Introduction
Mutations in subunit five of the conserved
oligomeric Golgi (COG) complex gives rise to type IIi congenital
disorders of glycosylation (CDG) (1). CDG is physiologically related to other
pathologies involving glycan defects and can lead to multiple organ
failure, dysmorphism, skeletal malformation, hormonal disorders and
coagulopathy (2). A total of eight
subunits, which are associated with the retrograde transport of
Golgi components, are present in the COG complex). Mutations in the
COG1, COG2, COG4, COG5, COG6, COG7, and COG8 genes have been
reported to cause CDG (COG-CDG), which can result in heterogeneous
clinical phenotypes ranging from severe multi-organ disorders to
moderate forms of neurological impairment (3,4).
Only 10 COG5-CDG cases have been reported worldwide
(2,5–8). Common
symptoms among these cases include neurological, morphological and
hepatic abnormalities (7,8). Defects in the COG5 protein are mainly
associated with abnormalities of the brain, liver, bladder and
auditory and visual systems (9).
Fung et al (5) described the
first Chinese patient with COG5-CDG. In this aforementioned study,
a female presented with mild neurohepatic disease and central and
peripheral neurological involvement.
Case report
In the current study, a case of COG5-CDG involved a
male Chinese patient with severe symptoms. To the best of our
knowledge, this is the second COG5-CDG case to be reported in
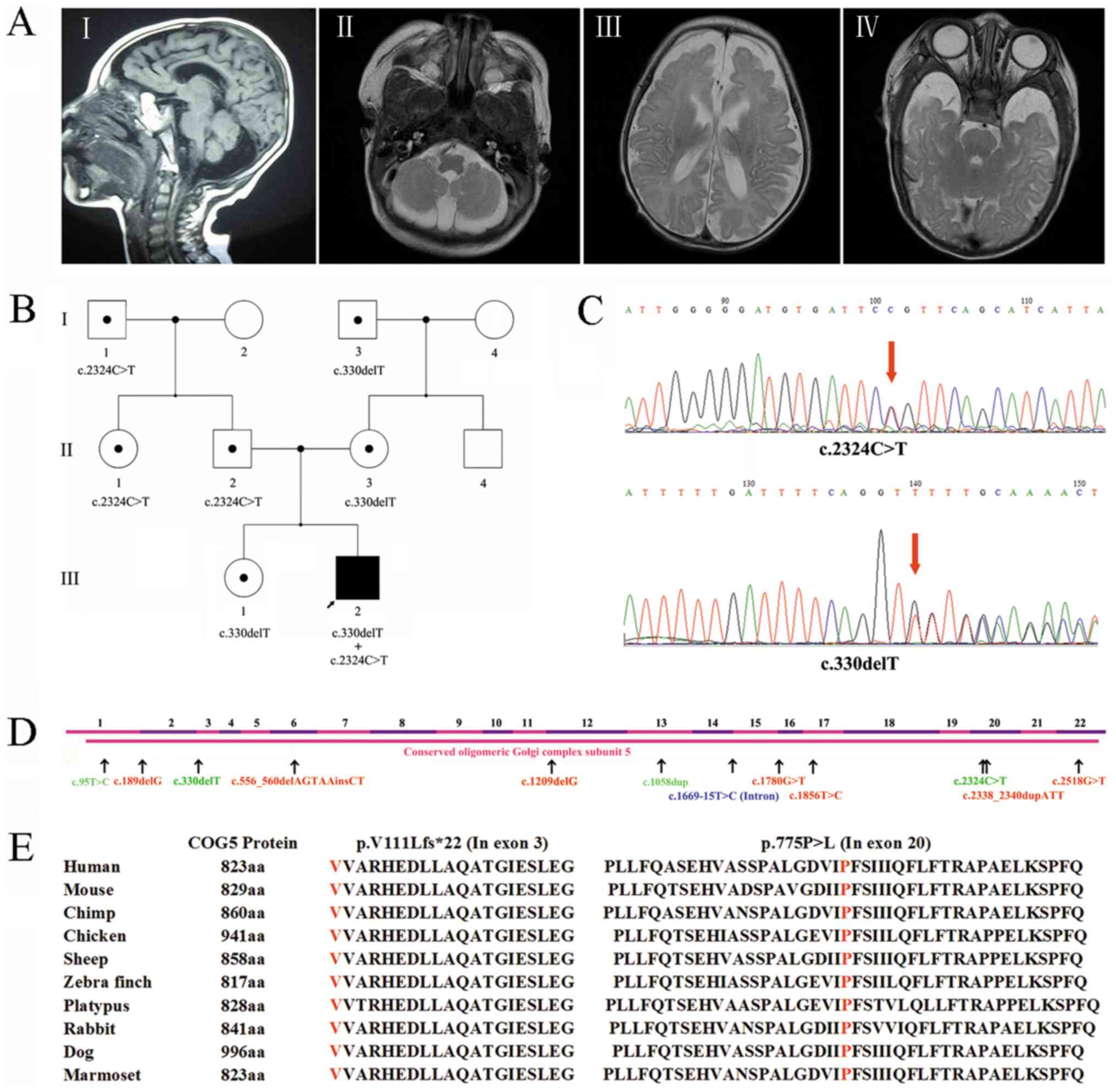
China. The 11-year-old patient (Fig.
