Introduction
In recent years, a vast amount of studies have
demonstrated that non-coding RNAs (ncRNAs) have important
biological functions. Depending on the length of the nucleic acid
strands, ncRNAs may be classified as small non-coding RNA or as
long non-coding RNA (lncRNA). The former may be further sub-divided
into microRNA (miRNA), piwi-interacting RNA and small interfering
RNA (siRNA) (1). miRNAs are a class
of small non-coding RNA with lengths of 18 to 25 nucleotides. They
bind to the 3′-untranslated regions (UTR) of target mRNA by
complete or incomplete complementary base pairing, resulting in
suppression of translation of the target gene, induction of RNA
silencing or mRNA degradation, and thereby regulation of gene
expression. Each miRNA is able to regulate hundreds of genes;
conversely, each gene may be the target of several miRNAs. In
short, miRNAs and their target genes form complex regulatory
networks that participate in biological processes, including
development, proliferation, differentiation and apoptosis (2,3). miRNAs
may be sub-divided into tumorigenic miRNA and tumor suppressor
miRNA, based on their role and their target genes in tumors.
Compared with miRNAs, lncRNAs have relatively long nucleotide
strands, and form specific and complex secondary structures that
not only provide several sites for protein binding, but also
interact specifically and dynamically with DNA and RNA through the
principle of complementary base pairing. In addition to being
directly involved in regulation of gene expression, lncRNAs may
also act as competing endogenous (ce)RNAs to compete with other RNA
transcripts for the same miRNAs, thus mediating the interaction and
regulation between miRNAs and mRNAs (4). The hypothesis of ceRNAs as novel
regulators of gene expression was proposed recently. Serving as
ceRNAs, lncRNAs, pseudogene transcripts and mRNA transcripts bind
to miRNA competitively with common miRNA response elements (MREs),
thereby regulating gene expression and cell function (5).
A large number of studies have confirmed that
regulatory ceRNA networks of lncRNAs, miRNAs and mRNAs are
associated with the pathogenesis of various cancer types, including
gastric, lung and prostate cancer (6–8).
However, integration analyses of lncRNA-associated ceRNA networks
in glioblastoma multiforme (GBM) are rare. In the present study,
the expression profiles of mRNAs, lncRNAs and miRNAs in GBM were
comprehensively analyzed using cohorts from The Cancer Genome Atlas
(TCGA) database and Gene Expression Omnibus (GEO), and a
GBM-specific and lncRNA-associated ceRNA network was then
constructed. All datasets of GBM and corresponding non-tumor
samples from TCGA were used to establish an lncRNA-associated ceRNA
network that is expected to clarify the interactions of the
lncRNA-miRNA-mRNA network in GBM, as well as the molecular
mechanisms involved in tumorigenesis and development of GBM.
Materials and methods
Analysis of expression profiles of
mRNAs, lncRNAs and miRNAs in GBM and adjacent normal tissues
The method by Fan and Liu (9) was adopted for the design of the present
study. Datasets including the quantified expression levels of mRNAs
and lncRNAs in GBM were obtained from TCGA (https://cancergenome.nih.gov/). To compare lncRNA and
mRNA expression signatures in GBM, all datasets that included GBM
and non-tumoral brain samples were selected. The edgeR package was
used to screen for differentially expressed mRNAs (DEmRNAs) and
DElncRNAs in GBM and adjacent normal tissues. The expression values
of lncRNAs and mRNAs were obtained by background correction and
quantile normalization (10).
DEmiRNAs were obtained by analyzing the dataset of GSE25631
(11) with GEO2R, a GEO online tool
(https://www.ncbi.nlm.nih.gov/geo/geo2r/). The
inclusion criteria were |log2 fold-change| (|log2FC|) <2 and a
false discovery rate (FDR)<0.01 (12). Subsequently, the gplots package in R
software was utilized to visualize meaningful RNAs with significant
differences.
Construction of a ceRNA network in
GBM
Interactions between DElncRNA and DEmiRNA were
predicted using the miRcode database (13). mRNAs targeted by DEmiRNAs were
retrieved from the databases TargetScan, miRTarBase and miRDB
(14–16).
DEmRNA candidates targeted by DEmiRNAs were
determined only when the mRNAs were recognized by all three
databases and then overlaid with DEmRNAs. Subsequently, a
lncRNA-miRNA-mRNA ceRNA network was constructed based on the
DEmiRNA-DElncRNA and DEmiRNA-DEmRNA interactions, and was
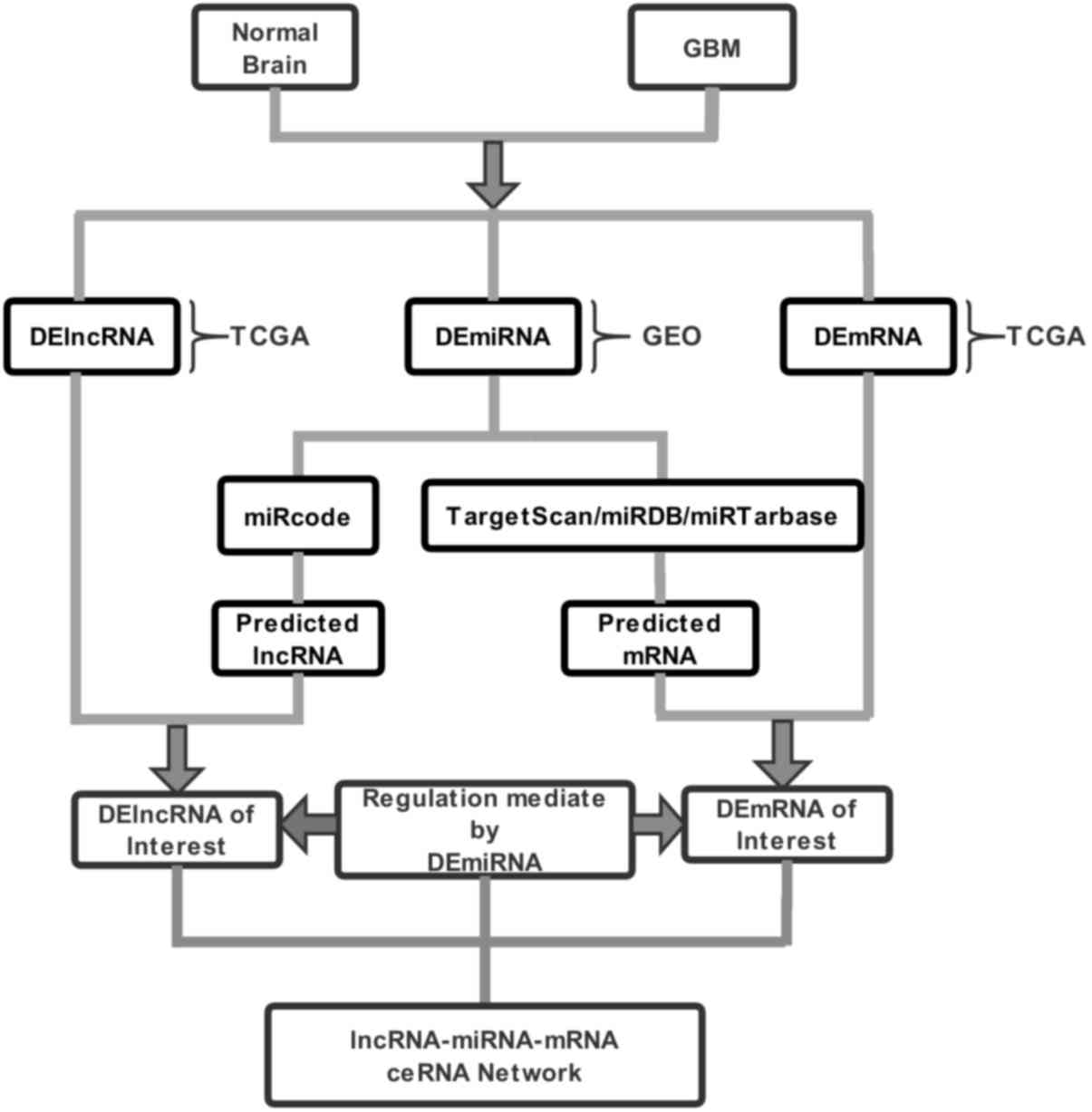
visualized using Cytoscape software (https://cytoscape.org/). The flow chart depicting the
strategy for the lncRNA-microRNA-mRNA ceRNA network analysis is
provided in Fig. 1. Pearson
correlation analysis was used to assess the correlations among the
expression levels of the ceRNAs.
