Introduction
MicroRNAs (miRNAs/miRs) are endogenous non-coding
RNA molecules, 19–25 nucleotides in length, that exert
post-transcriptional inhibitory effects on gene expression
(1). To date, abnormal miRNA
expression has been observed in a number of diseases, including
cancer, systemic lupus erythematosus (SLE), autoimmune diabetes,
atherosclerosis and rheumatoid arthritis (RA) (2–4). In
particular, miR-146 is a miRNA that serves an important role in the
regulation of the immune response by targeting genes to mediate
downstream pathophysiological processes, with the interleukin
(IL)-1 receptor-associated kinase l (IRAK1)/tumor necrosis factor
(TNF) receptor-associated factor 6 (TRAF6) TRAF6/NF-κB axis as the
most important target (5). It has
been reported that miR-146a inhibits the activation of donor T
cells in acute graft-vs.-host disease by targeting TRAF6, leading
to reduced TNF transcription (6).
Human miR-146 exists in two forms, miR-146a and miR-146b, the
former of which has a seed sequence of 5′-UGAGAACUGAAUUCCAUGGGUU-3′
and is highly expressed in macrophages, mononuclear cells and T
lymphocytes (7).
SLE is a complex chronic autoimmune inflammatory
disease that is caused by inflammatory responses mediated by the
lack of self-tolerance in the immune system. Innate and adaptive
immune responses appear to be involved in the development of SLE. A
variety of immune cell types, including monocytes, lymphocytes,
mediated by a number of cytokines, including type I interferon
(IFN) and IL-6, have been previously reported to serve their roles
in this process (8). Indeed,
previous studies have demonstrated that miR-146a expression is
reduced in the peripheral blood mononuclear cells (PBMCs) of
patients with SLE (9,10). A pilot study performed by Zhou et
al (unpublished data) revealed that miR-146a levels were lower
in the serum of patients with SLE. These findings suggested that
miR-146a may be associated with the development and progression of
SLE.
A previous study revealed that interleukin (IL)-1
receptor-associated kinase l (IRAK1) is a potential target of
miR-146a (11). IRAK1 is the
critical molecule in the Toll-like receptor (TLR)-4-myeloid
differentiation primary response 88 (MyD88) signaling pathway.
IRAK1 is phosphorylated after combining with My D88, and forms a
complex with TRAF6. This complex then activates inhibitor of
nuclear factor (NF)-κB kinase (IΚKβ) and induces the activation and
translocation of the NF-κB transcription factor into the nucleus.
Activated NF-κB subsequently induces the synthesis and secretion of
large quantities of inflammatory cytokines (12).
Immunosuppressive therapy is commonly applied during
the clinical management of patients with SLE. Under these
conditions, patients are predisposed to infection, including
nosocomial and latent infections, which in turn may exacerbate SLE
symptoms (13). Pathogen-associated
molecular patterns such as lipopolysaccharide (LPS) are the most
common etiological agents that induce inflammatory responses by
activating the TLR/IRAK1/NF-κB signaling pathway (14). Therefore, the aim of the present
study was to investigate the regulatory effects of miR-146a on
inflammatory responses by analyzing its effects on the expression
of IRAK1 and downstream NF-κB activation. In addition, the current
study aimed to investigate the possible infection-associated
mechanisms of miR-146a in the pathogenesis of SLE.
Materials and methods
Cell culture and transfection
The human acute monocytic leukemia THP-1 cell line
was purchased from the Cell Bank of the Chinese Academy of
Sciences. THP-1 cells were cultured using RPMI-1640 culture medium
supplemented with 10% fetal bovine serum (both Gibco; Thermo Fisher
Scientific, Inc.), 100 U/ml penicillin and 100 mg/ml streptomycin
at 37°C and 5% CO2. Cells in the logarithmic growth
phase were seeded into 6-well plate (1×106 cells/well)
and incubated overnight. They were subsequently transfected with 50
nM miR-146a mimic (cat. no. miR 10000449), 50 nM negative control
(NC; cat. no. miR 01101) mimic, 100 nM miR-146a inhibitor or (cat.
no. miR 20000449) 100 nM inhibitor NC (cat. no. miR 02101). All
from Guangzhou RiboBio Co., Ltd.) using
Lipofectamine®2000 (Invitrogen; Thermo Fisher
Scientific, Inc.), according to the manufacturer's protocol. For
the blank control, corresponding quantities of Opti-MEM (Gibco;
Thermo Fisher Scientific, Inc.) solution were used in place of RNA
during transfection (15).
Stimulation of cells with LPS and cell
grouping
At 24 h following transfection, THP-1 cells were
washed with Hank's balanced salt solution (Beijing Solarbio Science
& Technology Co., Ltd.) before the culture medium was replaced
with fresh RPMI-1640 medium. A final concentration of 1 µg/ml LPS
(Sigma-Aldrich; Merck KGaA) was subsequently added into the culture
medium, followed by incubation for a further 3 h. The cells were
then collected for the measurement of IRAK1 and p68 expression,
whilst the supernatant of the culture medium was collected for the
measurement of cytokine levels, as described previously (15). The cells were divided into the
following experimental groups: Blank control, no RNA transfection
or LPS treatment; LPS group, treated with LPS only; mimic group,
miR-146 amimic+LPS; NC mimic group, NC mimic + LPS; inhibitor
group, miR-146a inhibitor + LPS; and inhibitor NC group, inhibitor
NC + LPS.
Reverse transcription-quantitative PCR
(RT-qPCR)
For the measurement of miRNA expression, small RNA
was extracted from cells using the mirVana PARIS kit (Applied
Biosystems; Thermo Fisher Scientific, Inc.). cDNA was then reverse
transcribed from small RNA using the TaqMan® MicroRNA RT
kit (Applied Biosystems; Thermo Fisher Scientific, Inc.) according
to manufacturer's protocol. The thermocycling conditions were as
follows: 16°C for 30 min, 42°C for 30 min, 85°C for 5 min and 4°C
hold. miR-146a expression levels were measured using
TaqMan® MicroRNA Assay kit (Assay ID, 000468; Applied
Biosystems; Thermo Fisher Scientific, Inc.), the primers, which are
patented, were provided by the manufacturer as part of the kit. U6
expression were used for the normalization of miRNA expression.