1B) was the second child of a Chinese couple. During the
gestation period, a TORCH examination was performed and the results
were negative, and ultrasound results were normal except for
polyhydramnios, which was noted in the second trimester. The
patient was born at full-term via caesarean section. Upon physical
examination, the patient's head and feet were slightly smaller than
average, the small forehead and an umbilical hernia was discovered.
An MRI of the brain at 1.5 months revealed brain hypotrophy and
thinning of the corpus callosum. At 6.5 months, the patient
exhibited slow responses, glassy eyes, and appeared epileptic with
the presence of mild microcephaly. The patient feet were still
slightly smaller than average, and the upper limbs appeared
hypermyotonic. The patient was unable to turn over, crawl, or grasp
objects. An additional MRI was performed at 12 months and revealed
cerebellar atrophy (Fig. 1A).
Fig. 1A-I and 1A-II images revealed cerebellar hemisphere
and vermis hypoplasia at midst sagittal plane and cerebellum level
respectively, while Fig. 1A-III
revealed the bilateral temporal lobes and Fig. 1A-IV showed frontal lobes hypogenesis.
The patients' symptoms were still present at 18 months and the
patient was unable walk and, even when holding onto a desk, could
only stand for short periods of time. Speech development was
absent, although according to the parents, the patient was able to
understand a few words. All other family members presented a normal
phenotype (Table I).
 | Table I.Phenotypes of patient and the family
members. |
Table I.
Phenotypes of patient and the family
members.
| Family | Mutation | Mental
retardation | Delayed speech
development | Delayed motor
development | Cerebral/cerebellar
atrophy | Microcephaly | Hypotonia | Convulsions | Liver
involvement | Other |
|---|
| Patient III-2 | c.330delT | Severe | Absence of
speech | ++ | Both | + | + | + | + | Slightly smaller
feet |
|
| c.2324 C>T |
|
|
|
|
|
|
|
|
|
| Sister | c.330delT | − | − | − | − | − | − | − | − | − |
| III-1 |
| Father | c.2324 C>T | − | − | − | − | − | − | − | − | − |
| II-2 |
| Mother | c.330delT | − | − | − | − | − | − | − | − | − |
| II-3 |
| Aunt | c.2324 C>T | − | − | − | − | − | − | − | − | − |
| II-1 |
| Maternal uncle | − | − | − | − | − | − | − | − | − | − |
| II-4 |
| Grandfather | c.2324 C>T | − | − | − | − | − | − | − | − | − |
| I-1 |
| Grandmother | − | − | − | − | − | − | − | − | − | − |
| I-2 |
| Maternal
grandfather | c.330delT | − | − | − | − | − | − | − | − | − |
| I-3 |
| Maternal
grandmother | − | − | − | − | − | − | − | − | − | − |
| I-4 |
Total genomic DNA was extracted according to
standard protocols (TIANamp Genomic DNA kit; Tiangen Biotech Co.,
Ltd.) from peripheral blood leukocytes isolated from the patient
and his family members. Targeted enrichment of the whole exome DNA
was performed using the Nextera Rapid Capture Exome kit (Illumina,
Inc.) according to manufacturer's protocol. Whole exome libraries
were then sequenced on an Illumina HiSeq platform (Illumina, Inc.)
using a 2×100 bp sequencing protocol (10). The results indicated the presence of
potentially homozygous, compound heterozygous, and de novo
variants, which were selected for further investigation. Variants
with minor allele frequencies <0.01 in any of the dbSNP, ExAC,
1000 Genomes Project, gnomAD, or in-house databases were selected
for further exploration.
After data filtration, a potential compound
heterozygous mutation (NM_006348.3: c.330delT, p.V111Lfs*22;
c.2324C>T, p.P775L) was identified. Then the c.330delT mutation
was amplified using a specific forward (5′-TCCCGCTTACAACTCATCT-3′)
and reverse primer (5′-CCAATGGGGAAGTCGATGCT-3′), while c.2324C>T
mutation was amplified by specific primer pair: Forward
5′-TTCTCCCACCAAATCACCTA-3′ and reverse 5′-TGCTCTCGTGAACAAAAACTG-3′.