Analysis of the influence of important
RNAs in GBM with patient survival
For each specific DEmRNA and DElncRNA in the ceRNA
network, GBM patients were divided into high- and low-expression
groups using the median expression value as a cut-off, and the
Kaplan-Meier survival curves were plotted. Subsequently, the
differences in survival time between the high-expression group and
the low-expression group were evaluated by using the log-rank test.
The mRNAs and lncRNAs that were significantly associated with
survival of GBM patients were thereby identified. P<0.05 was
considered to indicate statistical significance.
Functional enrichment analysis
Functional enrichment analysis of DEmRNAs in the
ceRNA network was performed to reveal the functional significance
of these mRNAs in the genesis of GBM. An online tool, the Database
for Annotation, Visualization and Integrated Discovery (DAVID;
http://david.ncifcrf.gov) was used for gene
ontology (GO) functional enrichment analysis (17). Furthermore, Kyoto Encyclopedia of
Genes and Genomes (KEGG) pathway enrichment analysis was performed
by using the clusterprofiler package (18) in R software. P<0.05 was set as the
threshold for statistical significance in the GO and KEGG
enrichment analysis.
Results
Cancer-specific mRNAs, lncRNAs and
miRNAs in GBM
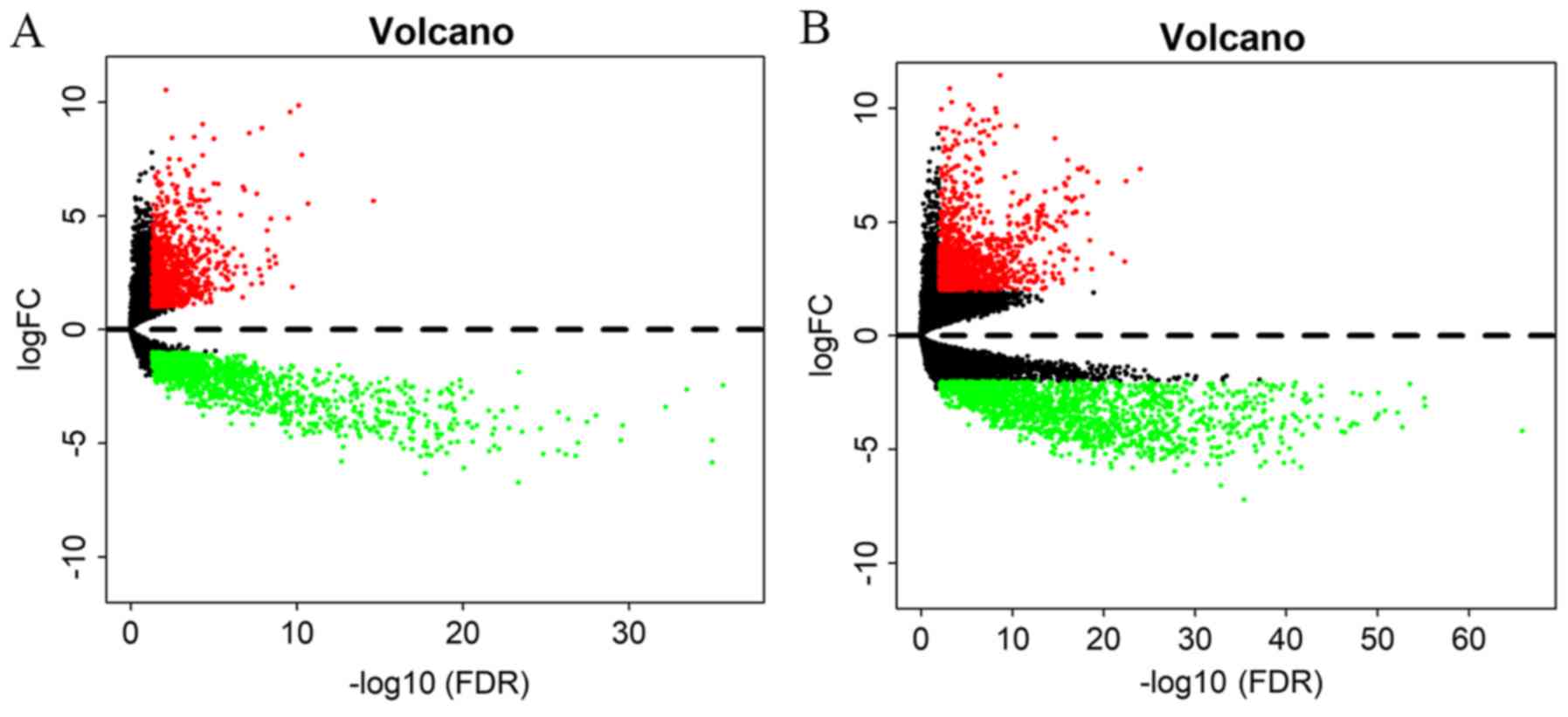
By applying the screening criteria, 2,932 DEmRNAs
between GBM and normal tissues (1,341 up- and 1,591 downregulated)
were identified. Furthermore, 2,291 DElncRNAs (981 up- and 1,310
downregulated) and 42 DEmiRNAs (27 up- and 15 downregulated) in GBM
vs. normal tissues were identified. All DElncRNAs and DEmRNAs were
presented in the two dimensions of -log10(FDR) and |logFC| through
a volcano plot. The expression levels of all of the lncRNAs and
mRNAs were standardized into the averages of samples (Fig. 2).
DElncRNA-DEmiRNA interaction predicted
with the miRcode database
The lncRNAs targeted by certain miRNAs were
predicted by using the miRcode database. Only interactions between
DElncRNAs and DEmiRNAs were selected for construction of the ceRNA
network. A total of 334 interaction pairs were identified between
the 159 DElncRNAs and 7 DEmiRNAs.
