Introduction
Diabetes mellitus is a chronic endocrine disorder,
characterized by hyperglycemia and other metabolic disorders of
fat, protein and electrolytes due to absolute or relative insulin
deficiency (1). Epidemiological data
suggest that diabetes-induced cardiovascular complications are a
leading cause of diabetes-related mortality and disability
(2). Diabetes-induced cardiovascular
complications include macrovascular and microvascular diseases
(3), of which vascular endothelial
cell apoptosis is considered to be an initial event and
pathological basis (4). However, the
underlying mechanism of diabetes-induced endothelial cell apoptosis
remains unknown.
Mitochondria, membrane-bound organelles located in
the cytoplasm of almost all eukaryotic cells, are commonly referred
to as cellular power plants critical to the production of energy in
the form of ATP (5). In addition,
mitochondria serve a central role in cell apoptosis (6). Damage to the mitochondria leads to
increased mitochondrial membrane permeability and subsequent
release of cytochrome c (7), an
essential component of the electron transport chain (8). Upon entering the cytoplasm, cytochrome
c interacts with apoptotic protease activating factor-1 that
triggers the activation of caspase cascades (9). Previous studies have confirmed that
hyperglycemia induces mitochondrial pathway-dependent apoptosis in
vascular endothelial cells (10),
however the underlying mechanisms remain unknown.
Increasing evidence suggests that voltage-dependent
anion channel (VDAC), an abundant protein located in the outer
mitochondrial membrane (11), serves
a role in the regulation of mitochondrial membrane permeability and
is essential for the release of cytochrome c during cell apoptosis
(12). The VDAC protein family
consists of 3 isoforms: VDAC1, VDAC2 and VDAC3, and vascular
endothelial cells mainly express VDAC1 (13). VDAC provides the pathway for the
movement of ATP and other small molecules out of the mitochondria
(14). Mitochondrial membrane
permeability is regulated by VDAC1 together with other factors
including hexokinase 2 (HK2), Bcl-2 and Bax (15). HK2 and Bcl-2 decreases, while Bax
increases mitochondrial permeability (16–18).
Once cells are exposed to apoptogenic factors, such as oxidative
stress, this leads to the VDAC1-mediated increase in mitochondrial
permeability, with the release of cytochrome c and subsequent cell
apoptosis (19).
The present study investigated the pro-apoptotic
effect of high glucose on human umbilical vein endothelial cells
(HUVECs), a commonly used cell line in diabetic vascular research
(20) and the potential underlying
mechanisms. The results of the current study indicated that high
glucose may induce HUVEC apoptosis via downregulation of HK2 and
Bcl-2 proteins, leading to decreased interactions with VDAC1.
Furthermore, increased interactions between Bax and VDAC1 lead to
the subsequent release of cytochrome c. Taken together, the results
of the current study may provide new approaches and potential
targets for the prevention of diabetes-induced cardiovascular
complications.
Materials and methods
Cell culture
HUVECs were purchased from the American Type Culture
Collection. HUVECs were cultured in Dulbecco's modified Eagle's
medium (DMEM; Thermo Fisher Scientific, Inc.) containing 5.5 mM
glucose with 10% fetal bovine serum (FBS; Thermo Fisher Scientific,
Inc.), 100 U/ml penicillin and 100 mg/ml streptomycin (Thermo
Fisher Scientific, Inc.) at 37°C in a 5% CO2-humidified
incubator. MG132 was utilized to prevent the potential
proteasome-induced degradation of protein. HUVEC cells were
pretreated with 50 µM MG132 (Selleck Chemicals) for 8 h at 37°C in
a 5% CO2-humidified incubator before culture in DMEM
containing 5.5, 16.5 or 33 mM glucose for a further 72 h at 37°C.
To examine the effect of cyclosporine A (CyA), HUVECs were treated
with 10 mM CyA (EMD Millipore) for 12 h at 37°C in a 5%
CO2-humidified incubator. Following the 12-h treatment,
HUVECs were cultured in DMEM containing 5.5 or 33 mM glucose for a
further 72 h.
Cell infection
HUVEC cells were seeded into 6-well plates at a
density of 4×105 cells/well overnight at 37°C. Virus
particles (2×107)/well [HK2 adenovirus or the control
virus; OBiO Technology (Shanghai) Corp., Ltd.] were subsequently
added and incubated for 2 h at 37°C. Cells were cultured in fresh
DMEM containing 5.5 or 33 mM glucose for a further 72 h at 37°C in
a 5% CO2-humidified incubator.
Vectors
pGL3-HK2, pGL3-Bcl-2, pRL-TK-Renilla plasmid and pCF
CREB, pCMV4 and pSV-SPORT expression vectors were gifts provided by
Professor Dongping Wei (The First Affiliated Hospital of Nanjing,
Nanjing, China). The mammalian expression vector pcDNA3.1 was
purchased from Thermo Fisher Scientific (San Jose, CA, USA). The
HK2 adenovirus and the control virus were purchased from Obio
Technology Co., Ltd. (Shanghai, China). The pSV Sport PPARγ
(Addgene plasmid no. 8886) and pCMV4 p65 (Addgene plasmid no.
21966) plasmids were gifted by Dr Bruce Spiegelman (Dana-Farber
Cancer Institute, Harvard Medical School) and Dr Warner Greene
(Howard Hughes Medical Institute, Department of Medicine, Duke
University Medical Center), respectively.
Cell transfection
HUVEC cells were seeded into 6-well plates at a
density of 4×105 cells/well overnight at 37°C. Cells
were subsequently transfected with pCF CREB (pcDNA3.1), pCMV4-p65
(pCMV4), pSV SPORT PPARγ (pSV SPORT) and flag-tagged PPARγ plasmids
(1 µg/well) using Lipofectamine® 2000 (Thermo Fisher
Scientific, Inc.). A ratio of 1 µg plasmid to 2 µl liposome was
then diluted in serum-free DMEM. After 4 h, cells were cultured in
10% FBS with DMEM for a further 48 h at 37°C in a 5%
CO2-humidified incubator prior to subsequent
experimentation.
Cell viability assay
Cell viability was examined by MTT assay. HUVEC
cells were seeded into 96-well plates at a density of
2×104 cells/well and cultured for 24 h. Subsequently,
cells were cultured in fresh DMEM containing 5.5, 16.5 or 33 mM
glucose for a further 72 h or cells were infected with the HK2
adenovirus or the control virus following culture in fresh DMEM
containing 5.5, 16.5 or 33 mM glucose for a further 72 h. Following
incubation, MTT solution (Beyotime Institute of Biotechnology) was
added to each well at a final concentration of 5 mg/ml and cells
were incubated for 1 h at 37°C. The formazan precipitate was
dissolved in 200 µl DMSO (Thermo Fisher Scientific, Inc.) and the
absorbance was measured at 570 nm using a microplate reader
(SpectraFluor; Tecan Group, Ltd.). Cell survival was expressed as a
percentage of the absorbance in the control wells.