PCR was conducted with Premix Taq (TaKaRa Taq Version 2.0; R004A).
The primer extension was cycled for 35 cycles with 15 sec at 95°C,
15 sec at 53°C (c.330delT)/55°C (c.2324C>T), and 15 sec at 72°C.
Following PCR amplification, Sanger sequencing confirmed that
NM_006348.3: c.2324C>T was inherited paternally and that I-1 and
II-1 were carriers of this mutation (Fig. 1B and C). NM_006348.3: c.330delT was
inherited maternally, while I-3 and III-1 were carriers of this
variant (Fig. 1B and C). Therefore,
the results revealed that the patient carried a frameshift
mutation, c.330delT (p.V111Lfs*22), and a missense mutation, c.2324
C>T (p.P775L), in the COG5 gene. Due to both variants involving
the same transcript (NM_006348.3), the presence of a compound
heterozygous mutation was observed.
To the best of our knowledge, in the 1000 Genomes
project (http://www.internationalgenome.org/), the Exome
Aggregation Consortium (http://exac.broadinstitute.org/), the gnomAD
(http://gnomad-old.broadinstitute.org/) and our
in-house database, neither variant had previously been described.
The NM_006348.3: c.2324 C>T, p.P775L mutation was recently
reported to be pathogenic (8). This
was supported by the American College of Medical Genetics (ACMG)
guidelines, which classified this variant as pathogenic
(categories, PM2, PM3, PP3 and PP5) (11). This mutation is highly conserved
among a variety of species (Fig. 1E;
http://genome.ucsc.edu/). The prediction,
regarding the effect of this mutation, with SIFT (PROVEAN v1.1.3)
and PolyPhen-2 (version 2.2) software indicated that the protein
variant was ‘deleterious’ and ‘probably damaging’, respectively.
The NM_006348.3: c.330delT variant (p.V111Lfs*22) is a frameshift
mutation that leads to the production of an aberrant and truncated
protein of 132 amino acids. This frameshift variant may result in
nonsense-mediated decay, leading to a premature termination codon
(PTC) at exon 4 (22 exons in total). According to the ACMG
guidelines, this frameshift mutation was placed into the PVS1 and
PM2 categories, thereby classifying it as a likely pathogenic
variant (11).
Discussion
Defects of the COG complex indirectly affect
glycosylation through the altered trafficking of
glycosyltransferases (4). Therefore,
the identification of a variety of forms of CDG, which are caused
by COG deficiency, founded a new understanding of CDG pathogenesis.
The COG complex is composed of eight subunits (named COG1-8), which
are separated into two sub-complexes: Lobe A (COGs 1–4) and lobe B
(COGs 5–8) (12,13). It has been suggested that comparable
molecular defects may be more detrimental in lobe A COG-CDG than in
lobe B COG-CDG (12).
Including the present case, twelve COG5 mutations
have been identified worldwide (Table
II). Most of these mutations (9/12) are located in exons, while
one mutation is a homozygous intronic substitution that leads to
COG5 exon skipping (Fig. 1D). Due to
the COG5 protein being highly conserved among numerous species
(Fig. 1E), the full-length protein
is important for its function, while lobe A appears to be essential
for maintaining overall Golgi structure and lobe B impacts
recognition and vesicle tethering to the right Golgi compartment
(14). In the current case, the
c.330delT frameshift mutation led to a patched (PTC) of COG5
protein translation, which generated a truncated 133-amino acid
protein. According to the mechanism of action, the location of the
PTC may lead to nonsense mediated decay. This hypothesis could not
be tested in the current study due to a limited sample quantity.
However, it is possible that this mutation may cause a loss of
function in COG5. The c.2324C>T missense mutation causes an
amino acid substitution at residue 755, from proline to
leucine.
| Table II.Clinical features of a patient with
COG5 mutations. |
Table II.