Prediction of DEmRNAs targeted by
DEmiRNAs
The 7 DEmiRNAs identified in the abovementioned
steps were inputted into the TargetScan, miRTarBase and miRDB
databases to search for their target mRNAs. In all three databases,
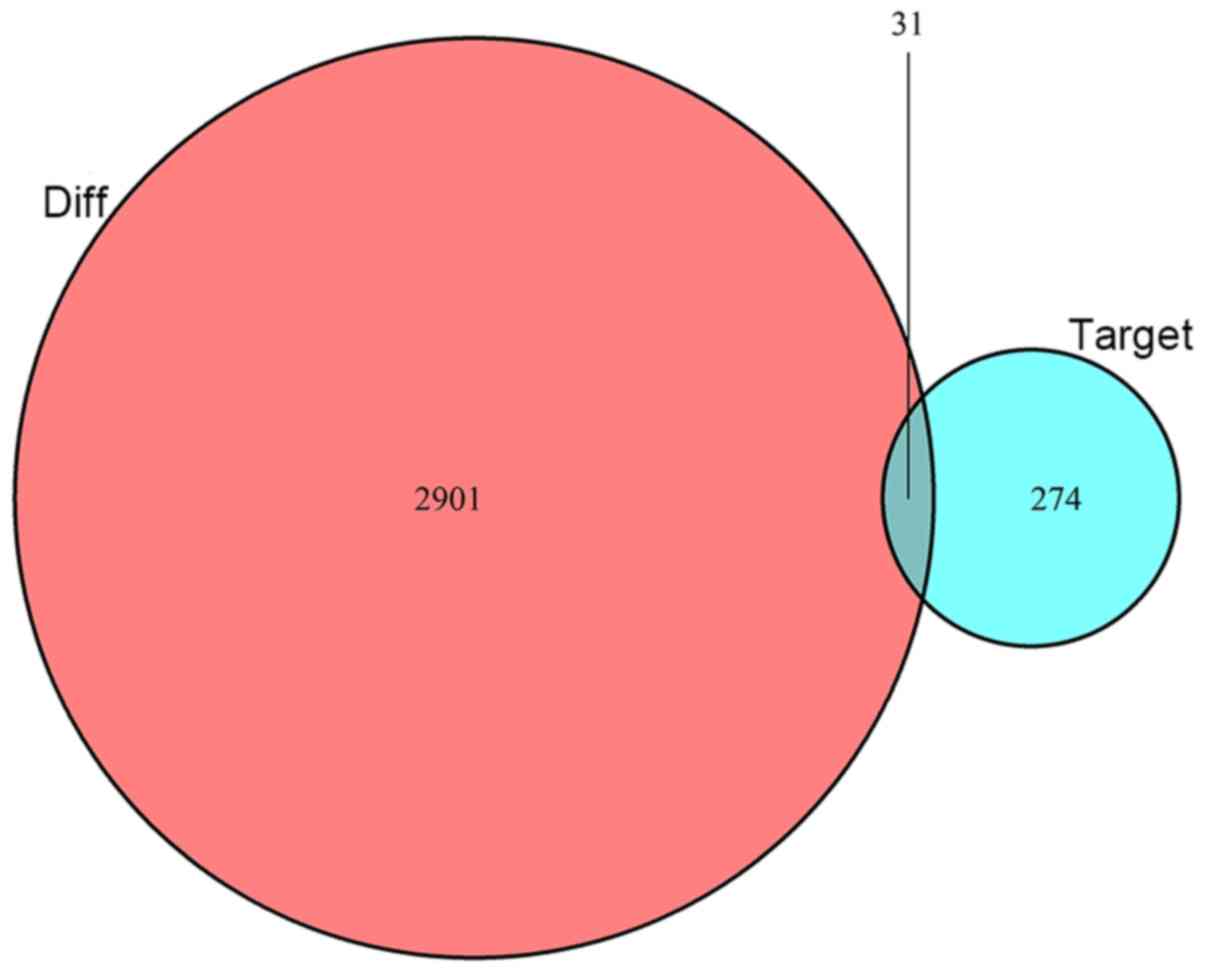
a total of 305 mRNAs were identified as targets of these DEmiRNAs.
These 305 candidate mRNAs were intersected with 2,932 DEmRNA
candidates predicted by miRcode with 31 DEmRNAs differentially
expressed and shared as targets (Fig.
3). To emphasize the ceRNA characteristics of the network, only
DEmiRNAs interacting with DEmRNAs and DElncRNAs were introduced
into the ceRNA network. Finally, 7 DEmiRNAs, 31 DEmRNAs and 159
DElncRNAs were selected for construction of the ceRNA network.
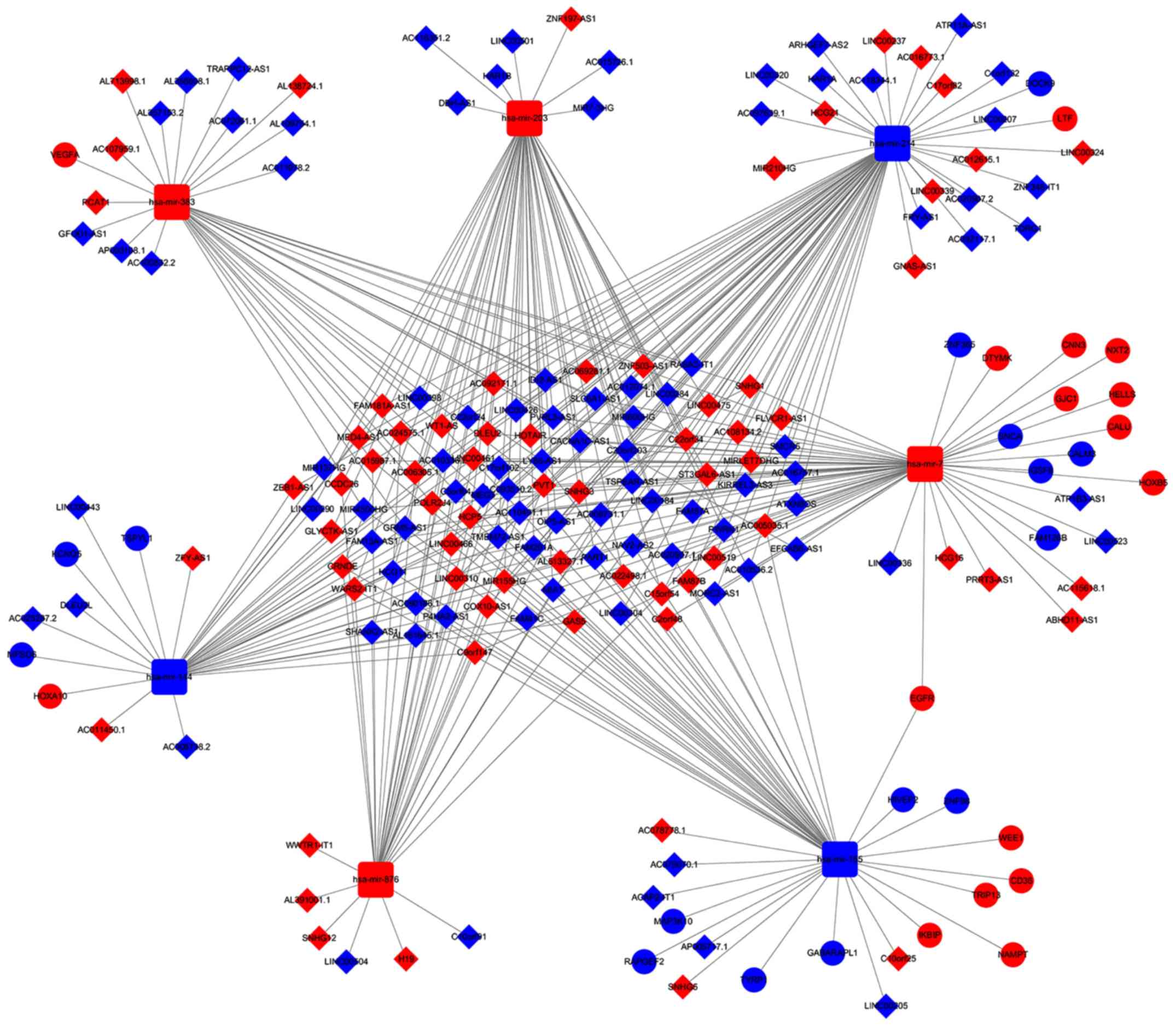
ceRNA network in GBM
According to the abovementioned data and results, a
lncRNA-miRNA-mRNA ceRNA network incorporating 197 molecules and 367
interactions (335 DEmiRNA-DElncRNA and 32 DEmiRNA-DEmRNA
interactions) was constructed. The top eight DElncRNAs targeted by
most DEmiRNAs are provided in Table
I. Only five of seven DEmiRNAs with their target DEmRNAs in the
ceRNA network are presented in Table
II as two DEmiRNAs did not have target DEmRNAs. The network was
visualized by Cytoscape and displayed in Fig. 4. The ceRNA network suggests the
possibility of indirect interactions between DElncRNAs and DEmRNAs.