To determine mRNA expression levels, total RNA was
extracted from cells using TRIzol® reagent (Invitrogen;
Thermo Fisher Scientific, Inc.), and then reverse transcription was
performed using High-Capacity cDNA Reverse Transcription Kits
(Applied Biosystems; Thermo Fisher Scientific, Inc.) according to
manufacturer's protocol. The thermocycling conditions used was as
follows: 25°C for 10 min, 37°C for 120 min, 85°C for 5 min and 4°C
hold. qPCR was performed to measure IRAK1 mRNA levels in cells
using PowerUp™ SYBR™ Green Master Mix (Applied Biosystems; Thermo
Fisher Scientific, Inc.). GAPDH was used as an internal reference.
The sequences of the primers used were as follows: IRAK1 forward,
5′-GTGCTAGAGACCTTGGCTG-3′ and reverse, 5′-TGTGCTCTGGGTGCTTCTC-3′;
and GAPDH forward, 5′-CCATGTTCGTCATGGGTGT-3′ and reverse,
5′-TGAGTCCTTCCACGATACC-3′.
Both qPCR assays aforementioned were performed using
ABI Real Time PCR System 7500 (Applied Biosystems; Thermo Fisher
Scientific, Inc.) using the following thermocycling protocol:
Initiation reaction at 50°C for 2 min and 10 min at 95°C, followed
by 40 cycles of 95°C for 15 sec and 60°C for 1 min. The relative
expression levels of miR-146a and IRAK1 were determined using the
2−ΔΔCq method (16).
Western blotting
Following treatment with LPS for 3 h, the cells were
collected and total protein was extracted using RIPA buffer
(Pierce; Thermo Fisher Scientific, Inc.) supplemented with a
cocktail of protease and phosphatase inhibitors (Sigma-Aldrich;
Merck KGaA). Proteins from the nuclear fraction were extracted
using a Nuclear Protein Extraction Kit (cat. R0050; Beijing
Solarbio Science & Technology Co., Ltd.), according to the
manufacturer's protocols. Protein concentrations were determined
using a bicinchoninic acid assay protein quantification kit
(Pierce; Thermo Fisher Scientific, Inc.). The protein samples were
then heat-denatured, and 5 µg protein/lane was separated on a 10%
SDS-PAGE gel, prior to transferral to polyvinylidene difluoride
membranes. The membranes were subsequently blocked in 5% bovine
serum albumin (Sigma-Aldrich; Merck KGaA), which was prepared in a
TBS-T buffer (50 mM Tris, pH 8.0, and 150 mM NaCl, containing 0.05%
Tween-20) and incubated overnight at 4°C. Subsequently, the
membranes were incubated with primary antibodies against anti-IRAK1
(cat. no. SC-5288; dilution, 1:400), anti-NF-κB p65 (cat. no.
SC-8008; dilution, 1:400), anti-β-actin (cat. no. SC-47778;
dilution, 1:1,000) (all from Santa Cruz Biotechnology, Inc.) and
anti-histone H3 (cat. no. 4499; dilution, 1:2,000; Cell Signaling
Technology, Inc.) at 4°C overnight. Following washing with TBS-T
for 20 min, the membranes were incubated with the corresponding
horseradish peroxidase-conjugated secondary antibodies (cat. nos.
7076 and 7074; dilution 1:2,000; Cell Signaling Technology, Inc.)
for 1 h at room temperature. The membranes were visualized using a
Millipore Enhanced Chemiluminescence system (EMD Millipore; Merck
KGaA) and analyzed using Image Lab (version 3.0; Bio-Rad
Laboratories, Inc.). β-actin and histone H3 were used as internal
controls. Relative protein expression, normalized to the internal
control, is presented as a ratio of the blank control group.
ELISA
Cells in the logarithmic growth phase were seeded
into a 24-well plate with a density of 2×105 cells/well.
Subsequently, they were transfected with miR-146a mimic, mimic NC,
miR-146a inhibitor or and inhibitor NC. For the blank control, the
same amount of the Opti-MEM solution was used instead of RNA in the
transfection. After 24 h of transfection, the THP-1cells were
washed with Hank's balanced salt solution, and the culture medium
was replaced with RPMI-1640 medium. LPS (Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany) was added into culture medium at a final
concentration of 1 µg/ml. Following treatment with LPS for 12 h,
the culture medium was collected to measure IL-6 and TNF-α levels
using ELISA kits (D6050 and DTA00D, R&D Systems, Inc.),
according to manufacturer's protocols.
Dual-Luciferase reporter assay
Target Scan bioinformatics software (version 7.2;
www.targetscan.org) was used to predict the
potential targets of miR-146a by typing the term ‘miR-146-5p’ into
the search engine. The software revealed two conserved sites in the
3′-untranslated region (3′-UTR) of IRAK1. A wild-type IRAK1 mRNA
3′-UTR sequence containing the miR-146a target site was cloned into
the XbaI site downstream of the firefly luciferase gene in
the pGL-3-promoter vector (Promega Corporation) to obtain the
wt-IRAK1-3′-UTR vector. In a parallel experiment, the IRAK1 mRNA
3′-UTR sequence was replaced by a mutant sequence (synthesized by
ObiO Technology Co., Ltd.) to obtain the mut-IRAK1-3′-UTR vector.
The THP-1 cells were seeded into 96-well plates (4×104
cells/well) and cultured for 24 h. Subsequently, the cells were
co-transfected with 1 µg plasmids and 100 nM miR-146a mimic using
Lipofectamine 2000 (Invitrogen; Thermo Fisher Scientific, Inc.),
according to the manufacturer's protocol. An empty pGL-3-promoter
vector was used as the blank control, whilst an NC mimic was used
as the negative control. The cells were subsequently lysed using
Glo Lysis Buffer (Promega Corporation) at 24 h following
transfection, and luciferase activity was measured using a
Dual-Luciferase Reporter Assay system (Promega Corporation),
according to the manufacturer's protocol. All luciferase activities
were normalized to that of Renilla luciferase activity.