Clinical features of a patient with
COG5 mutations.
|
|
|
|
| Rymen et al
2012 (6) |
|
|
|---|
|
|
|
|
|
|
|
|
|---|
| Clinical
features | Present study | Paesold-Burda et
al 2009 (2) | Fung et al
2012 (5) | Case 1 | Case 2 | Case 3 | Case 4 | Case 5 | Case 6 | Chérot et al
2018 (8) | Kim et al
2017 (7) |
|---|
| Ethnic origin | Chinese | Iraqi | Chinese | Moroccan | Moroccan | Moroccan | Chinese | Italian | Belgian | NA | Korea |
| Consanguinity | − | + | − | + | + | + | − | + | − | − | + |
| Mutation sites |
|
|
|
|
|
|
|
|
|
|
|
| Allele
1 | c.330delT | Homozygous | c.556_560
delAGTAAinsCT | Homozygous | Homozygous | NA | c.556_560del
AGTAAinsCT | c.189delG | Homozygous | c.2324 C>T | Heterozygous |
| Allele
2 | c.2324 C>T |
c.1669-15T>C | c.1856T>C | c.2518G>T | c.2518G>T | NA | c.95T>G |
c.2338_2340dupATT | c.1780G>T | c.1508dup | c.1209delG |
| Mental
retardation | Severe | Moderate | Mild | Moderate | Severe | Severe | Mild | Severe | Severe | NA |
Mild-to-moderate |
| Delayed speech
development | Absence of
speech | + | + | ++ | ++ | ++ | + | ++ | ++ | NA | + |
| Delayed motor
development | ++ | + | + | ++ | ++ | ++ | + | ++ | ++ | NA | + |
| Cerebral/cerebellar
atrophy cerebellar atrophy | Both | Cerebellar | − | − | NA | − | − | Both | − | NA | Diffuse
isolated |
| Microcephaly | + | + | − | + | + | + | − | ++ | ++ | NA | − |
| Hypotonia | + | + | + | + | + | + | + | ++ | ++ | NA | − |
| Convulsions | + | − | − | − | − | − | − | − | + | NA | − |
| Short stature | − | − | − | + | + | + | − | + | + | NA | − |
| Liver
involvement | + | − | + | − | − | − | + | + | − | NA | − |
| Deafness | − | − | − | − | − | − | − | + | + | NA | − |
| Blindness | − | − | − | − | − | − | − | + | + | NA | − |
| Neurogenic
bladder | − | − | − | − | − | − | − | + | + | NA | − |
| Other | Slightly smaller
feet | − | Contractures | − | − | − | Contractures | − | Wrinkled skin | NA | Progressive ataxia;
Scoliosis; |
The c.2324C>T mutation was detected in the
patient's father. Having received two different COG5 mutations from
each parent, a compound heterozygous mutation was therefore
observed in this case. Abnormalities in glycoconjugate synthesis,
intracellular protein sorting and protein secretion have been
revealed to be caused by COG defects. COG serves an important role
in the regulation, compartmentalization or activity of multiple
Golgi glycosylation enzymes (15,16). As
a subunit of COG, although less severe, COG5 deficiency in HeLa
cells causes the dilation of a number of Golgi cisternae, defects
in global glycosylation and damage to intracellular processing
(1). Additionally, MALDI TOF
analysis revealed hypoglycosylation and the lack of a terminal
sialic acid of serum transferrin in patients with COG5-CDG
(5,6). Therefore, it is suggested that the
novel COG5 compound heterozygous mutations may lead to
glycosylation defects in the proband, resulting in COG5-related
CDG, but this needs to be investigated.
The COG5-8 gene exhibits a relative tolerance to
loss of function variants (12), and
its phenotypic severity is in line with the tolerance to other COG
gene variants. COG5 mutations have been indicated to lead to
relatively mild phenotypes. In the current case report, the patient
exhibited phenotypes of mental retardation, absence of speech,
delayed motor development, cerebral and cerebellar atrophy,
microcephaly, hypotonia, convulsions, liver involvement and
slightly smaller feet. Compared with lobe A, the patient exhibited
a milder phenotype. However, in comparison with the previously
reported COG5-CDG case (6–8), the patient showed severe central and
peripheral neurological symptoms.
In conclusion, a case of COG5-CDG in a Chinese
patient is reported, in which relatively severe clinical symptoms
were present. Further investigation into the relationship between
the phenotype and genotype of COG5-CDG will facilitate a better
understanding of this rare disease.
Acknowledgements
Not applicable.
Funding
The present study was jointly supported by grants
from the Natural Science Foundation of China (grant no. 81701462)
and the Obstetric Diseases Translational Medicine Research Center
Project of Liaoning Province (grant no. 2014225007).
Availability of data and materials
All data generated or analyzed during the present
study are included in this published article.
Authors' contributions
SWY, LYG and YL contributed to the conception,
design and performed experiments. YL and HQ contributed to the
analysis of the data, and the drafting and revising of the
manuscript.. YZhao, YZhang, CXL, HKJ and YL were responsible for
clinical diagnosis. YM, LYK and BL contributed to the acquisition
of the data and data analysis and interpretation. All authors are
accountable for all aspects of the work and read and approved the
final version of the manuscript.