In order to confirm this, a Pearson correlation analysis was
performed, revealing a positive correlation of the expression
levels of certain RNAs (Figs. 5 and
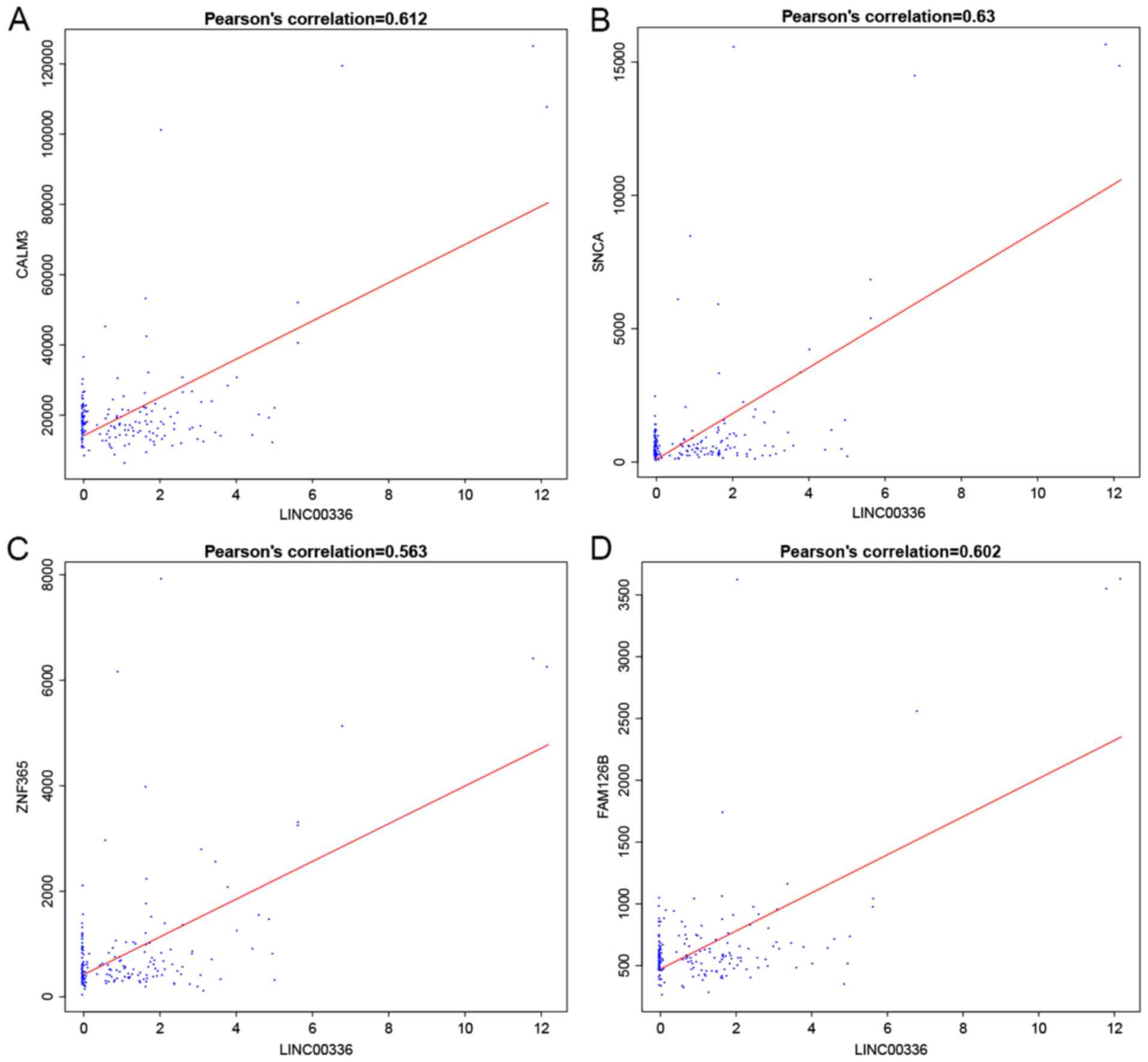
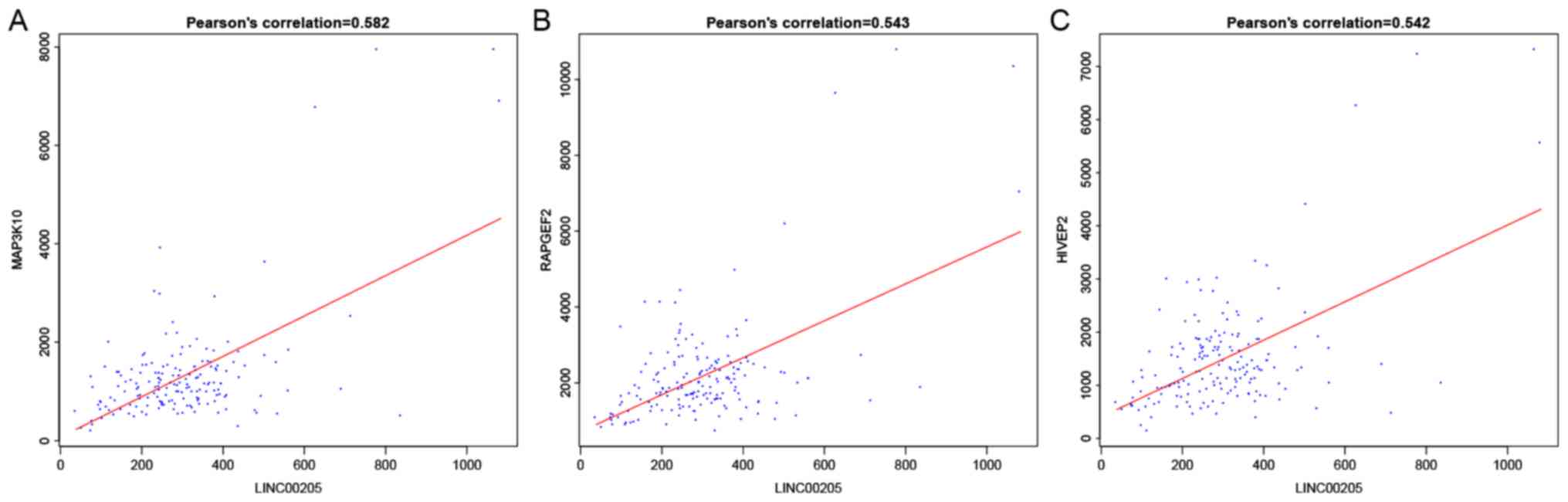
6). For instance, LINC00336
interacted with calmodulin (CALM3), synuclein α (SNCA), zinc finger
protein 365 (ZNF365) and family with sequence similarity 126 member
B (FAM126B), which was mediated by LINC00205, and Homo
sapiens (has)-miR-7 interacted with mitogen-activated protein
kinase kinase kinase 10 (MAP3K10), HIV type I enhancer binding
protein 2 (HIVEP2) and Rap guanine nucleotide exchange factor 2
(RARGEF2), which was mediated by hsa-miR-155 (Figs. 5 and 6).
 | Table I.Top 8 DElncRNAs putatively targeted by
most DEmiRNAs in the competing endogenous RNA network. |
Table I.
Top 8 DElncRNAs putatively targeted by
most DEmiRNAs in the competing endogenous RNA network.
| DElncRNAs | DEmiRNAs |
|---|
| HOTAIR | hsa-miR-203,
hsa-miR-214 |
| H19 | hsa-miR-876 |
| WT1-AS | hsa-miR-155,
hsa-miR-203, hsa-miR-383 |
| AC022498.1 | hsa-miR-155 |
| LINC00466 | hsa-miR-155,
hsa-miR-214, hsa-miR-144 |
| WARS2-IT1 | hsa-miR-876,
hsa-miR-144, hsa-miR-155, hsa-miR-203 |
| LINC00475 | hsa-miR-203,
hsa-miR-214 |
| CRNDE | hsa-miR-876,
hsa-miR-144, hsa-miR-155, hsa-miR-203 |
| Table II.DEmiRNAs with their target DEmRNAs in
the competing endogenous RNA network. |
Table II.