Statistical analysis
The data are presented as the mean ± standard
deviation. One-way analysis of variance followed by the least
significance difference post hoc test was performed to determine
statistical significance among different groups using the SPSS
software package (version 16.0; SPSS Inc.). P<0.05 was
considered to indicate a statistically significant difference.
Results
miR-146a regulates IRAK1 expression in
THP-1 cells
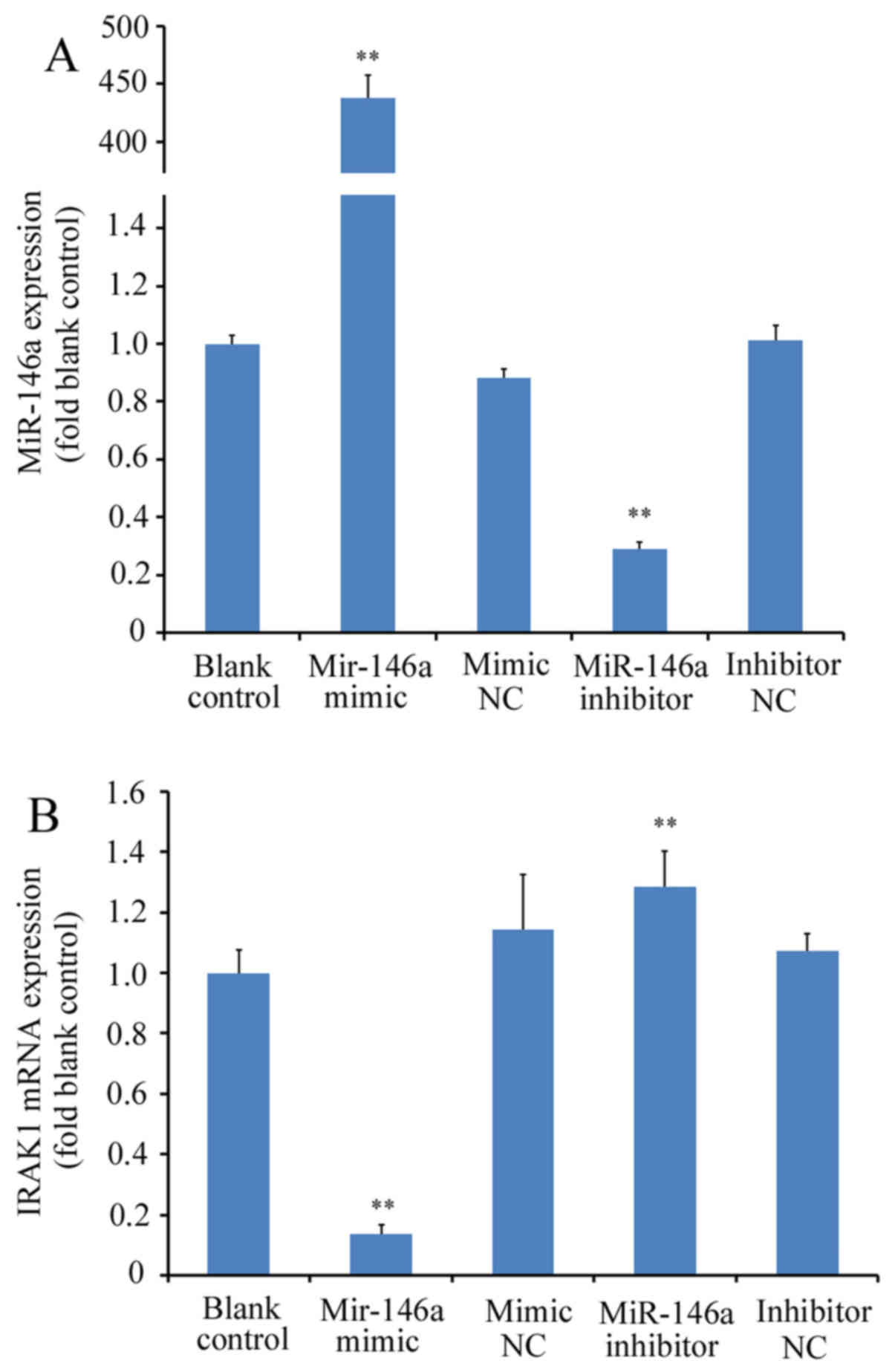
miR-146a mimicor miR-146a inhibitor were first
transfected into THP-1 cells prior to RT-qPCR analysis.
Transfection with miR-146a mimic significantly increased miR-146a
expression (>400-fold) whilst transfection with miR-146a
inhibitor significantly reduced miR-146a expression (by ~70%); with
differences observed compared with the blank control or
corresponding NC groups (P<0.01; Fig.
1A). IRAK1 mRNA expression was subsequently measured in THP-1
cells transfected with miR-146a mimic or inhibitor. miR-146a
overexpression using the miR-146a mimic led to a significant
reduction in IRAK1 mRNA expression (by ~88% relative to blank
control; P<0.01 vs. mimic NC), whereas transfection with the
miR-146a inhibitor led to a significant increase in IRAK1
expression (by ~20% relative to blank control; P<0.01 vs.
