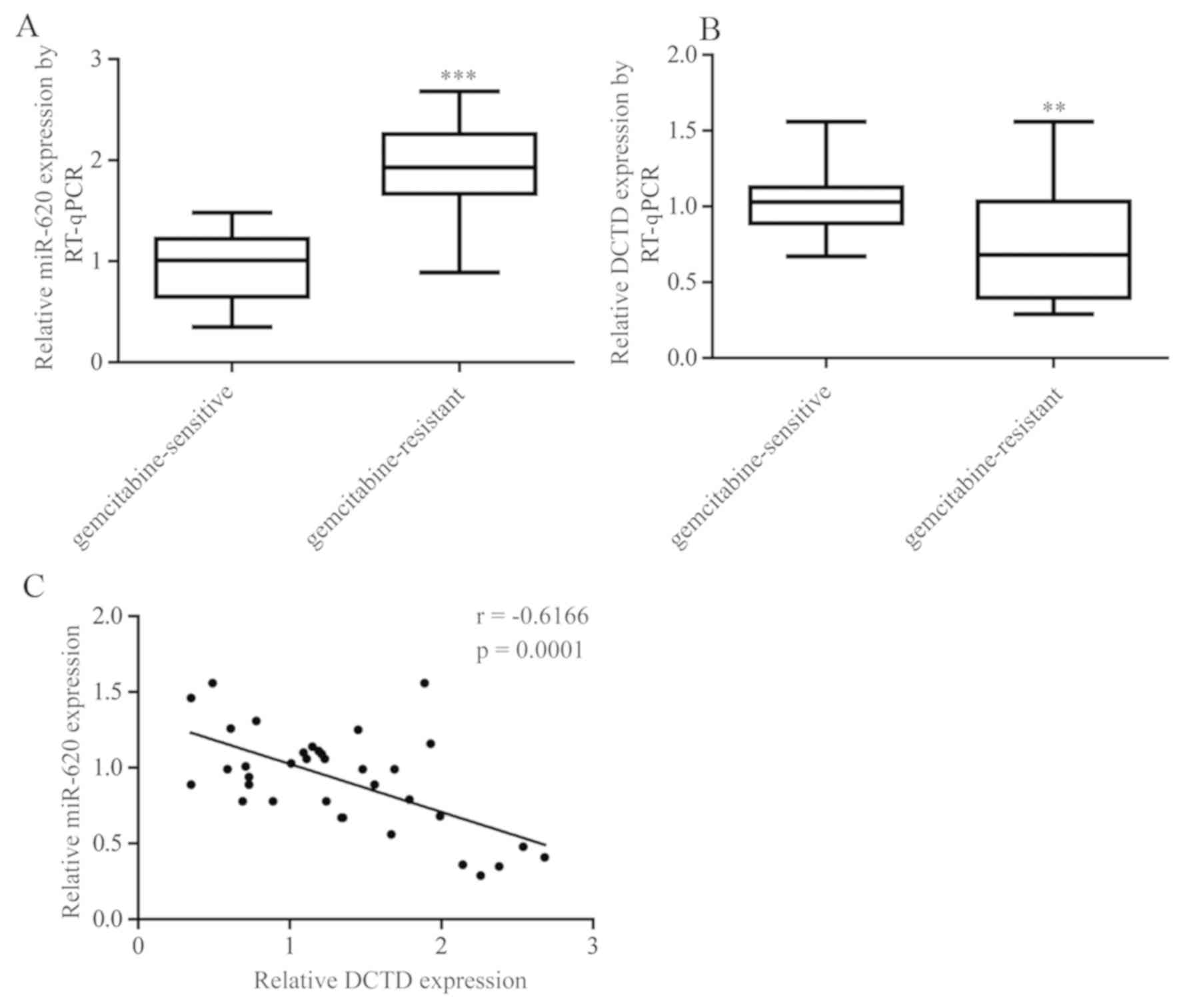
The expression of miR-620 and DCTD was assessed in
tumor tissues obtained from 21 patients with gemcitabine-sensitive
TNBC and pCR (pathologic complete response) following
gemcitabine-based neoadjuvant chemotherapy and 15 patients with
gemcitabine-resistant TNBC (the tumor size was the same or larger
at the time of surgical removal). As expected, miR-620 expression
levels were significantly increased in tumor tissues from patients
with gemcitabine-resistant TNBC whereas DCTD mRNA expression levels
were significantly decreased (Fig. 6A
and B). Furthermore, a negative correlation between miR-620 and
DCTD mRNA expression was observed (Fig.
6C).
Discussion
As a first-line drug for chemotherapy in patients
with recurrent or metastatic cancer, gemcitabine can improve
prognosis of patients with TNBC with a response rate as high as
78.6% (21). However, TNBC cells
gradually bypass the desired cell apoptosis and develop
chemoresistance, which typically leads to patient fatality
(22). A previous study indicated
that the metabolic pathway of gemcitabine is involved in
gemcitabine resistance (23). In the
present study, it was revealed that miR-620 could negatively
regulate DCTD, which is a key gene in gemcitabine metabolism, to
contribute to gemcitabine resistance in TNBC.