Cell apoptosis analysis
Cell apoptosis was examined by Hoechst 33258
staining. Cells were washed three times with PBS and fixed in 4%
paraformaldehyde for ~12 h at 4°C. Cells were then washed a further
three times with PBS and incubated with 10 µg/ml Hoechst 33258
(Beyotime Institute of Biotechnology) was added to each sample for
5 min at room temperature. Cells were washed with PBS and the
coverslips were mounted with glycerol and images were captured
using a fluorescence microscope (magnification, ×200) with an
excitation wavelength at 346 nm and an emission wavelength at 460
nm.
Isolation of mitochondria
HUVEC cells were resuspended in mitochondria
isolation reagents (Cell mitochondria separation kit; cat. no.
c3601; Beyotime Institute of Biotechnology) and incubated on ice
for 15 min. Following homogenization, the cell lysate was
centrifuged at 600 × g for 10 min at 4°C, and the supernatant was
collected and further centrifuged at 11,000 × g for 10 min at 4°C.
The remaining mitochondrial pellet was collected and used for
subsequent experimentation.
Flow cytometric analysis of
mitochondrial membrane potential using JC-1 staining
Following trypsinization, cells were washed twice
with PBS, resuspended in 1 ml JC-1 staining solution (1X; Beyotime
Institute of Biotechnology) and incubated at 37°C for 20 min.
Following incubation, cells were centrifuged at 600 × g for 10 min
at 4°C and resuspended in 500 µl staining buffer (cat. no. c2005;
Beyotime Institute of Biotechnology). Mitochondrial membrane
potential was subsequently examined via flow cytometric analysis
with FlowJo VX10 software (FlowJo LLC), where fluorescent emissions
were detected at 530 nm (JC-1 FL1; red) and 575 nm (JC-1 FL2;
green). The FL2/FL1 ratio represents mitochondrial membrane
potential.
Immunoprecipitation and western blot
analysis
HUVEC cells or mitochondria were lysed using
radioimmunoprecipitation assay buffer (20 mM Tris, 150 mM NaCl, 1%
Triton X-100 and 1 mM PMSF, pH 7.5) and incubated for 30 min on
ice. Samples were then centrifuged for 10 min at 13,800 × g at 4°C.
Lysates were resuspended in 3X loading buffer (2% SDS, 6%
β-mercaptoethanol, 50 mM Tris-HCl, 10% glycerol and 0.05%
bromophenol blue, pH 6.8). Cell lysates were incubated with primary
antibody (as stated below) for 2 h at 4°C followed by incubation
with protein A-sepharose beads (Sea Biotech, Shanghai, China) for 1
h at 4°C. Protein A-sepharose beads were washed three times with
lysis buffer and centrifuged at 4°C for 1 min at 6,200 × g. The
supernatant was discarded and 2X loading buffer (2% SDS, 6%
β-mercaptoethanol, 50 mM Tris-HCl, 10% glycerol and 0.05%
bromophenol blue; pH 6.8) was added to the protein A-sepharose
beads. Samples were then boiled at 95°C for 5 min. Protein quantity
was determined using a BCA protein assay kit (Thermo Fisher
Scientific, Inc.). Protein (25 µg) was separated by 10% SDS-PAGE
and transferred to PDVF membranes (EMD Millipore). Membranes were
then blocked with 5% skimmed milk in TBST for 1 h at room
temperature and incubated with the following primary antibodies:
HK2 (1:1,000; cat. no. 2867), β-actin (1:1,000; cat. no. 3700),
Bcl-2 (1:1,000; cat. no. 2872), Bax (1:1,000; cat. no. 5023),
cytochrome c (1:1,000; cat. no. 4272), cleaved caspase-3 (1:1,000;
cat. no. 9661), PPARγ (1:1,000; cat. no. 2435), Flag (1:1,000; cat.
no. 2368), phosphorylated-CREB (1:1,000; cat. no. 9198), CREB
(1:1,000; cat. no. 9197), phosphorylated-NF-κB (1:1,000; cat. no.
3033), NF-κB (1:1,000; cat. no. 8242; all Cell Signaling
Technology, Inc.), COX4 (1:1,000; cat. no. 202554; Abcam) and VDAC1
(1:1,000; cat. no. sc-390996; Santa Cruz Biotechnology, Inc.) for
12 h at 4°C. Following primary antibody incubation, membranes were
incubated with horseradish peroxidase-conjugated secondary
antibodies for 1 h at room temperature, including anti-rabbit IgG
(1:5,000; cat. no. 7074) and anti-mouse IgG (1:5,000; cat. no.
7076; both Cell Signaling Technology, Inc.). Protein bands were
visualized using the ECL Western Blotting Substrate (EMD Millipore)
and Tanon-5200 Chemiluminescence Imager (Tanon Science and
Technology Co., Ltd.).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from HUVECs using the
TRIzol® reagent (Thermo Fisher Scientific, Inc.).
Reverse transcription was performed using a reverse transcription
kit (Vazyme Biotech Co., Ltd.). qPCR was subsequently performed
using a StepOnePlus™ Real-Time PCR System (Applied Biosystems;
Thermo Fisher Scientific, Inc.) with THUNDERBIRD®
SYBR® qPCR master mix (Vazyme Biotech Co., Ltd.). The
Thermocycling conditions were as follows: Initial polymerase
activation step at 95°C for 30 sec; followed by 40 cycles at 95°C
for 10 sec and 60°C for 30 sec. A final stage was performed at 95°C
for 15 sec, 60°C for 60 sec and 95°C for 15 sec. The following
primer sequences were used for qPCR: HK2 forward,
5′-TGCCACCAGACTAAACTAGACG-3′ and reverse,
5′-CCCGTGCCCACAATGAGAC-3′; PPARγ forward,
5′-GGGATCAGCTCCGTGGATCT-3′ and reverse,
5′-TGCACTTTGGTACTCTTGAAGTT-3′; Bcl-2 forward,
5′-GGTGGGGTCATGTGTGTGG-3′ and reverse,
5′-CGGTTCAGGTACTCAGTCATCC-3′; and GAPDH forward,
5′-GGAGCGAGATCCCTCCAAAAT-3′ and reverse,
5′-GGCTGTTGTCATACTTCTCATGG-3′. The relative expression levels of
target genes were quantified using the 2−ΔΔCq method
(21) and normalized to the internal
reference gene GAPDH.
Dual-luciferase reporter assay
HUVEC cells were seeded onto 24-well plates at a
density of 1×105 cells/well and co-transfected with
luciferase reporter plasmids 200 ng pGL3-HK2, pGL3-Bcl-2 or 10 ng
pRL-TK-Renilla and 100 ng pSV SPORT PPARγ, pSV SPORT, pCF CREB,
pcDNA3.1, pCMV4-p65 or pCMV4 expression vectors in 250 ml of
serum-free DMEM using Lipofectamine® 2000 (Thermo Fisher
Scientific, Inc.) for 4 h at 37°C. Following incubation, HUVECs
were cultured in DMEM supplemented with 10% FBS for 24 h. Following
a 24-h incubation, transfected HUVECs were cultured in DMEM
containing 5.5 mM glucose for a further 48 h. Luciferase activities
were detected using the Dual-Luciferase Reporter assay system
(Promega Corporation, Madison, WI, USA) and Firefly activity was
normalized to Renilla luciferase activity. Three biological
replicates were performed in triplicate.