inhibitor NC; Fig. 1B). At the
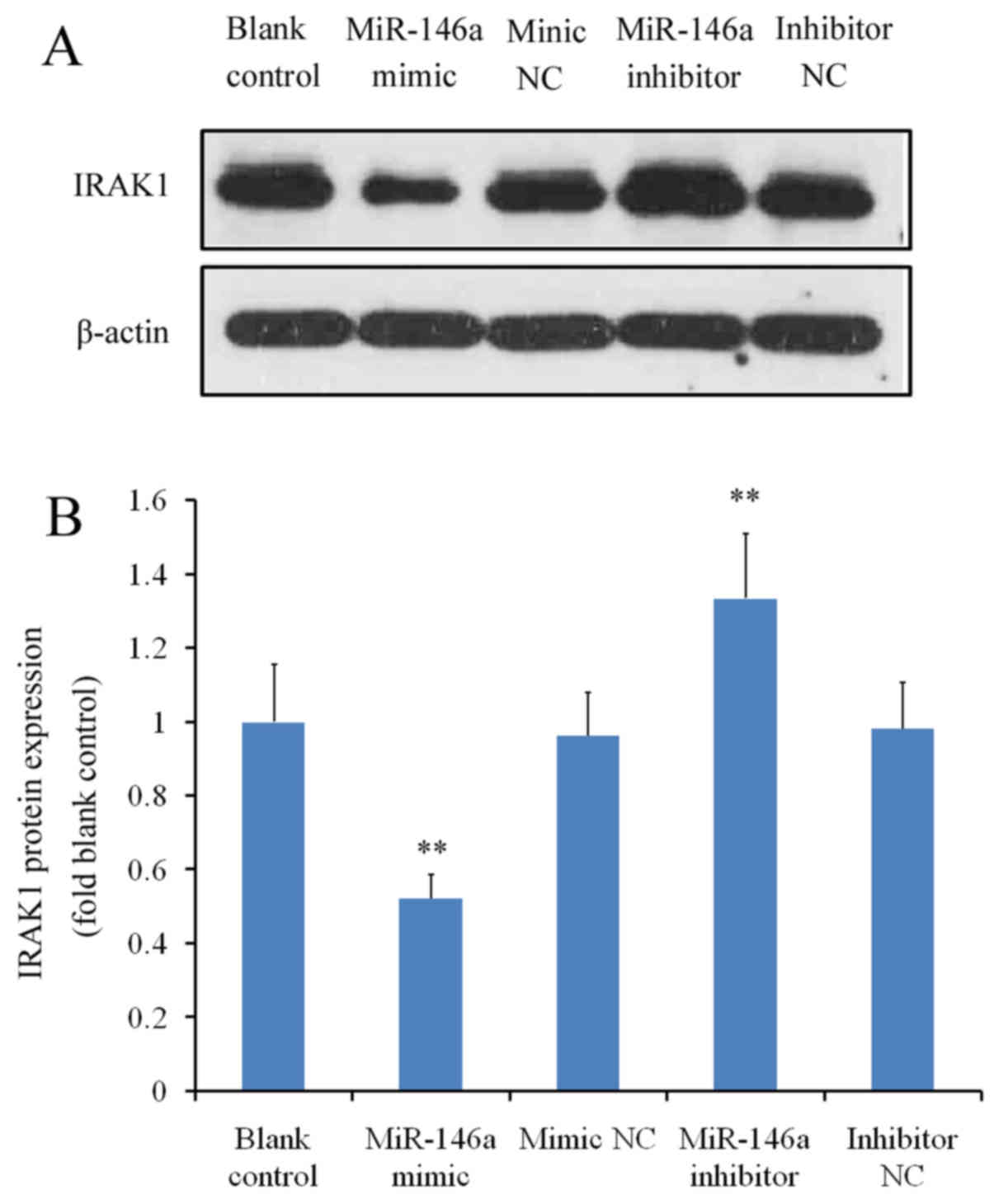
protein level, cells transfected with the miR-146a mimic exhibited
a significant reduction in IRAK1 expression compared with the mimic
NC, whereas miR-146a inhibitor transfection significantly increased
IRAK protein levels (P<0.01 vs. inhibitor NC; Fig. 2A and B). There were no significant
differences between the blank control and to two corresponding NC
groups (P>0.05 blank control vs. mimic NC and P>0.05 blank
control vs. inhibitor NC; Fig. 2A and
B).
miR-146a modulates NF-κB activation by
regulating IRAK1 expression under inflammatory conditions
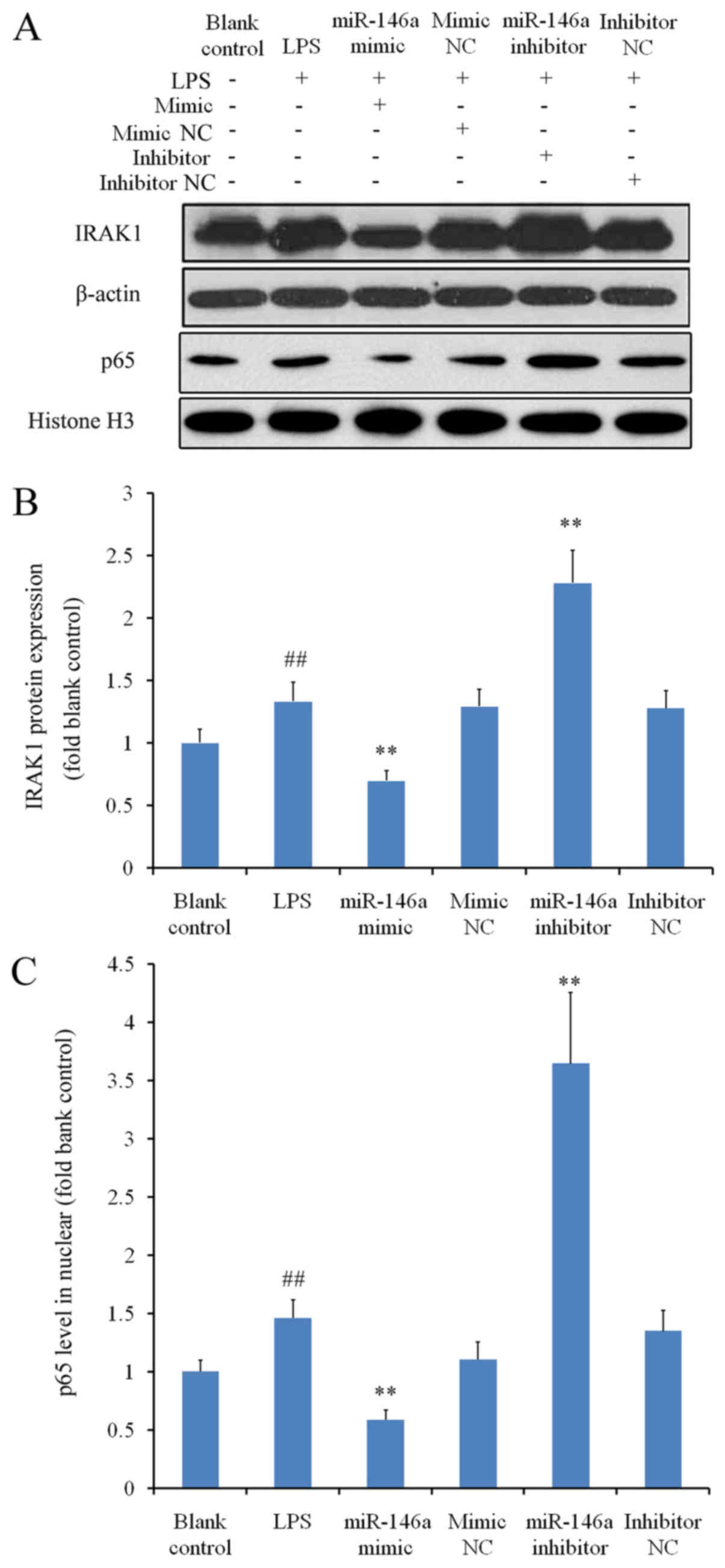
THP-1 cells were treated with LPS to induce
inflammation, which significantly augmented IRAK1 expression when
compared with the blank control group (P<0.01; Fig. 3A and B). Under inflammatory
conditions, transfection with miR-146a mimic significantly reduced
IRAK1 expression, whereas transfection with miR-146a inhibitor
significantly increased IRAK1 levels compared with the
corresponding NC groups (P<0.01; Fig.
3B). As NF-κB is a downstream molecule of IRAK1 (17), western blot analysis was subsequently
performed to measure the nuclear protein levels of p65; a subunit