DEmiRNAs with their target DEmRNAs in
the competing endogenous RNA network.
| DEmiRNAs | DEmRNAs |
|---|
| hsa-miR-155 | HIVEP2, ZNF98, NAMPT,
WEE1, TYRP1, TRIP13, IKBIP, GABARAPL1, CD36, MAP3K10, EGFR,
RAPGEF2 |
| hsa-miR-7 | DTYMK, FAM126B, GJC1,
ZNF365, IGSF8, NXT2, CALU, CNN3, CALM3, SNCA, HELLS, EGFR,
HOXB5 |
| hsa-miR-144 | KCNQ5, HOXA10,
TSPYL1, MFSD6 |
| hsa-miR-214 | LTF, DOCK9 |
| hsa-miR-383 | VEGFA |
| hsa-miR-876 | – |
| hsa-miR-203 | – |
Survival analysis
In the Kaplan-Meier analysis, a total of 14
DElncRNAs and 2 DEmRNAs were identified to be significantly
associated with overall survival (Figs.
7 and 8). Among these, the
expression of AC022498.1, mediator complex subunit 4
(MED4)-antisense 1 (AS1), zinc finger E-box binding homeobox 1
(ZEB1)-AS1 and zinc finger protein 197 (ZNF197)-AS1 was positively
associated with survival (Fig. 7);
GBM patients with higher expression levels of these RNAs in their
tumor tissues tended to have a longer survival time compared with
those with lower expression levels of these RNAs. Furthermore, the
two DEmRNAs and the remaining 10 DElncRNAs were deemed risky and
negatively associated with overall survival time.
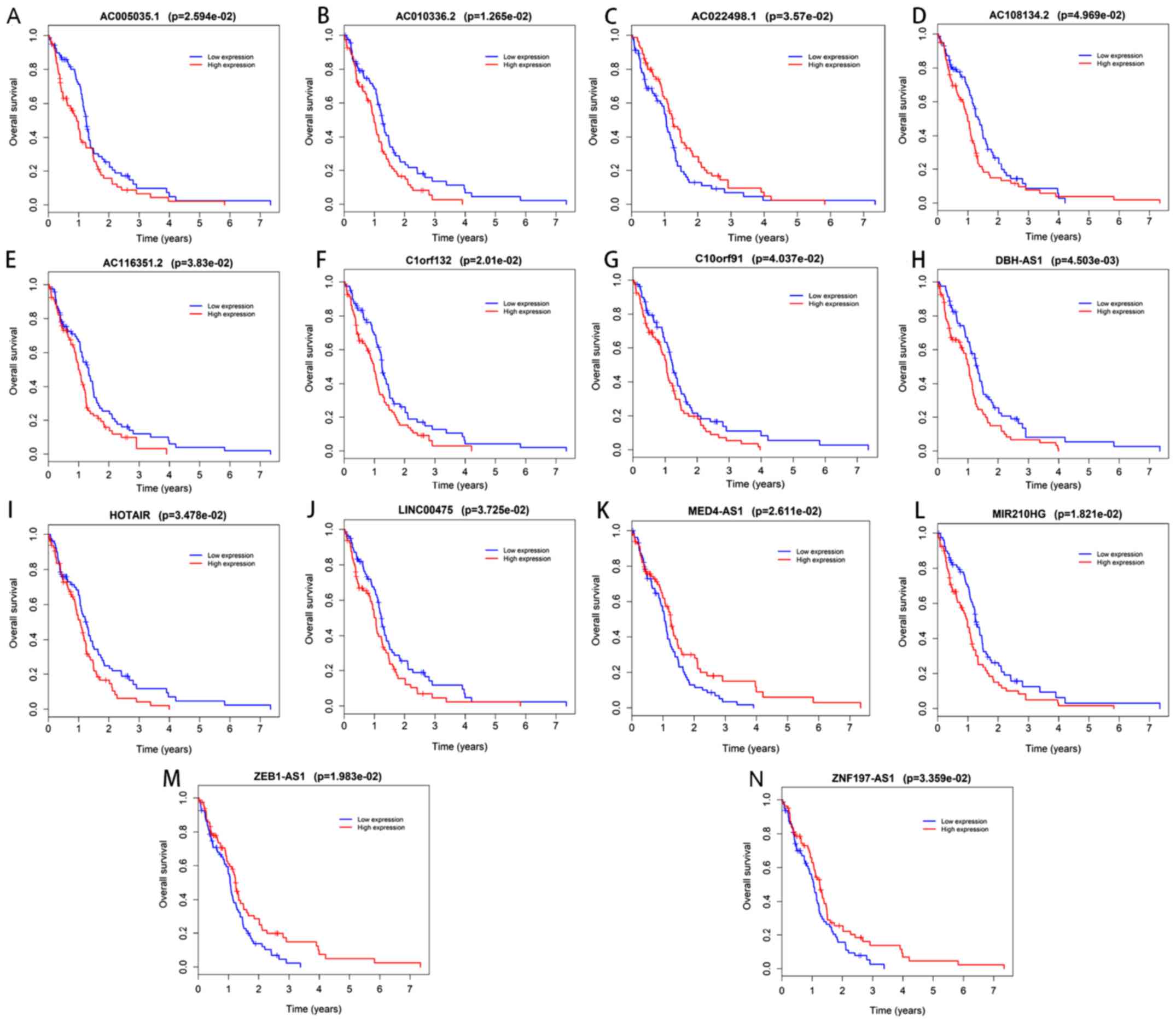 | Figure 7.Kaplan-Meier survival curves for the
14 differentially expressed long non-coding RNAs significantly
associated with overall survival of glioblastoma multiforme
patients. Kaplan-Meier survival curves for (A) AC005035.1, (B)
AC010336.2, (C) AC022498.1, (D) AC108134.2, (E) AC116351.2, (F)
C1orf132, (G) C10orf91, (H) DBH-AS1, (I) HOTAIR, (J) LINC00475, (K)
MED4-AS1, (L) MIR210HG, (M) ZEB1-AS1 and (N) ZNF197-AS1. LINC00475,
long intergenic non-protein coding RNA 475; AS1, antisense 1; DBH,
dopamine β-hydroxylase; MED4, mediator complex subunit 4; MIR210HG,
microRNA 210 host gene; ZEB1, zinc finger E-box binding homeobox 1;
ZNF197, zinc finger protein 197. |
Functional enrichment analysis
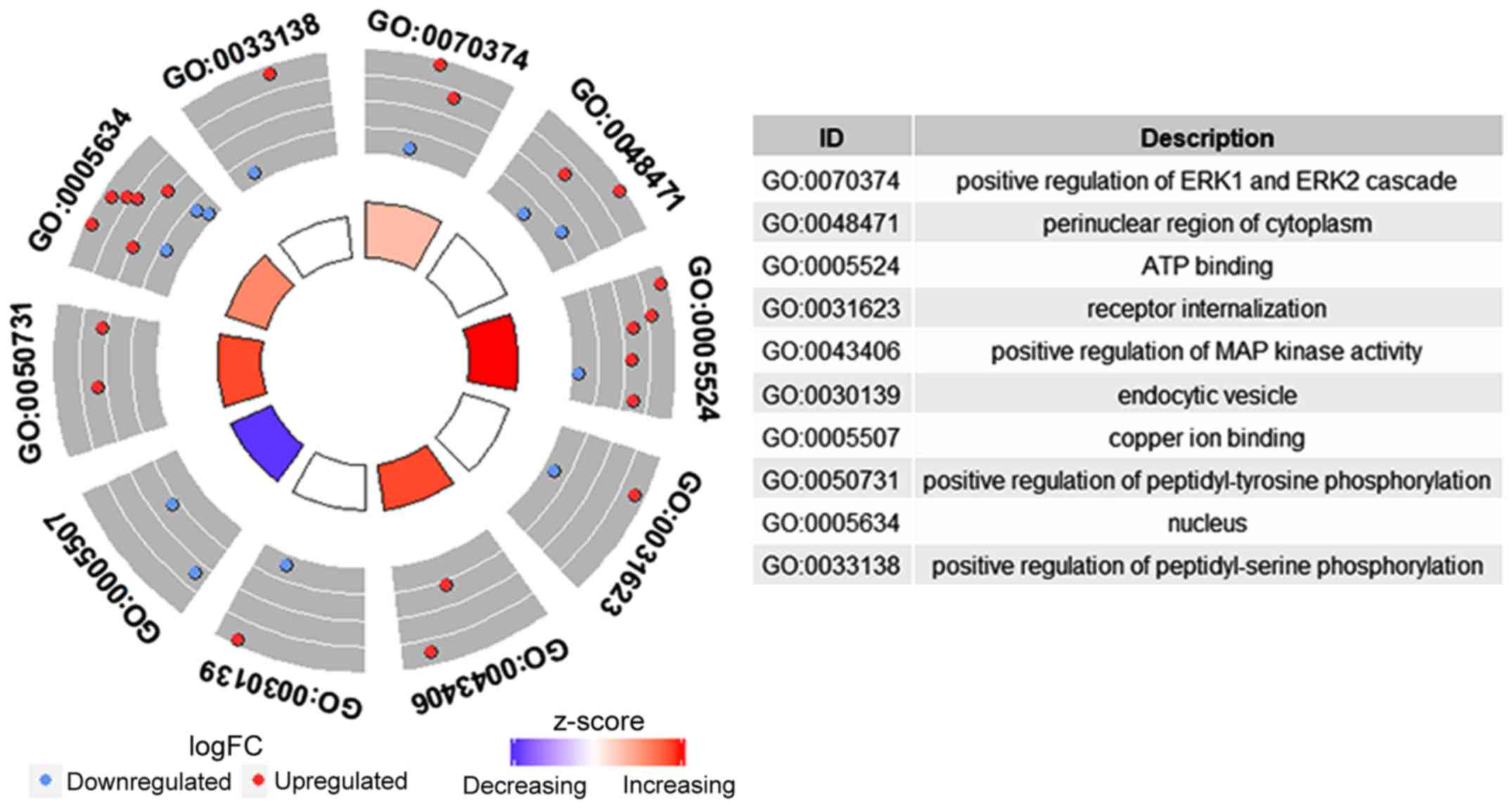
Functional enrichment analysis based on DEmRNAs in