Statistical analysis
Data are presented as the mean ± standard error.
Comparisons between groups were analyzed via one-way analysis of
variance (GraphPad, Inc.). P<0.05 was considered to indicate a
statistically significant result. Three biological replicates were
performed in triplicate.
Results
High glucose induces apoptosis in
HUVECs
HUVECs exposed to high concentrations (16.5 and 33
mM) of glucose demonstrated a significantly decreased cell
viability, increased cell apoptosis and cleaved caspase-3 levels,
compared with HUVECs exposed to a low (5.5. mM) glucose
concentration (Fig. 1A-C). In
addition, the mitochondrial membrane potential in HUVECs exposed to
high concentrations of glucose was significantly decreased compared
with HUVECs exposed to a low glucose concentration (Fig. 1D). These results suggest that
exposure to high glucose may induce cell apoptosis in a
mitochondria-dependent manner via impairment of mitochondrial
structure and function. The change in mitochondrial permeability
was confirmed by a reduced mitochondrial expression level and an
unchanged cellular expression level of cytochrome c (Fig. 1E and F), which is released from the
mitochondria into the cytoplasm when mitochondrial function is
disrupted (22). Since mitochondrial
permeability is controlled by VDAC1, which is regulated by Bcl-2,
Bax and HK2, Bcl-2, Bax and HK2 protein expression (19) and subcellular distribution were
examined. HUVECs exposed to high glucose concentrations
demonstrated decreased mitochondrial and cellular protein
expression levels of HK2 and Bcl-2 compared with HUVECs exposed to
a low glucose concentration (Fig. 1E and
F). Although the mitochondrial protein level of VDAC1, as well
as the cellular protein levels of VDAC1 and Bax remained unchanged,
the mitochondrial protein level of Bax was increased in HUVECs
exposed to high concentrations of glucose (Fig. 1E and F). Immunoprecipitation studies
revealed that exposure to high glucose enhanced the interaction
between VDAC1 and Bax, and reduced the interaction between VDAC1
and HK2 as well as Bcl-2 in HUVECs (Fig.
1G).
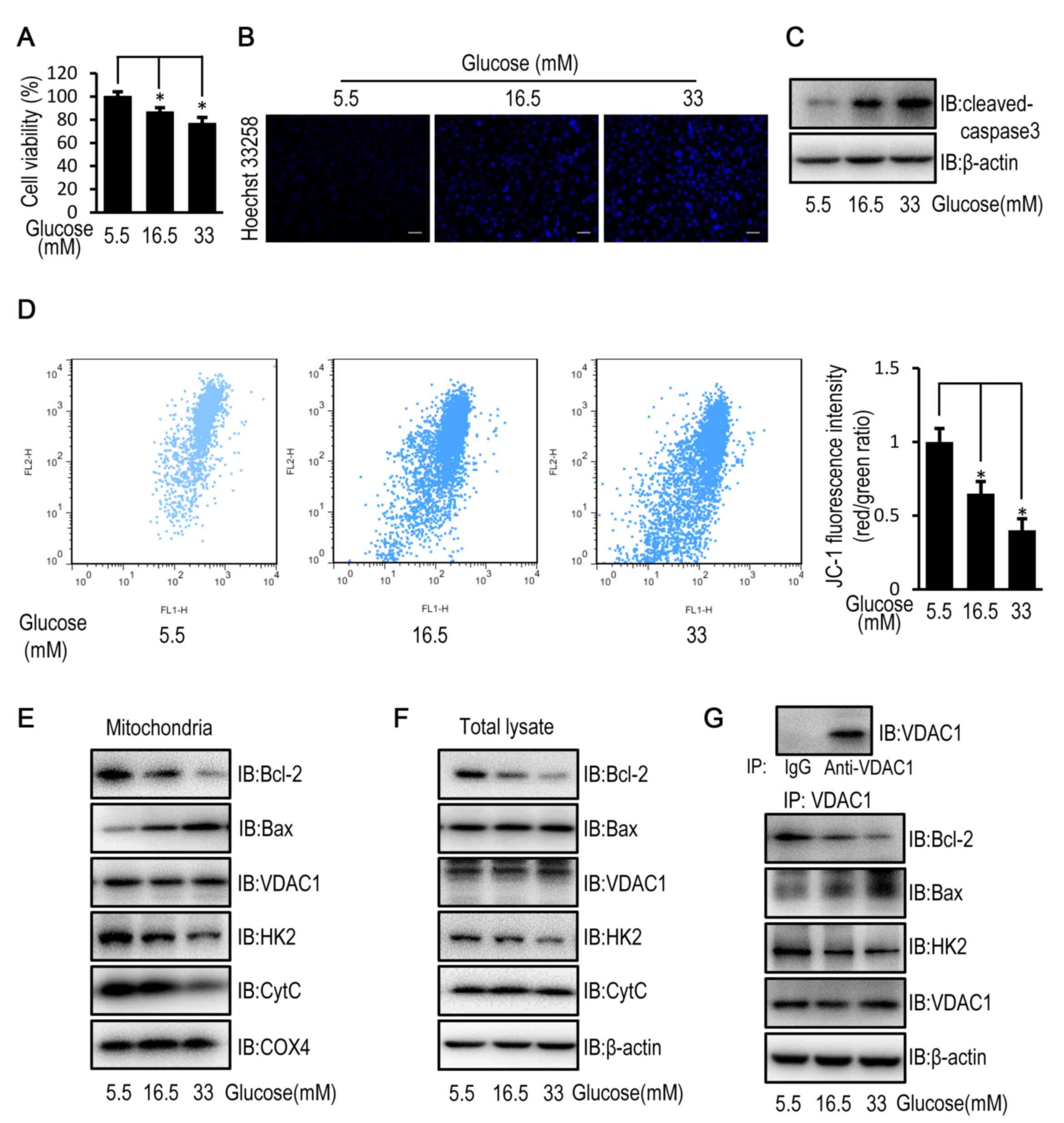 | Figure 1.High glucose induces apoptosis in
HUVECs by disturbing the mitochondrial membrane potential. HUVECs
were cultured in DMEM containing 5.5, 16.5 or 33 mM glucose for 72
h. (A) MTT assay was used to examine cell viability of HUVECs. (B)
Cell apoptosis was examined in HUVECs stained with the fluorescent
nuclear dye Hoechst 33258. Scale bar=200 mm. (C) The protein
expression level of cleaved caspase-3 was determined by western
blot analysis in HUVECs. (D) Mitochondrial membrane potential was
examined in HUVECs following JC-1 staining and flow cytometric
analysis for fluorescence emission at 530 nm (JC-1 FL1) and 575 nm
(JC-1 FL2). The FL2/FL1 ratio represents mitochondrial membrane
potential. (E) The protein expression level of Bcl-2, Bax, VDAC1,
HK2, CytC and COX4 were determined by western blot analysis in
mitochondria isolated from HUVECs. (F) The protein expression
levels of Bcl-2, Bax, VDAC1, HK2 and CytC were determined by
western blot analysis in HUVECs. (G) The protein expression level
of Bcl-2, Bax, HK2 and VDAC1 were determined by western blot
analysis in HUVECs following immunoprecipitation with VDAC1
antibody. Data are presented as the mean ± standard error.