Various miRs, such as miR-608 and miR-145, have been
indicated to promote gemcitabine resistance (24,25). In
breast cancer, prostate cancer and pancreatic cancer cells, miR-620
upregulation could promote cell proliferation and decrease the
number of cells in the G2/M phase so as to contribute to
radiation resistance (26). In the
present work, an elevation of miR-620 in gemcitabine-resistant
MDA-MB-231 cells was indicated. Additionally, inhibition of miR-620
evoked cell apoptosis and cell growth arrest in MDA-MB-231rGEM1
cells treated with gemcitabine, which indicated that downregulation
of miR-620 could reverse gemcitabine resistance in MDA-MB-231rGEM1
cells. The data uncovered a critical role for miR-620 in driving
gemcitabine resistance in TNBC.
DCTD promotes catalysis of deamination and converts
dCMP to dUMP (17). In the
gemcitabine metabolic pathway, DCTD transfers gemcitabine
monophosphate to difluorodeoxyuridine monophosphate to inhibit
thymidylate synthetase (27).
Decreased DCTD expression leads to the reduction of gemcitabine
self-potential by interference of dNTP pool, and multi-factorial,
principal component analysis suggested that the low expression of
DCTD was associated with gemcitabine resistance in breast cancer
(16). High expression of DCTD has
also been linked to shortened overall survival in patients with
gliomas (28). In the present study,
a decrease of DCTD in gemcitabine-resistant MDA-MB-231 cells was
indicated. Further study revealed that DCTD was directly regulated
by miR-620 in MDA-MB-231rGEM1 cells. In addition, DCTD
overexpression could reverse gemcitabine resistance in
MDA-MB-231rGEM1 cells, inducing cell growth arrest and cell
apoptosis. Furthermore, there was a negative correlation between
miR-620 and DCTD in patients with TNBC. The present data extended
current knowledge on the regulation of DCTD, and further implied a
DCTD-miR-620 interplay during the development of gemcitabine
resistance.
In conclusion, the present findings suggested
miR-620 as a novel factor in promoting gemcitabine resistance in
TNBC. Furthermore, miR-620 may contribute to gemcitabine resistance
via directly targeting DCTD. Thus, the present work indicated that
miR-620 could be a predictor for gemcitabine sensitivity of
patients with TNBC and a therapeutic target for patients with
gemcitabine-resistant TNBC.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contribution
AZ and TT collected the clinical samples. CW, TT and
YW acquired and interpreted the data. CW and ZS wrote the
manuscript. ZS designed and supervised the study.
Ethics approval and consent to
participate
The current study was supervised by the Ethics
Committee of Liaocheng People's Hospital (Liaocheng, China). All
patients provided written consent prior to surgery.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
DeSantis C, Ma J, Bryan L and Jemal A:
Breast cancer statistics, 2013. CA Cancer J Clin. 64:52–62. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Dent R, Trudeau M, Pritchard KI, Hanna WM,
Kahn HK, Sawka CA, Lickley LA, Rawlinson E, Sun P and Narod SA:
Triple-negative breast cancer: Clinical features and patterns of
recurrence. Clin Cancer Res. 13:4429–4434. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wu ZH, Lin C, Liu MM, Zhang J, Tao ZH and
Hu XC: Src inhibition can synergize with gemcitabine and reverse
resistance in triple negative breast cancer cells via the AKT/c-Jun
pathway. PLoS One. 11:e01692302016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chen M, He M, Song Y, Chen L, Xiao P, Wan
X, Dai F and Shen P: The cytoprotective role of gemcitabine-induced
autophagy associated with apoptosis inhibition in triple-negative
MDA-MB-231 breast cancer cells. Int J Mol Med. 34:276–282. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
de Sousa Cavalcante L and Monteiro G:
Gemcitabine: Metabolism and molecular mechanisms of action,
sensitivity and chemoresistance in pancreatic cancer. Eur J
Pharmacol. 741:8–16. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Iorio MV and Croce CM: microRNA
involvement in human cancer. Carcinogenesis. 33:1126–1133. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hesse M and Arenz C: MicroRNA maturation
and human disease. Methods Mol Biol. 1095:11–25. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gommans WM and Berezikov E: Controlling
miRNA regulation in disease. Methods Mol Biol. 822:1–18. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Selcuklu SD, Donoghue MT, Rehmet K, de
Souza Gomes M, Fort A, Kovvuru P, Muniyappa MK, Kerin MJ, Enright
AJ and Spillane C: MicroRNA-9 inhibition of cell proliferation and
identification of novel miR-9 targets by transcriptome profiling in
breast cancer cells. J Biol Chem. 287:29516–29528. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhu M, Xu Y, Ge M, Gui Z and Yan F:
Regulation of UHRF1 by microRNA-9 modulates colorectal cancer cell
proliferation and apoptosis. Cancer Sci. 106:833–839. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Pouliot LM, Chen YC, Bai J, Guha R, Martin
SE, Gottesman MM and Hall MD: Cisplatin sensitivity mediated by
WEE1 and CHK1 is mediated by miR-155 and the miR-15 family. Cancer
Res. 72:5945–5955. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lv J, Xia K, Xu P, Sun E, Ma J, Gao S,
Zhou Q, Zhang M, Wang F, Chen F, et al: miRNA expression patterns
in chemoresistant breast cancer tissues. Biomed Pharmacother.