ceRNA networks provided 10 GO terms, including five terms in the
category biological function, three terms in the category cellular
component and two terms in the category molecular function. Certain
GO terms comprised transcriptional regulation, including negative
regulation and DNA template (GO:0045892) and transcriptional
activation activity and binding to transcription factors of RNA
polymerase II (GO:0001190) (Fig. 9).
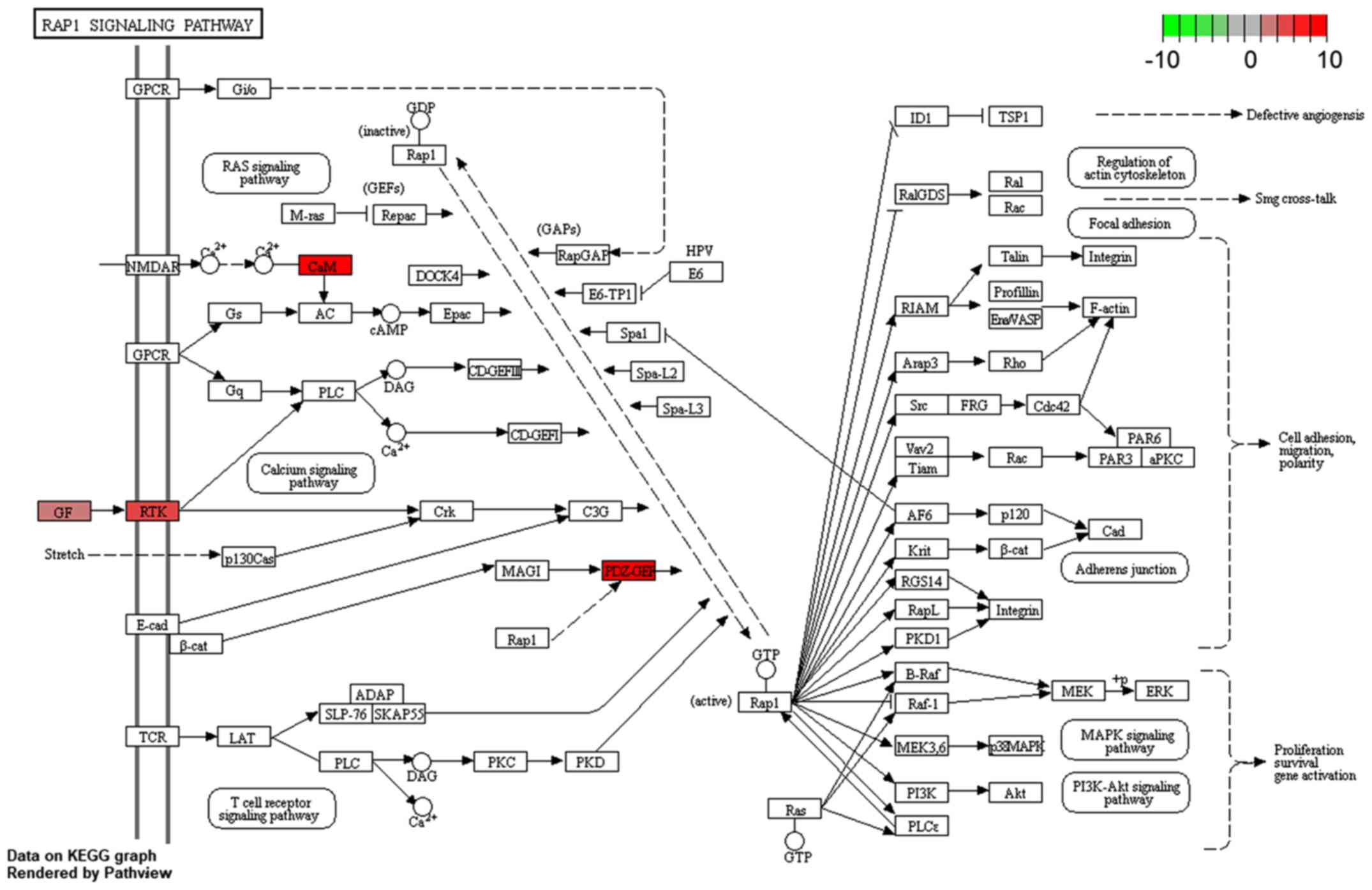
KEGG pathway enrichment analysis of all 31 DEmRNAs was also
performed. One KEGG pathway, hsa04015: Rap1 signaling pathway, was
identified to be significantly enriched (Fig. 10). Within this pathway, calmodulin 3
(CaM) and rap guanine nucleotide exchange factor 2 (PDZ-GEF) genes
were enriched significantly.
Discussion
Gliomas are central nervous system tumors with a
high incidence and the highest mortality rate among brain tumors.
The median survival time in patients with glioblastoma is 12–15
months (19). Tumor-associated
ceRNAs regulate the occurrence and development of tumors. ceRNAs,
including pseudogenes and lncRNAs, may be potential oncogenes or
tumor suppressor genes involved in biological processes of the
tumors.
miRNA sponges are synthetic transcripts that link
several copies of MRE and clones to viral vectors. As specific
inhibitors of miRNAs, they inhibit the activity of miRNAs (20). ceRNAs may be thought of as endogenous
sponges; they have substantial advantages of inhibiting several
MREs of various miRNAs compared to miRNA sponges. miRNA sponges and
endogenous sponges of ceRNA may become targets for the RNA-based
treatment of diseases including cancer in the future (21).
The theory of ceRNA will challenge certain
traditional theories. For instance, mRNA requires to be translated
into protein prior to exerting certain biological functions, or
only affect functions of the coding region while neglecting those
in the UTR (as binding sites of miRNA may appear in the UTR, the
entire function of a gene may be ignored).