*P<0.05 as indicated. HUVEC, human umbilical vein endothelial
cell; DMEM, Dulbecco's modified Eagle's medium; Bax, Bcl-2
associated X; VDAC1, voltage-dependent anion channel 1; HK2,
hexokinase 2; CytC, cytochrome c; COX4, cytochrome c oxidase
subunit 4; IB, immunoblotting. |
HK2 overexpression partially reverses
high glucose-induced cell apoptosis
Protein expression levels of HK2 were increased in
HUVECs following transfection with HK2 adenovirus compared with
control adenovirus (Fig. 2A). When
compared with control virus infected HUVECs, HK2 overexpression
partially reversed the decrease in cell viability, enhanced cell
apoptosis and activation of caspase-3, and reduced mitochondrial
membrane potential of HUVECs following exposure to high levels of
glucose (Fig. 2B-E). In addition,
HK2 overexpression partially reversed the decreased mitochondrial
and cellular protein expression levels of HK2 and Bcl-2 as well as
the increased mitochondrial protein expression level of Bax
observed in HUVECs compared with control virus infected HUVECs
following exposure to high levels of glucose. Furthermore, HK2
overexpression suppressed cytochrome c release (Fig. 2F and G). Furthermore,
immunoprecipitation assays revealed that HK2 overexpression
enhanced the interactions between VDAC1 and HK2 as well as Bcl-2,
and attenuated the interactions between VDAC1 and Bax (Fig. 2H). CyA, a known inhibitor of
mitochondrial permeability transition (23), was used to examine the effect of
mitochondrial permeability on cell viability and apoptosis in
HUVECs exposed to a high concentration of glucose. Treatment with
CyA partially reversed the decrease in cell viability and the
increase in cell apoptosis in HUVECs exposed to a high glucose
concentration, similarly to the aforementioned HK2 overexpression
(Fig. 2I and J).
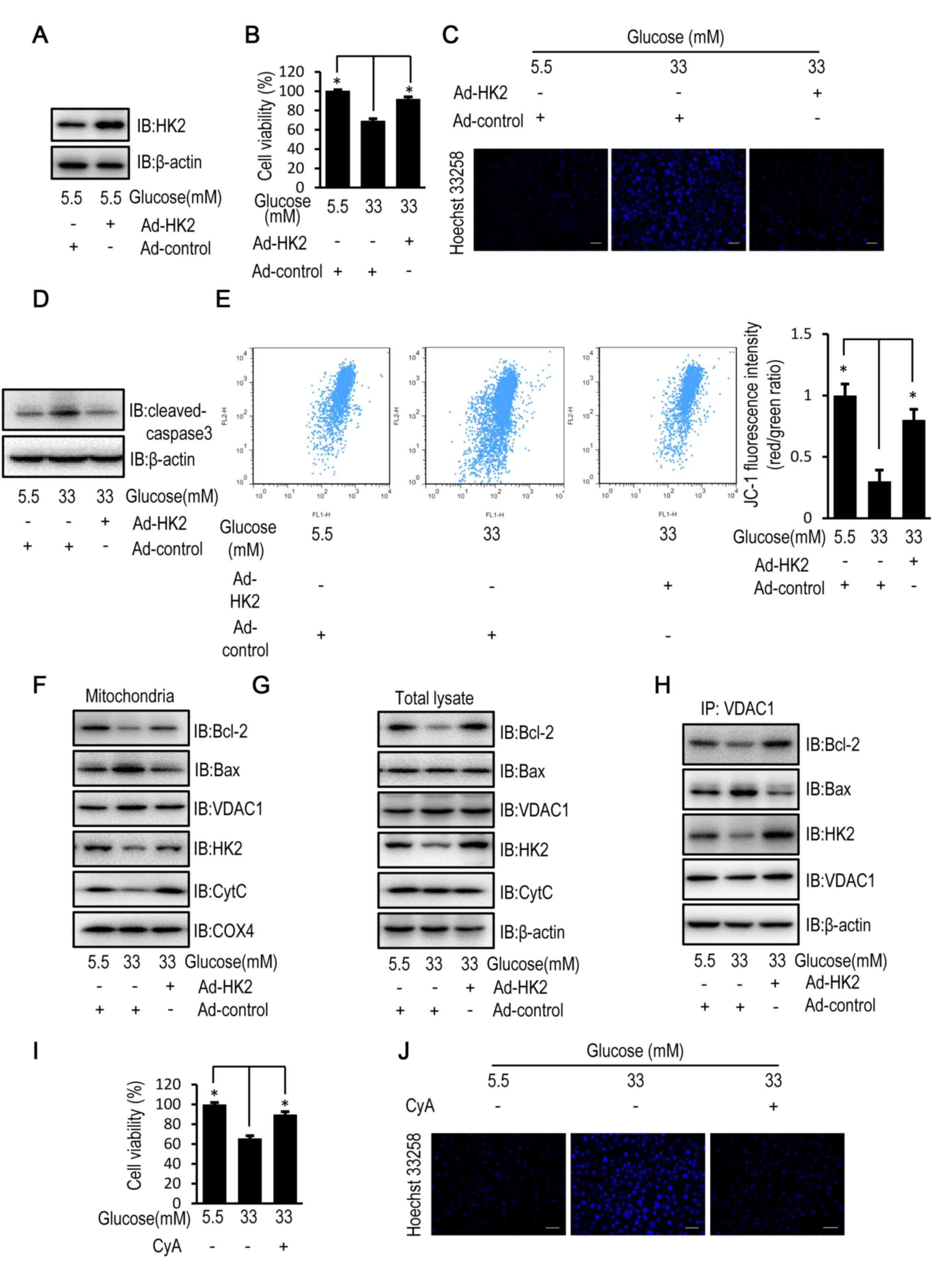 | Figure 2.HK2 overexpression reverses high
glucose-induced apoptosis in HUVECs. HUVECs were transfected with
HK2 adenovirus or control virus for 24 h. Following transfection,
HUVECs were cultured in DMEM containing 5.5, 16.5 or 33 mM glucose
for a further 72 h. (A) The protein expression level of HK2 was
determined by western blot analysis in HUVECs. (B) MTT assay was
used to examine cell viability of HUVECs. (C) Cells apoptosis was
examined in HUVECs stained with the fluorescent nuclear dye Hoechst
33258. Scale bar=200 mm. (D) The protein expression level of
cleaved caspase-3 was determined by western blot analysis in
HUVECs. (E) Mitochondrial membrane potential was examined in HUVECs
following JC-1 staining and flow cytometric analysis for
fluorescence emission at 530 nm (JC-1 FL1) and 575 nm (JC-1 FL2).