of NF-κB. Nuclear p65 levels were significantly higher in the LPS
group compared with those in the blank control group (P<0.01;
Fig. 3C). The changes in nuclear
NF-κB p65 protein levels in cells transfected with either miR-146a
mimic or miR-146a inhibitor mirrored those of IRAK1 levels in the
corresponding treatment groups (Fig. 3B
and C). These results suggested that miR-146a regulates NF-κB
activation in the presence of LPS, by modulating the expression of
IRAK1.
miR-146a inhibits IL-6 and TNF-α
secretion
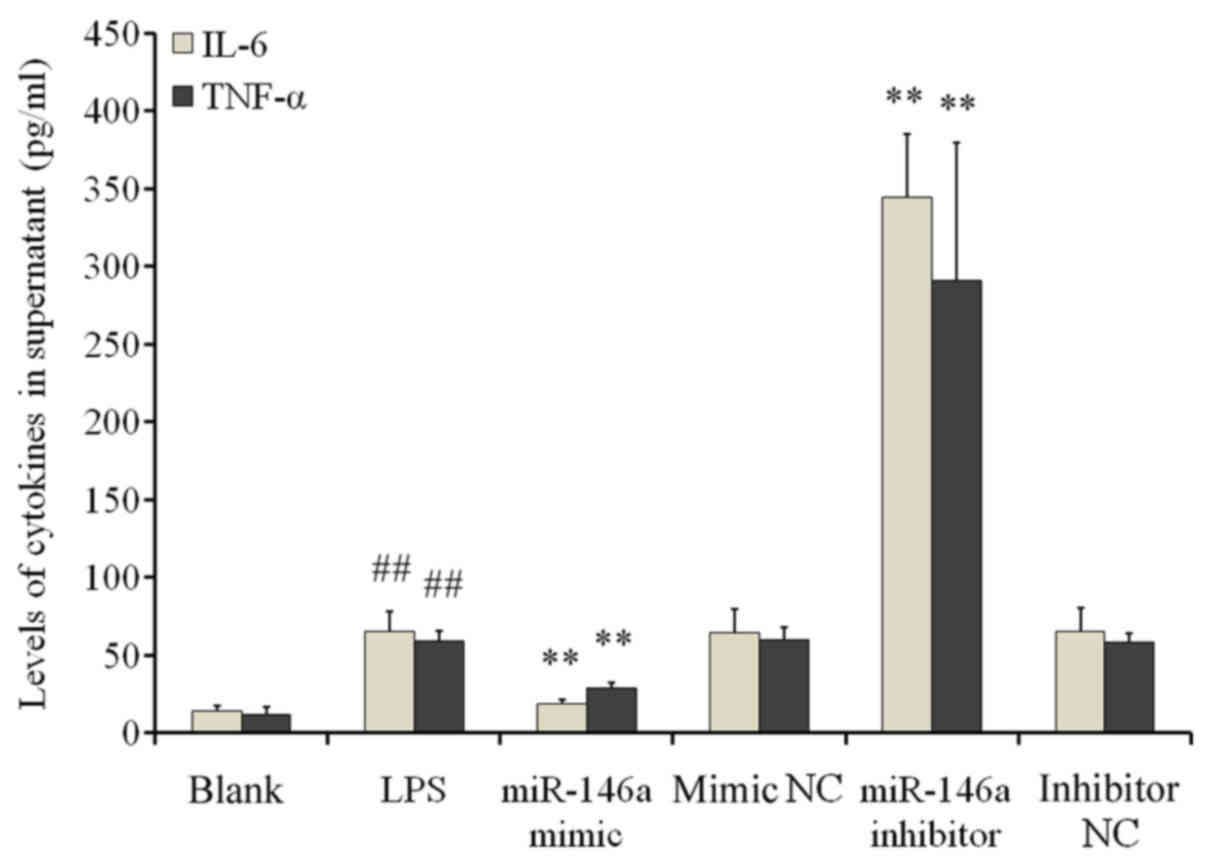
ELISA results demonstrated significantly increased
IL-6 and TNF-α levels in the supernatant of cells in the LPS group
when compared with cells in the blank control group (P<0.01;
Fig. 4). miR-146a overexpression
significantly reduced IL-6 and TNF-α secretion compared with the NC
mimic groups (P<0.01). By contrast, transfection with the
miR-146a inhibitor significantly increased IL-6 and TNF-α secretion
when compared with the inhibitor NC group (P<0.01; Fig. 4). These findings suggested that LPS
stimulation induced inflammatory responses in THP-1 cells by
increasing the secretion of inflammatory cytokines; a process that
is negatively regulated by miR-146a.