Ethics approval and consent to
participate
All procedures performed in studies involving human
participants were in accordance with the Ethical Standards of the
Institutional and/or National Research Committee and in accordance
with the 1964 Helsinki Declaration and its later amendments or
comparable ethical standards. Informed consent was obtained from
the legal guardians of the patient in this study.
Patient consent to publication
A written informed consent for the publication of
the images was provided by the patient's guardians.
Competing interests
The authors declare that they have no conflicts of
interest.
References
|
1
|
Oka T, Vasile E, Penman M, Novina CD,
Dykxhoorn DM, Ungar D, Hughson FM and Krieger M: Genetic analysis
of the subunit organization and function of the conserved
oligomeric golgi (COG) complex: Studies of COG5- and COG7-deficient
mammalian cells. J Biol Chem. 280:32736–32745. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Paesold-Burda P, Maag C, Troxler H,
Foulquier F, Kleinert P, Schnabel S, Baumgartner M and Hennet T:
Deficiency in COG5 causes a moderate form of congenital disorders
of glycosylation. Hum Mol Genet. 18:4350–4356. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ungar D, Oka T, Brittle EE, Vasile E,
Lupashin VV, Chatterton JE, Heuser JE, Krieger M and Waters MG:
Characterization of a mammalian Golgi-localized protein complex,
COG, that is required for normal Golgi morphology and function. J
Cell Biol. 157:405–415. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Barone R, Fiumara A and Jaeken J:
Congenital disorders of glycosylation with emphasis on cerebellar
involvement. Semin Neurol. 34:357–366. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Fung CW, Matthijs G, Sturiale L, Garozzo
D, Wong KY, Wong R, Wong V and Jaeken J: COG5-CDG with a mild
neurohepatic presentation. JIMD Rep. 3:67–70. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Rymen D, Keldermans L, Race V, Régal L,
Deconinck N, Dionisi-Vici C, Fung CW, Sturiale L, Rosnoblet C,
Foulquier F, et al: COG5-CDG: Expanding the clinical spectrum.
Orphanet J Rare Dis. 7:942012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kim YO, Yun M, Jeong JH, Choi SM, Kim SK,
Yoon W, Park C, Hong Y and Woo YJ: A mild form of COG5 defect
showing early-childhood-onset Friedreich's-Ataxia-Like phenotypes
with isolated cerebellar atrophy. J Korean Med Sci. 32:1885–1890.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cherot E, Keren B, Dubourg C, Carré W,
Fradin M, Lavillaureix A, Afenjar A, Burglen L, Whalen S, Charles
P, et al: Using medical exome sequencing to identify the causes of
neurodevelopmental disorders: Experience of 2 clinical units and
216 patients. Clin Genet. 93:567–576. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Jaeken J and Péanne R: What is new in CDG?
J Inherit Metab Dis. 40:569–586. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bronner IF, Quail MA, Turner DJ and
Swerdlow H: Improved protocols for Illumina sequencing. Curr Protoc
Hum Genet. 80:18.2.1–42. 2014. View Article : Google Scholar
|
|
11
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the American College
of Medical Genetics and Genomics and the Association for Molecular
Pathology. Genet Med. 17:405–424. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Haijes HA, Jaeken J, Foulquier F and van
Hasselt PM: Hypothesis: Lobe A (COG1-4)-CDG causes a more severe
phenotype than lobe B (COG5-8)-CDG. J Med Genet. 55:137–142. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ungar D, Oka T, Vasile E, Krieger M and
Hughson FM: Subunit architecture of the conserved oligomeric Golgi
complex. J Biol Chem. 280:32729–32735. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Peanne R, Legrand D, Duvet S, Mir AM,
Matthijs G, Rohrer J and Foulquier F: Differential effects of lobe
A and lobe B of the Conserved Oligomeric Golgi complex on the
stability of {beta}1,4-galactosyltransferase 1 and
{alpha}2,6-sialyltransferase 1. Glycobiology. 21:864–876. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kingsley DM, Kozarsky KF, Segal M and
Krieger M: Three types of low density lipoprotein
receptor-deficient mutant have pleiotropic defects in the synthesis
of N-linked, O-linked, and lipid-linked carbohydrate chains. J Cell
Biol. 102:1576–1585. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Reddy P and Krieger M: Isolation and
characterization of an extragenic suppressor of the low-density
lipoprotein receptor-deficient phenotype of a Chinese hamster ovary
cell mutant. Mol Cell Biol. 9:4799–4806. 1989. View Article : Google Scholar : PubMed/NCBI
|