In the present study, large cohorts from TCGA and
GEO databases were used to identify DElncRNAs, DEmRNAs and DEmiRNAs
between GBM and normal tissues. A lncRNA-miRNA-mRNA ceRNA network
was then built to provide an integrated view of the
ceRNA-regulatory crosstalk among GBM-specific RNA transcripts.
Functional enrichment analysis further revealed the GO terms and
pathways associated with DEmRNAs that have a role in the
development of GBM.
In the present study, 14 DElncRNAs, namely
AC005035.1, AC010336.2, AC022498.1, AC108134.2, AC116351.2,
C1orf132, C10orf91, DBH-AS1, HOTAIR, LINC00475, MED4-AS1, MIR210HG,
ZEB1-AS1 and ZNF197-AS1, were identified to be significantly
associated with the prognosis of GBM patients from the ceRNA
network.
Of the 14 lncRNAs, all except AC022498.1, MED4-AS1,
ZEB1-AS1 and ZNF197-AS1, may be considered risk factors, as the
patients with high expression levels of these 10 markers had a
shorter lifespan than those with lower levels. While little is
known about the functions of these 10 DElncRNAs, the results
suggest that they have a certain predictive value regarding
prognosis and further study of the involvement of the 10 lncRNAs in
GBM in other directions is warranted.
As the hub element of the ceRNA network, miRNA has a
crucial role in the interaction among various RNA transcripts.
LINC00336 interacts with CALM3, FAM126, SNCA and ZNF365 with the
mediation of hsa-miR-7, while LINC00205 interacts with HIVEP2,
MAP3K10 and RAPGEF2 with the mediation of hsa-miR-155. Of note,
hsa-mir-7 and hsa-miR-155 have been previously reported to be
involved in the pathogenesis of GBM (22,23). All
of the important DEmiRNAs had promising potential as biomarkers for
survival prediction in GBM. Hsa-miR-155 has been reported as a key
miRNA in breast cancer (24). As for
hsa-miR-7, circular RNA U2 small nuclear RNA auxiliary factor 1 was
indicated to promote human glioma by de-repressing
neuro-oncological ventral antigen 2 by sponging hsa-miR-7-5p
(25). While in the study by Cao
et al (26), the target
miRNAs were obtained only by predicting miRNA-lncRNA interactions
through databases, the present study identified DEmiRNAs by using
GEO2R to analyze the dataset GSE25631 on GBM.
mRNA constitutes another important part of the ceRNA
network that directly targets miRNAs or interacts indirectly with
lncRNAs mediated by miRNAs. Comparable to lncRNA and miRNAs,
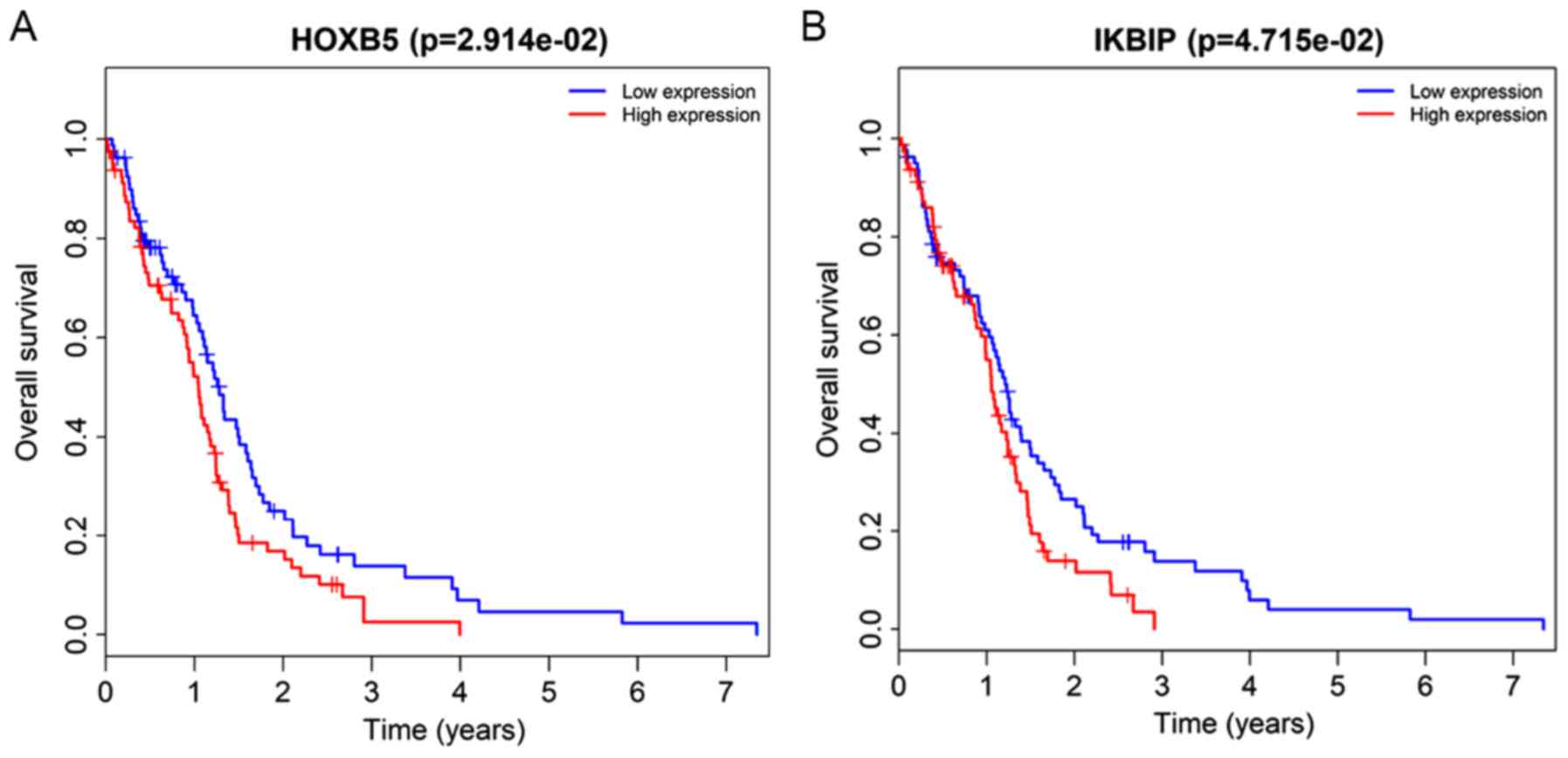
certain mRNAs also correlate with survival in GBM patients,
including HOXB5 and inhibitor of nuclear factor κB
kinase-interacting protein (IKBIP). Patients with high expression
levels of these two mRNAs have shorter survival times than those
with low levels. HOXB5 has been reported to promote cell
proliferation, migration and invasion in lung cancer,
retinoblastoma and breast cancer (27–29).
IKBIP is the target of tumor protein 53 with a pro-apoptotic
function (30). To the best of our
knowledge, the present study was the first to report the
association of these two key mRNAs with the prognosis of GBM
patients.
Functional enrichment analysis revealed that certain
GO terms and pathways associated with transcriptional regulation
and tumorigenesis, including the Rap1 signaling pathways, were
enriched by the DEmRNAs. The close association between enriched
KEGG pathways and the ceRNA network demonstrates the credibility of
the results.
Of note, the present study had certain limitations.
Due to the in silico nature of the study, there was a lack
of experimental validation in vitro and in vivo. The
present results and conclusions may serve as a foundation for the
establishment of mechanistic hypotheses as a basis for further
experiments on clinical samples and cell lines.
In conclusion, in the present study, GBM-associated
lncRNAs, miRNAs and mRNAs were identified using cohorts from TCGA
and GEO databases. A ceRNA network associated with lncRNAs was
successfully constructed, providing perceptiveness into the newly
proposed crosstalk among distinct types of RNA transcripts. A
significant correlation between overall survival and clinical
characteristics in patients with GBM may be established by
analyzing key lncRNAs in future study. The present study enhances
the understanding of the biological mechanisms of ceRNAs and helps
to clarify the pathogenesis of GBM.