(F) The protein expression levels of Bcl-2, Bax, VDAC1, HK2, CytC
and COX4 were determined by western blot analysis in mitochondria
isolated from HUVECs. (G) The protein expression levels of Bcl-2,
Bax, VDAC1, HK2 and CytC were determined by western blot analysis
in HUVECs. (H) The protein expression levels of Bcl-2, Bax, HK2 and
VDAC1 were determined by western blot analysis in HUVECs following
immunoprecipitation with VDAC1 antibody. HUVECs were treated with
10 mM CyA for 12 h. Following 12-h treatment, HUVECs were cultured
in DMEM containing 5.5 or 33 mM glucose for a further 72 h. (I) MTT
assay was used to examine cell viability of HUVECs following
treatment with CyA. (J) Cells apoptosis was examined in HUVECs
stained with the fluorescent nuclear dye Hoechst 33258 following
treatment with CyA. Scale bar=200 µm. Data are presented as the
mean ± standard error. *P<0.05 as indicated. HK2, hexokinase 2;
HUVEC, human umbilical vein endothelial cell; DMEM, Dulbecco's
modified Eagle's medium; Bax, Bcl-2 associated X; VDAC1,
voltage-dependent anion channel 1; CytC, cytochrome c; COX4,
cytochrome c oxidase subunit 4; IB, immunoblotting. |
High glucose-induced Bcl-2
downregulation is reversed by HK2 upregulation
HK2 overexpression partially restored the relative
mRNA expression level of Bcl-2 in HUVECs exposed to high
concentrations of glucose (Fig. 3A).
Luciferase assays confirmed the transcriptional regulation of Bcl-2
by CREB and NF-κB p65 in HUVECs (Fig.
3B). Furthermore, the phosphorylation level of CREB and NF-κB,
as an indicator of activation, was examined by western blot
analysis in HUVECs. HK2 overexpression reversed the high
glucose-induced effect on the activation status of phosphorylated
CREB and NF-κB in HUVECs (Fig.
3C).
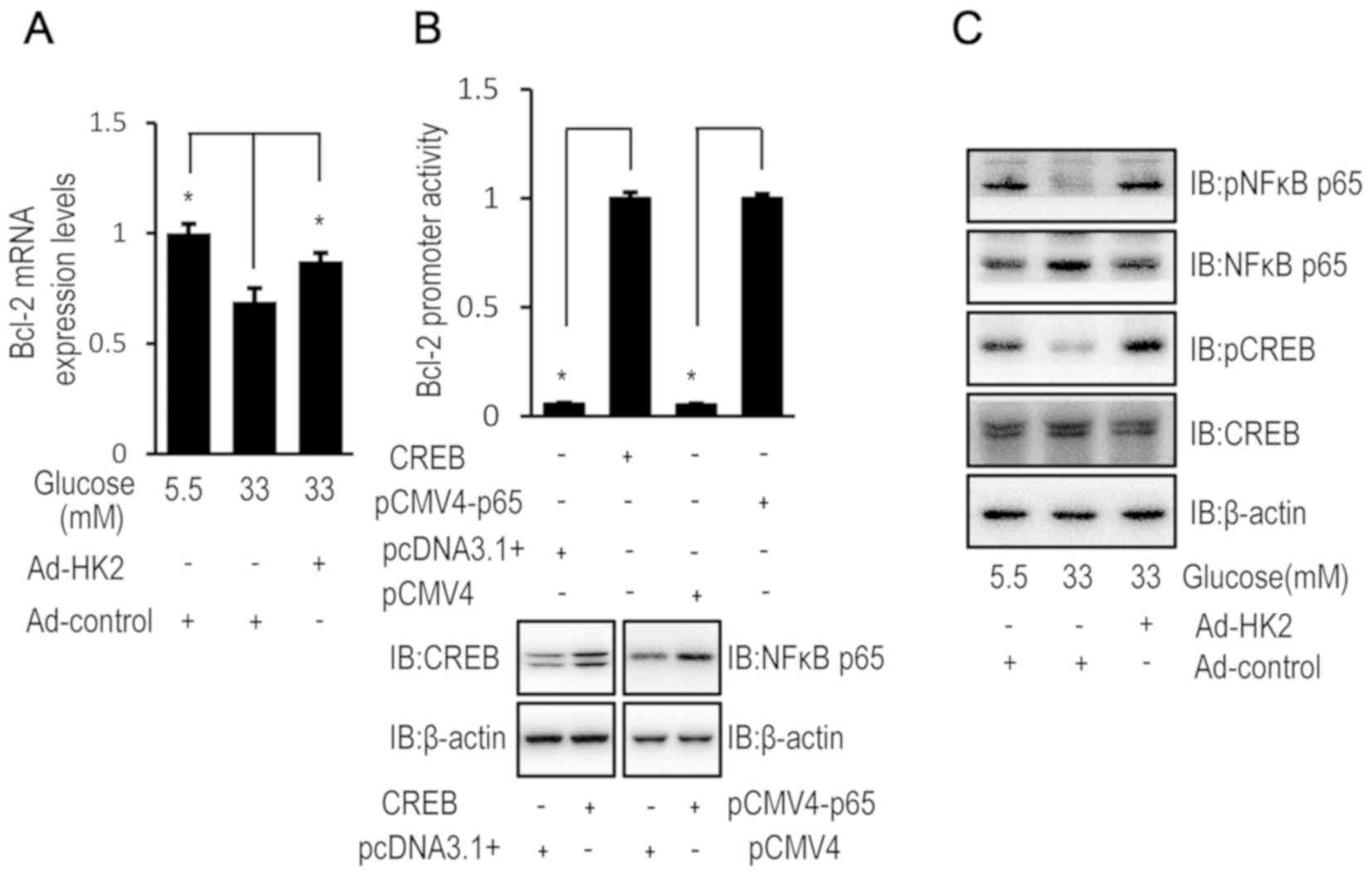 | Figure 3.High glucose downregulates Bcl-2
expression via HK2 expression. HUVECs were transfected with HK2
adenovirus or control virus for 24 h. Following transfection,
HUVECs were cultured in DMEM containing 5.5, 16.5 or 33 mM glucose
for 72 h. (A) The relative mRNA expression level of Bcl-2 was
determined by RT-qPCR in HUVECs. (B) HUVECs were co-transfected
with pCF CREB (or pcDNA3.1) and pGL3-Bcl-2 (or pRL-TK-Renilla) or
pCMV4-p65 (NF-κB) (or pCMV4) and pGL3-Bcl-2 (or pRL-TK-Renilla) for
48 h and luciferase activity was detected using the Dual-Luciferase
reporter assay. The protein expression of CREB or NF-κB were
increased in HUVECs following transfection with pCF CREB or
pCMV4-p65 compared with pcDNA3.1 or pCMV4, respectively. (C) The
protein expression levels of p-NFκB, NFκB, p-CREB and CREB were
determined by western blot analysis in HUVECs. Data are presented
as the mean ± standard error. *P<0.05 as indicated. HK2,
hexokinase 2; HUVEC, human umbilical vein endothelial cell; DMEM,
Dulbecco's modified Eagle's medium; NFκB, nuclear factor-κB; CREB,
cyclic AMP response element binding protein; RT-qPCR, reverse
transcription-quantitative polymerase chain reaction; IB,
immunoblotting. |
High glucose reduces HK2 transcription
via downregulation of PPARγ in HUVECs
To investigate the mechanisms underlying the
inhibitory effect of high glucose on HK2 expression, the relative
mRNA and protein expression levels of HK2 were determined by
RT-qPCR and western blotting, respectively, in HUVECs exposed to
high and low glucose concentrations. The relative mRNA and protein
expression levels of HK2 were decreased in HUVECs exposed to high
glucose concentrations compared with HUVECs exposed to a low
glucose concentration (Fig. 4A and
B). In addition, when MG132, a cell-permeable proteasome
inhibitor, was used to block HK2 protein degradation, a high
glucose-induced decrease in HK2 protein expression level was
observed (Fig. 4B). These results
suggest that high glucose may regulate HK2 expression at a
transcriptional level. Furthermore, PPARγ is reported to be a
transcription factor for HK2 (24).