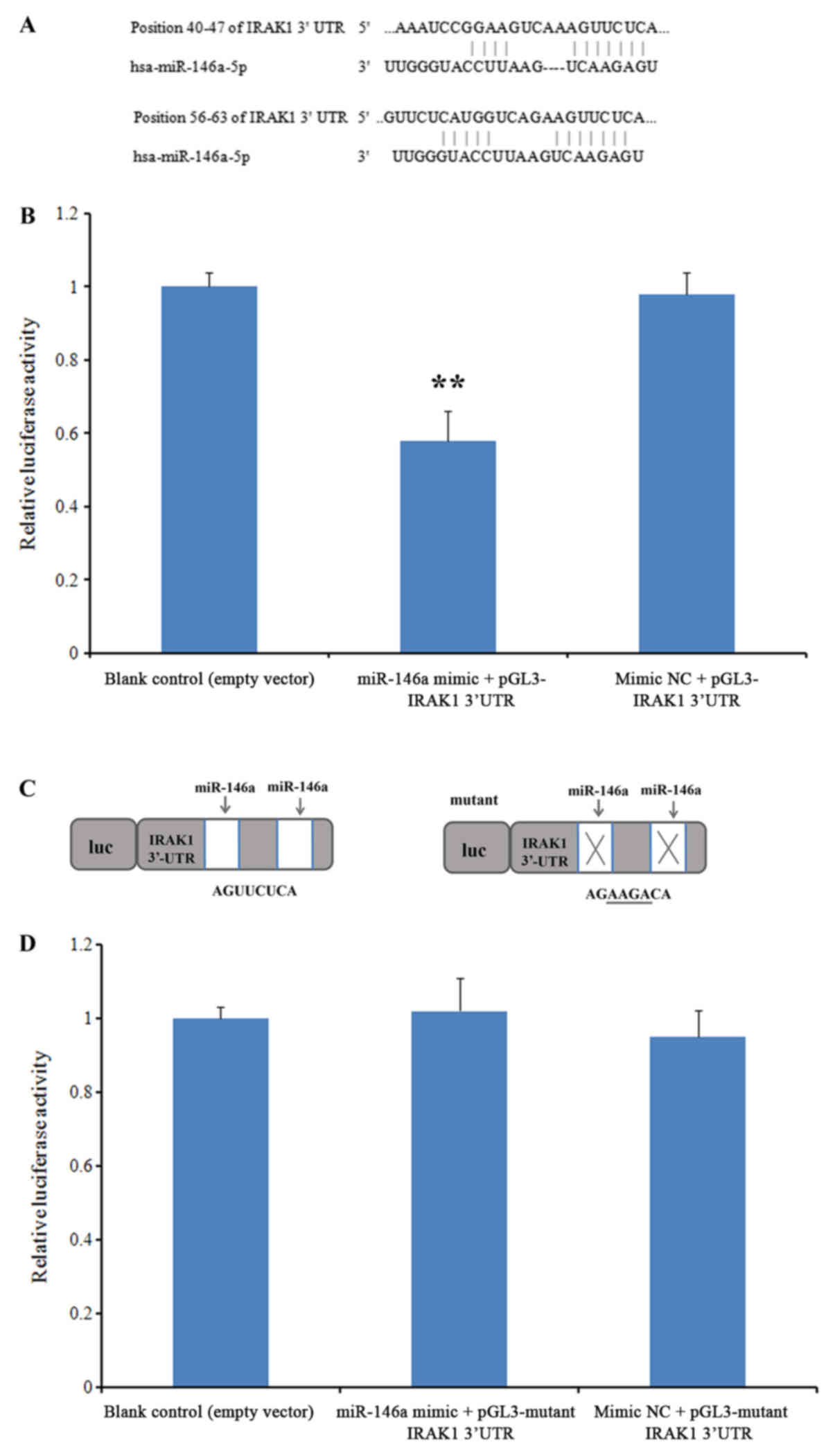
IRAK1 is a direct target of
miR-146a
To investigate whether miR-146a can interact with
the IRAK1 mRNA 3′-UTR, a luciferase reporter assay was performed.
Prior to this assay, the predicted targets of this miRNA were
revealed using TargetScan (version 7.2). Two evolutionarily
conserved potential miR-146a target sites were discovered in the
3′-UTR of IRAK1 mRNA (positions 40–47 and 56–63; Fig. 5A). In the group co-transfected with
miR-146a mimics and the wild-type 3′-UTR IRAK1 sequence, luciferase
activity was significantly repressed when compared with the blank
control group, while no significant difference was observed in
cells co-transfected with the NC mimic (P<0.01; Fig. 5B). When the IRAK1 3′-UTR sequence was
replaced with a mutant sequence (Fig.
5C), no significant differences were observed in the luciferase
activity of cells co-transfected with the miR-146a mimic when
compared with the blank control group (Fig. 5D). These results suggested that
miR-146a directly targets the IRAK1 3′-UTR.
Discussion
SLE is a chronic diffuse connective tissue disease
that is caused by inflammatory responses associated with
autoimmunity (18). Although the
mechanisms underlying SLE pathogenesis are unclear, the current
consensus is that SLE may be associated with disturbances in the
immune response, predisposing genetic factors, sexual hormone
levels and exogenous stimulation (19). Dai et al (19) reported significant differences in the
expression profiles of miRNAs between PBMCs derived from patients
with SLE and healthy controls. Specifically, seven miRNAs were
discovered to be downregulated whilst nine miRNAs were found to be
upregulated in patients with SLE. Tang et al (20) discovered that the expression of
miR-146a in PBMCs was significantly lower in patients with SLE
compared with the control group, further suggesting that miR-146a
negatively regulates the IFN-1 signaling pathway. It has been
established that type I IFN serves a key role in SLE (20,21).