Acknowledgements
Not applicable.
Funding
The project was supported by the Science and
Technology Project of Shenyang (grant no. 18-014-4-03) and the
Science and Technology Project of the Education Department of
Liaoning Province (grant no. LFWK201705).
Availability of data and materials
The datasets generated and/or analyzed during the
current study are available from the corresponding author on
reasonable request.
Authors' contributions
GL conceived and designed the study. SL performed
the data mining, acquisition and analysis. GL and SL wrote and
approved the final manuscript.
Ethical approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Qi P and Du X: The long non-coding RNAs, a
new cancer diagnostic and therapeutic gold mine. Mod Pathol.
26:155–165. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Amirkhah R, Schmitz U, Linnebacher M,
Wolkenhauer O and Farazmand A: MicroRNA-mRNA interactions in
colorectal cancer and their role in tumor progression. Genes
Chromosomes Cancer. 54:129–141. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wang C, Hu DZ and Liu JZ: Identification
of critical TF-miRNA-mRNA regulation loops for colorectal cancer
metastasis. Genetics Mol Res. 14:5485–5495. 2015. View Article : Google Scholar
|
|
4
|
Zhang C, Wang C, Jia Z, Tong W, Liu D, He
C, Huang X and Xu W: Differentially expressed mRNAs, lncRNAs, and
miRNAs with associated co-expression and ceRNA networks in
ankylosin spondylitis. Oncotarget. 8:113543–113557. 2017.PubMed/NCBI
|
|
5
|
Salmena L, Poliseno L, Tay Y, Kats L and
Pandolfi PP: A ceRNA hypothesis: The Rosetta stone of a hidden RNA
language? Cell. 146:353–358. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Liu K, Guo L, Guo Y, Zhou B, Li T, Yang H,
Yin R and Xi T: AEG-1 3-untranslated region functions as a ceRNA in
inducing epithelial-mesenchymal transition of human non-small cell
lung cancer by regulating miR-30a activity. Eur J Cell Biol.
94:22–31. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Peng W, Si S, Zhang Q, Li C, Zhao F, Wang
F, Yu J and Ma R: Long non-coding RNA MEG3 functions as a competing
endogenous RNA to regulate gastric cancer progression. J Exp Clin
Cancer Res. 34:792015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Johnsson P, Ackley A, Vidarsdottir L, Lui
WO, Corcoran M, Grandér D and Morris KV: A pseudogene
long-noncoding-RNA network regulates PTEN transcription and
translation in human cells. Nat Struct Mol Biol. 20:440–446. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Fan Q and Liu B: Comprehensive analysis of
a long noncoding RNA-associated competing endogenous RNA network in
colorectal cancer. Onco Targets Ther. 11:2453–2466. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Du Z, Fei T, Verhaak RG, Su Z, Zhang Y,
Brown M, Chen Y and Liu XS: Integrative genomic analyses reveal
clinically relevant long noncoding RNAs in human cancer. Nat Struct
Mol Biol. 20:908–913. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang W, Zhang J, Hoadley K, Kushwaha D,
Ramakrishnan V, Li S, Kang C, You Y, Jiang C, Song SW, et al:
MiR-181d: A predictive glioblastoma biomarker that downregulates
MGMT expression. Neuro Oncol. 14:712–719. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Robinson MD, McCarthy DJ and Smyth GK:
EdgeR: A bioconductor package for differential expression analysis
of digital gene expression data. Bioinformatics. 26:139–140. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jeggari A, Marks DS and Larsson E:
MiRcode: A map of putative microRNA target sites in the long
non-coding transcriptome. Bioinformatics. 28:2062–2063. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Agarwal V, Bell GW, Nam JW and Bartel DP:
Predicting effective microRNA target sites in mammalian mRNAs.
Elife. 4:2015. View Article : Google Scholar
|
|
15
|
Chou CH, Chang NW, Shrestha S, Hsu SD, Lin
YL, Lee WH, Yang CD, Hong HC, Wei TY, Tu SJ, et al: MiRTarBase
2016: Updates to the experimentally validated miRNA-target
interactions database. Nucleic Acids Res. 44:D239–D247. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wong N and Wang X: MiRDB: An online
resource for microRNA target prediction and functional annotations.
Nucleic Acids Res. 43:D146–D152. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Dennis G Jr, Sherman BT, Hosack DA, Yang
J, Gao W, Lane HC and Lempicki RA: DAVID: Database for annotation,
visualization, and integrated discovery. Genome Biol. 4:R602003.
View Article : Google Scholar
|
|
18
|
Yu G, Wang L-G, Han Y and He Q-Y:
ClusterProfiler: An R package for comparing biological themes among
gene clusters. OMICS. 16:284–287. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Roy S, Lahiri D, Maji T and Biswas J:
Recurrent glioblastoma: Where we stand. South Asian J Cancer.
4:163–173. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ebert MS, Neilson JR and Sharp PA:
MicroRNA sponges: Competitive inhibitors of small RNAs in mammalian
cells. Nat Methods. 4:721–726. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Brown BD, Cantore A, Annoni A, Sergi LS,
Lombardo A, Della Valle P, DAngelo A and Naldini L: A
microRNA-regulated lentiviral vector mediates stable correction of
hemophilia B mice. Blood. 110:4144–4152. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang X, Zhang X, Hu S, Zheng M, Zhang J,
Zhao J, Zhang X, Yan B, Jia L, Zhao J, et al: Identification of
miRNA-7 by genome-wide analysis as a critical sensitizer for
TRAIL-induced apoptosis in glioblastoma cells. Nucleic Acids Res.
45:5930–5944. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yu J, Cai X, He J, Zhao W, Wang Q and Liu
B: Microarray-based analysis of gene regulation by transcription
factors and microRNAs in glioma. Neurol Sci. 34:1283–1289. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liang F, Yang M, Tong N, Fang J, Pan Y, Li
J and Zhang X: Identification of six key miRNAs associated with
breast cancer through screening large-scale microarray data. Oncol
Lett. 16:4159–4168. 2018.PubMed/NCBI
|
|
25
|
Li G, Huang M, Cai Y, Yang Y, Sun X and Ke
Y: Circ-U2AF1 promotes human glioma via derepressing
neuro-oncological ventral antigen 2 by sponging hsa-miR-7-5p. J
Cellular Physiol. 234:9144–9155. 2019. View Article : Google Scholar
|
|
26
|
Cao Y, Wang P, Ning S, Xiao W, Xiao B and
Li X: Identification of prognostic biomarkers in glioblastoma using
a long non-coding RNA-mediated, competitive endogenous RNA network.
Oncotarget. 5:41737–41747. 2016.
|
|
27
|
Lee JY, Kim JM, Jeong DS and Kim MH:
Transcriptional activation of EGFR by HOXB5 and its role in breast
cancer cell invasion. Biochem Biophys Res Commun. 18:2924–2930.
2018. View Article : Google Scholar
|
|
28
|
Xu H, Zhao H and Yu J: HOXB5 promotes
retinoblastoma cell migration and invasion via ERK1/2
pathway-mediated MMPs production. Am J Transl Res. 15:1703–1712.
2018.
|
|
29
|
Zhang B, Li N and Zhang H: Knockdown of
homeobox B5 (HOXB5) inhibits cell proliferation, migration, and
invasion in non-small cell lung cancer cells through inactivation
of the Wnt/β-catenin pathway. Oncol Res. 19:37–44. 2018. View Article : Google Scholar
|
|
30
|
Hofer-Warbinek R, Schmid JA, Mayer H,
Winsauer G, Orel L, Mueller B, Wiesner CH, Binder BR and de Martin
R: A highly conserved proapoptotic gene, IKIP, located next to the
APAF1 gene locus, is regulated by p53. Cell Death Differ.
11:1317–1325. 2004. View Article : Google Scholar : PubMed/NCBI
|