In the current study, luciferase activity confirmed the
transcriptional regulation of HK2 by PPARγ (Fig. 4C). Although HUVECs exposed to high
glucose concentrations demonstrated decreased mRNA and protein
expression levels of endogenous PPARγ (Fig. 4D and E), high glucose had no obvious
effect on the protein expression level of flag-tagged PPARγ level
in HUVECs (Fig. 4F).
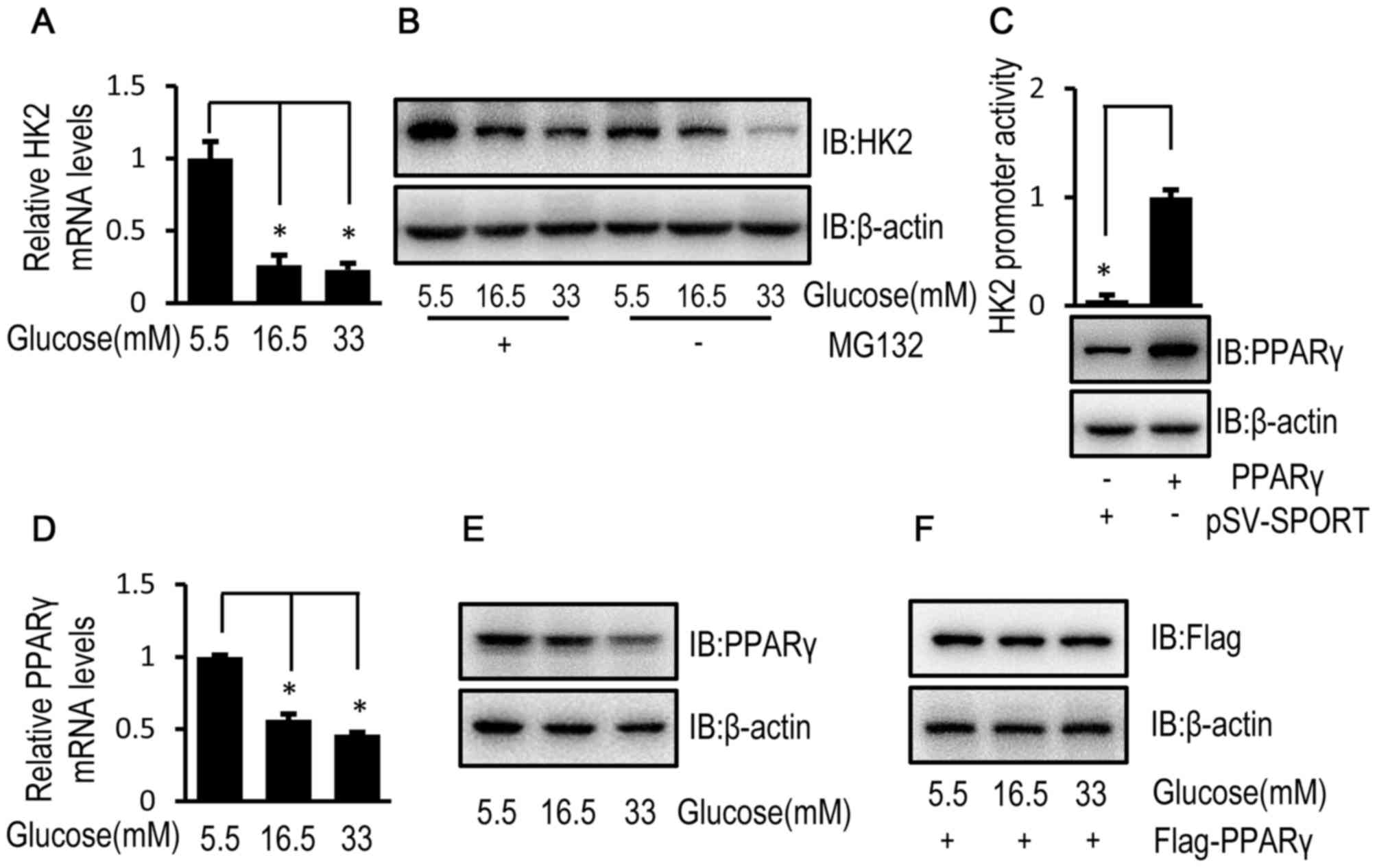 | Figure 4.High glucose inhibits HK2
transcription of HK2 via PPARγ downregulation in HUVECs. (A) HUVECs
were cultured in DMEM containing 5.5, 16.5 or 33 mM glucose for 72
h and the relative mRNA expression level of HK2 was determined by
RT-qPCR. (B) HUVECs were cultured in DMEM containing 5.5, 16.5 or
33 mM glucose for 72 h followed by treatment with MG132 (50 µM) for
8 h. Following 8-h treatment, the protein expression level of HK2
was determined by western blot analysis. (C) HUVECs were
co-transfected with pSV SPORT PPARγ (or pSV-SPORT) and pGL3-HK2 (or
pRL-TK-Renilla) into cells for 48 h and luciferase activity was
detected using the Dual-Luciferase assay. HUVECs were cultured in
DMEM containing 5.5, 16.5 or 33 mM glucose for 72 h. (D) The
relative mRNA expression level of PPARγ was determined by RT-qPCR
in HUVECs. (E) The protein expression level of PPARγ was determined
by western blot analysis in HUVECs. (F) HUVECs were transfected
with the Flag tagged PPARγ for 24 h. Following transfection, HUVECs
were cultured in medium containing 5.5, 16.5 or 33 mM glucose for
72 h. The protein expression level of Flag was determined by
western blot analysis in HUVECs. Data are presented as the mean ±
standard error. *P<0.05 as indicated. HK2, hexokinase 2; PPARγ,
peroxisome proliferator-activated receptor γ; HUVEC, human
umbilical vein endothelial cell; DMEM, Dulbecco's modified Eagle's
medium; RT-qPCR, reverse transcription-quantitative polymerase
chain reaction; MG132, cell-permeable proteasome inhibitor; IB,
immunoblotting. |
Discussion
Hyperglycemia-induced apoptosis of vascular
endothelial cells serves a role in the pathogenesis of both
macrovascular and microvascular complications of diabetes (25). In the present study, an in
vitro model of chronic hyperglycemia was established in HUVECs
following exposure to high glucose, in order to investigate the
potential mechanisms and therapeutic targets for diabetes-induced
vascular complications.