Immune complexes of autoantibodies against self-nucleoproteins and
self-DNA from lupus patients are known to induce IFN-α production
in plasmacytoid dendritic cells (22). Once induced, type I IFN binds to its
receptors to initiate downstream signaling by activation of STAT
proteins, ultimately leading to the transcription of target genes
(20). Reduced expression of
miR-146a in patients with SLE and correlation between miR-146a
levels and disease activity have been previously reported (20,23).
miR-146a has been demonstrated to inhibit the type I IFN pathway by
downregulating IFN regulatory factor 5 (IRF-5) and STAT-1 (20).
IRAK1, one of the regulatory targets of miR-146a, is
a critical signaling molecule involved in the TLR4 signal
transduction pathway. When the TLR is activated by pathogens or
antigen-antibody complexes, it activates the associated adaptor
protein IRAKI by the downstream My D88-dependent signaling pathway,
leading to the activation of TRAF6, IΚKβ and eventually NF-κB
(24,25). The findings of the present study
demonstrated that miR-146a overexpression may downregulate the
expression of IRAK1, reduce NF-κB activation and inhibit the
secretion of pro-inflammatory cytokines, a signaling cascade that
may serve as one of the mechanisms underlying miR-146a-mediated
regulation of inflammatory responses in SLE.
Patients with SLE are predisposed to infection due
to alterations in the immune system as a result of
immunosuppressive therapy. As a result, infections may exacerbate
SLE by promoting inflammatory responses. The development and
progression of SLE is associated with enhanced inflammatory
responses and dysregulated pro-inflammatory cytokines secretion
(26). IL-6 and TNF-α expression are
generally increased in patients with SLE compared with healthy
individuals (27–29). The levels of IL-6 and TNF-α are
positively associated with disease progression and serum levels of
anti-double-stranded (ds) DNA antibody (29,30).
IL-6 is an important type II T helper cell cytokine that is
secreted by a variety of cell types, including T cells and
monocytes, and stimulates the production of anti-ds DNA autoimmune
antibodies in patients with SLE. This subsequently leads to the
production of IL-6, and the cycle begins again (30,31).
TNF-α activates a number of inflammatory cells, which infiltrate
multiple organs and induce inflammation-associated damage (30–32). The
activation of NF-κB promotes the expression of pro-inflammatory
cytokines including IL-6 and TNF-α (33). The results of the present study
indicated that miR-146a may inhibit the activation of NF-κB,
thereby decreasing the expression of IL-6 and TNF-α. Similarly, Du
et al (34) demonstrated that
miR-146 a suppresses the β-glucan-induced production of IL-6 and
TNF-α by inhibiting the dectin-1/tyrosine-protein kinase SYK/NF-κB
signaling pathway. These observations suggest that the
downregulation of miR-146a may eliminate its regulatory effects on
the secretion of pro-inflammatory cytokines, leading to an increase
in IL-6 and TNF-α levels. This may promote the development of SLE,
particularly under conditions of infection. Ultimately, miR-146a
may regulate IRAK1 expression and inhibit the activation of
inflammatory signals, leading to the secretion of pro-inflammatory
cytokines that may be involved in SLE pathogenesis.
Although an association between miR-146a and
inflammation was observed in vitro in the present study, the
clinical significance of miR-146a would need to be addressed in
future studies. Previous studies have observed aberrant miR-146a
expression in blood cells, including PBMCs, T and B cells (35–37).
Procedures involving cell isolation and extraction are complex, and
therefore not suitable for clinical detection. Alternatively, it
has been clearly demonstrated that miRNA remains stable in the
serum and other bodily fluids (38).
Therefore, the serum levels of miR-146a between patients with SLE
and healthy individuals, and the potential application of serum
miRNA as a tool for monitoring SLE in a clinical setting, remain
potential avenues for further investigation. The in vitro
experiments performed in the present study revealed that miR-146a
may regulate the expression of IL-6 and TNF-α in THP-1 cells;
therefore, in vivo miR-146a levels in patients with SLE may
be associated with the levels of IL-6 and TNF-α. A clinical trial
is currently in progress involving >120 patients with SLE, in
which the patients will be divided into active and inactive groups
according to the Systemic Lupus Erythematosus Disease Activity
Index. In addition, the role of the miR-146a/IRAK1/NF-κB axis in
vivo will need to be investigated. Finally, the association
between miR-146a and other autoimmune diseases, including RA and
Sjogren's syndrome, are also interesting topics of future
study.
Acknowledgements
Not applicable.
Funding
This study was supported by the National High
Technology Research and Development Program (grant no.
2015AA021107), the Natural Science Foundation of Tianjin (grant
nos. 17JCYBJC27500 and 17JCYBJC26200), the National Natural Science
Foundation of China (grant no. 81602403) and the Clinical Research
Foundation of Tianjin Fist Central Hospital (grant no.
CF201812).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
HM and CZ designed the study, analyzed the data, and
contributed significantly to the preparation of the manuscript. LZ
and KW performed the reverse transcription-quantitative PCR and
western blot experiments. QQ and MW performed the cell culture
experiments. PS and LY performed the ELISA experiments.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ranganathan K and Sivasankar V:
MicroRNAs-biology and clinical applications. J Oral Maxillofac
Pathol. 18:229–234. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Pileczki V, Cojocneanu-Petric R, Maralani
M, Neagoe IB and Sandulescu R: MicroRNAs as regulators of apoptosis
mechanisms in cancer. Clujul Med. 89:50–55. 2016.PubMed/NCBI
|
|
3
|
Miao CG, Yang YY, He X, Huang C, Huang Y,
Zhang L, Lv XW, Jin Y and Li J: The emerging role of microRNAs in
the pathogenesis of systemic lupus erythematosus. Cell Signal.