68:935–942. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Park EY, Chang E, Lee EJ, Lee HW, Kang HG,
Chun KH, Woo YM, Kong HK, Ko JY, Suzuki H, et al: Targeting of
miR34a-NOTCH1 axis reduced breast cancer stemness and
chemoresistance. Cancer Res. 74:7573–7582. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chen L and Bourguignon LY: Hyaluronan-CD44
interaction promotes c-Jun signaling and miRNA21 expression leading
to Bcl-2 expression and chemoresistance in breast cancer cells. Mol
Cancer. 13:522014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chen X, Wang YW, Xing AY, Xiang S, Shi DB,
Liu L, Li YX and Gao P: Suppression of SPIN1-mediated PI3K-Akt
pathway by miR-489 increases chemosensitivity in breast cancer. J
Pathol. 239:459–472. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Dorman SN, Baranova K, Knoll JH, Urquhart
BL, Mariani G, Carcangiu ML and Rogan PK: Genomic signatures for
paclitaxel and gemcitabine resistance in breast cancer derived by
machine learning. Mol Oncol. 10:85–100. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Eriksson S, Skog S, Tribukait B and
Jäderberg K: Deoxyribonucleoside triphosphate metabolism and the
mammalian cell cycle. Effects of thymidine on wild-type and dCMP
deaminase-deficient mouse S49 T-lymphoma cells. Exp Cell Res.
155:129–140. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Xu YZ and Plunkett W: Modulation of
deoxycytidylate deaminase in intact human leukemia cells. Action of
2′,2′-difluorodeoxycytidine. Biochem Pharmacol. 44:1819–1827. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tsaur I, Makarević J, Juengel E, Gasser M,
Waaga-Gasser AM, Kurosch M, Reiter M, Wedel S, Bartsch G, Haferkamp
A, et al: Resistance to the mTOR-inhibitor RAD001 elevates integrin
α2- and β1-triggered motility, migration and invasion of prostate
cancer cells. Br J Cancer. 107:847–855. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lee SY, Im SA, Park YH, Woo SY, Kim S,
Choi MK, Chang W, Ahn JS and Im YH: Genetic polymorphisms of
SLC28A3, SLC29A1 and RRM1 predict clinical outcome in patients with
metastatic breast cancer receiving gemcitabine plus paclitaxel
chemotherapy. Eur J Cancer. 50:698–705. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
O'Reilly EA, Gubbins L, Sharma S, Tully R,
Guang MH, Weiner-Gorzel K, McCaffrey J, Harrison M, Furlong F, Kell
M and McCann A: The fate of chemoresistance in triple negative
breast cancer (TNBC). BBA Clin. 3:257–275. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Rosell R, Cobo M, Isla D, Camps C and
Massuti B: Pharmacogenomics and gemcitabine. Ann Oncol. 17 (Suppl
5):v13–v16. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Rajabpour A, Afgar A, Mahmoodzadeh H,
Radfar JE, Rajaei F and Teimoori-Toolabi L: MiR-608 regulating the
expression of ribonucleotide reductase M1 and cytidine deaminase is
repressed through induced gemcitabine chemoresistance in pancreatic
cancer cells. Cancer Chemother Pharmacol. 80:765–775. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhuang J, Shen L, Yang L, Huang X, Lu Q,
Cui Y, Zheng X, Zhao X, Zhang D, Huang R, et al: TGFβ1 promotes
gemcitabine resistance through regulating the
LncRNA-LET/NF90/miR-145 signaling axis in bladder cancer.
Theranostics. 7:3053–3067. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Huang X, Taeb S, Jahangiri S, Korpela E,
Cadonic I, Yu N, Krylov SN, Fokas E, Boutros PC and Liu SK: miR-620
promotes tumor radioresistance by targeting 15-hydroxyprostaglandin
dehydrogenase (HPGD). Oncotarget. 6:22439–22451. 2015.PubMed/NCBI
|
|
27
|
Ueno H, Kiyosawa K and Kaniwa N:
Pharmacogenomics of gemcitabine: Can genetic studies lead to
tailor-made therapy? Br J Cancer. 97:145–151. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hu H, Wang Z, Li M, Zeng F, Wang K, Huang
R, Wang H, Yang F, Liang T, Huang H and Jiang T: Gene expression
and methylation analyses suggest DCTD as a prognostic factor in
malignant glioma. Sci Rep. 7:115682017. View Article : Google Scholar : PubMed/NCBI
|