In the present study, high glucose-induced apoptosis
in HUVECs was associated with the release of mitochondrial
cytochrome c, as a result of the increase in mitochondrial membrane
permeability demonstrated by the reduced mitochondrial membrane
potential. VDAC1 plays a role in regulating mitochondrial membrane
permeability (26), following
exposure to high glucose, and its function is regulated by HK2,
Bcl-2 and Bax (27). While HK2 and
Bcl-2 reduce mitochondrial permeability, Bax increases
mitochondrial permeability (16–18). In
the current study, high glucose-induced downregulation of
mitochondrial and cellular HK2 and Bcl-2 expression, and therefore
decreased interactions with VDAC1. In addition, high glucose
induced upregulation of mitochondrial Bax by enhancing interactions
with VDAC1 without affecting total lysate Bax expression levels. As
HK2 and Bax competitively bind to VDAC1 (28), the decreased protein expression level
of HK2 is likely to be affect the interactions between Bax and
VDAC1. It was therefore hypothesized that downregulation of HK2 was
involved in high glucose-induced cell apoptosis. To examine whether
HK2 was involved in high glucose-induced cell apoptosis, HK2 was
overexpressed in HUVECs exposed to high glucose. HK2 overexpression
partially suppressed high glucose-induced cell apoptosis, by
reducing mitochondrial Bax and its interaction with VDAC1. The
antiapoptotic effect of HK2 may be achieved by directly competing
with Bax for binding to VDAC1, as there was no upregulation of
intracellular Bax observed. Furthermore, HK2 overexpression
increased the expression, mitochondrial abundance and interaction
of Bcl-2 with VDAC1. Bcl-2 is an apoptotic regulator which plays an
antiapoptotic role by binding to the N-terminal domain of VDAC1
(29). Therefore, upregulation of
Bcl-2 may also contribute to the antiapoptotic effect of HK2.
Consistent with the findings of the current study,
previous studies demonstrated that HK2 upregulated Bcl-2 expression
in several types of cancer cell lines (30), through the activation of CREB, a
Bcl-2 transcription factor, via phosphorylation (31). Despite the lack of studies regarding
the effect of long-term exposure to high glucose on CREB
phosphorylation, the current study indicated that high glucose
effectively attenuated CREB phosphorylation in HUVECs. In addition,
the current study demonstrated that high glucose attenuated
phosphorylation of NF-κB, another Bcl-2 transcriptional factor
(32). However, HK2 overexpression
partially reversed the high glucose-induced inhibition of NF-κB and
CREB phosphorylation. Therefore, the inhibition of CREB and NF-κB
phosphorylation by high glucose may reduce Bcl-2 expression,
however this was partially reversed by HK2 overexpression. The
underlying mechanism of CREB and NF-κB regulation by high glucose
and HK2 remains unknown.
To investigate the underlying mechanism by which
high glucose reduces HK2 expression in HUVECs, the mRNA and protein
levels of HK2 were examined. The current study demonstrated that
high glucose reduced the transcription of HK2 without affecting its
protein degradation rate. As high glucose may increase the
proteasome-mediated degradation of HK2, MG132 was used to prevent
this protein degradation, which resulted in a reduced difference of
protein levels between high and low glucose treatment. A previous
study revealed that PPARγ is an important transcription factor for
HK2 (25), which was confirmed in
the current study using a luciferase assay. As was the case with
HK2, high glucose reduced PPARγ expression by decreasing its mRNA
level. Therefore, high glucose-induced downregulation of PPARγ led
to suppressed HK2 transcription. However, the mechanism underlying
high glucose-mediated regulation of PPARγ expression in HUVECs
requires further investigation.
In conclusion, high glucose reduced HK2 expression
by suppressing the expression of the HK2 transcription factor
(PPARγ), which subsequently reduced Bcl-2 expression in HUVECs.
These changes weakened the interaction of mitochondrial VDAC1 with
HK2 and Bcl-2 leading to the enhanced binding of Bax to VDAC1,
which increased mitochondrial membrane permeability and induced
cell apoptosis. It is hypothesized that upregulating HK2 or PPARγ
may reduce vascular endothelial cell apoptosis and prevent
diabetes-induced vascular complications.
Acknowledgements
The authors would like to thank Dongping Wei (The
First Affiliated Hospital of Nanjing, Nanjing, China) for the
luciferase reporter plasmids pGL3-HK2 and pGL3-Bcl-2, and the pCF
CREB, pCMV4 and pSV-SPORT expression vectors. The authors would
also like to acknowledge Dr Bruce Spiegelman (Dana-Farber Cancer
Institute, Harvard Medical School) and Dr Warner Greene (Howard
Hughes Medical Institute, Department of Medicine, Duke University
Medical Center) pSV SPORT PPARγ and pCMV4 p65 expression vectors,
respectively.
Funding
The present study was funded by grants from the
Natural Science Foundation of Jiangsu Province of China (grant no.
BK20151015) and the Jiangsu Health International Exchange
Program.
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
JZ performed experiments and prepared the
manuscript. YG performed the cell apoptosis analysis. WG and XZ
performed the cell viability assay. MP designed and directed the
experiments, and wrote the manuscript. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
HK2
|
hexokinase 2
|
|
VDAC1
|
voltage-dependent anion channel 1
|
|
CyA
|
cyclosporine A
|
|
CREB
|
cyclic AMP response element binding
protein
|
|
NF-κB
|
nuclear factor-κB
|
|
PPARγ
|
peroxisome proliferator-activated
receptor γ
|
References
|
1
|
Wang N, Cheng J, Han B, Li Q, Chen Y, Xia
F, Jiang B, Jensen MD and Lu Y: Exposure to severe famine in the
prenatal or postnatal period and the development of diabetes in
adulthood: An observational study. Diabetologia. 60:262–269. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ervasti J, Virtanen M, Lallukka T, Pentti
J, Kjeldgård L, Mittendorfer-Rutz E, Tinghög P and Alexanderson K:
Contribution of comorbid conditions to the association between
diabetes and disability pensions: A population-based nationwide
cohort study. Scand J Work Environ Health. 42:209–216.
2016.PubMed/NCBI
|
|
3
|
Guo R, Liu B, Wang K, Zhou S, Li W and Xu
Y: Resveratrol ameliorates diabetic vascular inflammation and
macrophage infiltration in db/db mice by inhibiting the NF-κB
pathway. Diab Vasc Dis Res. 11:92–102. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wang Q, Zhang M, Torres G, Wu S, Ouyang C,
Xie Z and Zou MH: Metformin suppresses diabetes-accelerated
atherosclerosis via the inhibition of Drp1-mediated mitochondrial
fission. Diabetes. 66:193–205. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sturm A, Mollard V, Cozijnsen A, Goodman
CD and McFadden GI: Mitochondrial ATP synthase is dispensable in
blood-stage Plasmodium berghei rodent malaria but essential in the
mosquito phase. Proc Natl Acad Sci U S A. 112:10216–10223. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gottlieb RA: Cell death pathways in acute
ischemia/reperfusion injury. J Cardiovasc Pharmacol Ther.
16:233–238. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Balk J, Leaver CJ and Mccabe PF:
Translocation of cytochrome c from the mitochondria to the cytosol
occurs during heat-induced programmed cell death in cucumber
plants. FEBS Lett. 463:151–154. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wen Y, Li W, Poteet EC, Xie L, Tan C, Yan
LJ, Ju X, Liu R, Qian H, Marvin MA, et al: Alternative
mitochondrial electron transfer as a novel strategy for
neuroprotection. J Biol Chem. 286:16504–16515. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Shalaeva DN, Dibrova DV, Galperin MY and
Mulkidjanian AY: Modeling of interaction between cytochrome c and
the WD domains of Apaf-1: Bifurcated salt bridges underlying
apoptosome assembly. Biol Direct. 10:292015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Safi SZ, Batumalaie K, Mansor M, Chinna K,
Mohan S, Karimian H, Qvist R, Ashraf MA and Yan GO: Glutamine
treatment attenuates hyperglycemia-induced mitochondrial stress and
apoptosis in umbilical vein endothelial cells. Clinics (Sao Paulo).
70:569–576. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Shoshan-Barmatz V, Krelin Y and
Shteinfer-Kuzmine A: VDAC1 functions in Ca(2+) homeostasis and cell
life and death in health and disease. Cell Calcium. 69:81–100.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ben-Hail D, Begas-Shvartz R, Shalev M,
Shteinfer-Kuzmine A, Gruzman A, Reina S, De Pinto V and
Shoshan-Barmatz V: Novel compounds targeting the mitochondrial
protein VDAC1 inhibit apoptosis and protect against mitochondria
dysfunction. J Biol Chem. 291:24986–25003. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shoshan-Barmatz V and Mizrachi D: VDAC1
(voltage-dependent anion channel 1). Atlas Genet Cytogenet Oncol
Haematol. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shoshanbarmatz V and Mizrachi D: VDAC1:
From structure to cancer therapy. Front Oncol. 2:1642012.PubMed/NCBI
|
|
15
|
Shi Y, Chen J, Weng C, Chen R, Zheng Y,
Chen Q and Tang H: Identification of the protein–protein contact
site and interaction mode of human VDAC1 with Bcl-2 family
proteins. Biochem Biophys Res Commun. 305:989–996. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhou H, Zhang Y, Hu S, Shi C, Zhu P, Ma Q,
Jin Q, Cao F, Tian F and Chen Y: Melatonin protects cardiac
microvasculature against ischemia/reperfusion injury via
suppression of mitochondrial fission-VDAC1-HK2-mPTP-mitophagy axis.
J Pineal Res. 63:e124132017. View Article : Google Scholar
|
|
17
|
Sharpe JC, Arnoult D and Youle RJ: Control
of mitochondrial permeability by Bcl-2 family members. Biochim
Biophys Acta. 1644:107–113. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Pastorino JG, Chen ST, Tafani M, Snyder JW
and Farber JL: The overexpression of Bax produces cell death upon
induction of the mitochondrial permeability transition. J Biol
Chem. 273:7770–7775. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Abu-Hamad S, Arbel N, Calo D, Arzoine L,
Israelson A, Keinan N, Ben-Romano R, Friedman O and Shoshan-Barmatz
V: The VDAC1 N-terminus is essential both for apoptosis and the
protective effect of anti-apoptotic proteins. J Cell Sci.
122:1906–1916. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zitman-Gal T, Green J, Pasmanik-Chor M,
Golan E, Bernheim J and Benchetrit S: Vitamin D manipulates
miR-181c, miR-20b and miR-15a in human umbilical vein endothelial
cells exposed to a diabetic-like environment. Cardiovasc Diabetol.
13:82014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta) C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Gogvadze V, Orrenius S and Zhivotovsky B:
Multiple pathways of cytochrome c release from mitochondria in
apoptosis. Biochim Biophys Acta. 1757:639–647. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Varela AT, Gomes AP, Simões AM, Teodoro
JS, Duarte FV, Rolo AP and Palmeira CM: Indirubin-3′-oxime impairs
mitochondrial oxidative phosphorylation and prevents mitochondrial
permeability transition induction. Toxicol Appl Pharmacol.
233:179–185. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Panasyuk G, Espeillac C, Chauvin C,
Pradelli LA, Horie Y, Suzuki A, Annicotte JS, Fajas L, Foretz M,
Verdeguer F, et al: PPARγ contributes to PKM2 and HK2 expression in
fatty liver. Nat Commun. 3:6722012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Amore A, Cirina P, Conti G, Cerutti F,
Bagheri N, Emancipator SN and Coppo R: Amadori-configurated albumin
induces nitric oxide-dependent apoptosis of endothelial cells: A
possible mechanism of diabetic vasculopathy. Nephrol Dial
Transplant. 19:53–60. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zamarin D, García-Sastre A, Xiao X, Wang R
and Palese P: Influenza virus PB1-F2 protein induces cell death
through mitochondrial ANT3 and VDAC1. Plos Pathog. 1:e42005.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Huang L, Han J, Ben-Hail D, He L, Li B,
Chen Z, Wang Y, Yang Y, Liu L, Zhu Y, et al: A new fungal diterpene
induces VDAC1-dependent apoptosis in bax/bak-deficient cells. J
Biol Chem. 290:23563–23578. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tomasello MF, Guarino F, Reina S, Messina
A and De Pinto V: The voltage-dependent anion selective channel 1
(VDAC1) topography in the mitochondrial outer membrane as detected
in intact cell. Plos One. 8:e815222013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Monaco G, Decrock E, Arbel N, van Vliet
AR, La Rovere RM, De Smedt H, Parys JB, Agostinis P, Leybaert L,
Shoshan-Barmatz V and Bultynck G: The BH4 domain of anti-apoptotic
Bcl-XL, but not that of the related Bcl-2, limits the
voltage-dependent anion channel 1 (VDAC1)-mediated transfer of
pro-apoptotic Ca2+ signals to mitochondria. J Biol Chem.
290:9150–9161. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Rho M, Kim J, Jee CD, Lee YM, Lee HE, Kim
MA, Lee HS and Kim WH: Expression of type 2 hexokinase and
mitochondria-related genes in gastric carcinoma tissues and cell
lines. Anticancer Res. 27:251–258. 2007.PubMed/NCBI
|
|
31
|
Meller R, Minami M, Cameron JA, Impey S,
Chen D, Lan JQ, Henshall DC and Simon RP: CREB-mediated Bcl-2
protein expression after ischemic preconditioning. J Cereb Blood
Flow Metab. 25:234–346. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sheu ML, Ho FM, Yang RS, Chao KF, Lin WW,
Lin-Shiau SY and Liu SH: High glucose induces human endothelial
cell apoptosis through a phosphoinositide 3-kinase-regulated
cyclooxygenase-2 pathway. Arterioscler Thromb Vasc Biol.
25:539–545. 2005. View Article : Google Scholar : PubMed/NCBI
|