25:1828–1836. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sharma AR, Sharma G, Lee SS and
Chakraborty C: MiRNA-regulated key components of cytokine signaling
pathways and inflammation in rheumatoid arthritis. Med Res Rev.
36:425–439. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Habibi F, Ghadiri Soufi F, Ghiasi R,
Khamaneh AM and Alipour MR: Alteration in inflammation-related
miR-146a expression in NF-κB signaling pathway in diabetic rat
hippocampus. Adv Pharm Bull. 6:99–103. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Stickel N, Prinz G, Pfeifer D, Hasselblatt
P, Schmitt-Graeff A, Follo M, Thimme R, Finke J, Duyster J, Salzer
U and Zeiser R: MiR-146a regulates the TRAF6/TNF-axis in donor T
cells during GVHD. Blood. 124:2586–2595. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Taganov KD, Boldin MP and Baltimore D:
MicroRNAs and immunity: Tiny players in a big field. Immunity.
26:133–137. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Perl A: Pathogenic mechanisms in systemic
lupus erythematosus. Autoimmunity. 43:1–6. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang H, Peng W, Ouyang X, Li W and Dai Y:
Circulating microRNAs as candidate biomarkers in patients with
systemic lupus erythematosus. Transl Res. 160:198–206. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ji JD, Cha ES and Lee WJ: Association of
miR-146a polymorphisms with systemic lupus erythematosus: A
meta-analysis. Lupus. 23:1023–1030. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gao M, Wang X, Zhang X, Ha T, Ma H, Liu L,
Kalbfleisch JH, Gao X, Kao RL, Williams DL and Li C: Attenuation of
cardiac dysfunction in polymicrobial sepsis by MicroRNA-146a is
mediated via targeting of IRAK1 and TRAF6 expression. J Immunol.
195:672–682. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kaisho T and Aldra S: Toll-like receptor
function and signaling. J Allergy Clin Immunol. 117:979–988. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rodriguez-Pla A, Patel P, Maecker HT,
Rossello-Urgell J, Baldwin N, Bennett L, Cantrell V, Baisch J,
Punaro M, Gotte A, et al: IFN priming is necessary but not
sufficient to turn on a migratory dendritic cell program in lupus
monocytes. J Immunol. 192:5586–5598. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang P, Han X, Mo B, Huang G and Wang C:
LPS enhances TLR4 expression and IFN-γ production via the
TLR4/IRAK/NF-κB signaling pathway in rat pulmonary arterial smooth
muscle cells. Mol Med Rep. 16:3111–3116. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Nahid MA, Pauley KM, Satoh M and Chan EK:
MiR-146a is critical for endotoxin-induced tolerance: Implication
in innate immunity. J Biol Chem. 284:34590–34599. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chen Y, Zeng Z, Shen X, Wu Z, Dong Y and
Cheng JC: MicroRNA-146a-5p negatively regulates pro-inflammatory
cytokine secretion and cell activation in lipopolysaccharide
stimulated human hepatic stellate cells through inhibition of
Toll-like receptor 4 signaling pathways. Int J Mol Sci1. 17(pii):
E10762016. View Article : Google Scholar
|
|
18
|
Muñoz LE, Janko C, Schulze C, Schorn C,
Sarter K, Schett G and Herrmann M: Autoimmunity and chronic
inflammation-two clearance-related steps in the etiopathogenesis of
SLE. Autoimmun Rev. 10:38–42. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Dai Y, Huang YS, Tang M, Lv TY, Hu CX, Tan
YH, Xu ZM and Yin YB: Microarray analysis of microRNA expression in
peripheral blood cells of systemic lupus erythematosus patients.
Lupus. 16:939–946. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tang Y, Luo X, Cui H, Ni X, Yuan M, Guo Y,
Huang X, Zhou H, de Vries N, Tak PP, et al: MicroRNA-146A
contributes to abnormal activation of the type I interferon pathway
in human lupus by targeting the key signaling proteins. Arthritis
Rheum. 60:1065–1075. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wigren M, Nilsson J and Kaplan MJ:
Pathogenic immunity in systemic lupus erythematosus and
atherosclerosis: Common mechanisms and possible targets for
intervention. J Intern Med. 278:494–506. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Means TK, Latz E, Hayashi F, Murali MR,
Golenbock DT and Luster AD: Human lupus autoantibody-DNA complexes
activate DCs through cooperation of CD32 and TLR9. J Clin Invest.
115:407–417. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Husakova M: MicroRNAs in the key events of
systemic lupus erythematosus pathogenesis. Biomed Pap Med Fac Univ
Palacky Olomouc Czech Repub. 160:327–342. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Fang H, Wang PF, Zhou Y, Wang YC and Yang
QW: Toll-like receptor 4 signaling in intracerebral
hemorrhage-induced inflammation and injury. J Neuroinflammation.
10:272013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
He X, Zheng Y, Liu S, Shi S, Liu Y, He Y,
Zhang C and Zhou X: MiR-146a protects small intestine against
ischemia/reperfusion injury by down-regulatingTLR4/TRAF6/NF-κB
pathway. J Cell Physiol. 233:2476–2488. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Nahid MA, Satoh M and Chan EK: MicroRNA in
TLR signaling and endotoxin tolerance. Cell Mol Immunol. 8:388–403.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Willis R, Seif AM, McGwin G Jr,
Martinez-Martinez LA, González EB, Dang N, Papalardo E, Liu J, Vilá
LM, Reveille JD, et al: Effect of hydroxychloroquine treatment on
pro-inflammatory cytokines and disease activity in SLE patients:
Data from LUMINA (LXXV), a multiethnic US cohort. Lupus.
21:830–835. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ripley BJ, Goncalves B, Isenberg DA,
Latchman DS and Rahman A: Raised levels of interleukin 6 in
systemic lupus erythematosus correlate with anaemia. Ann Rheum Dis.
64:849–853. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sabry A, Sheashaa H, EI-Husseini A,
Mahmoud K, Eldahshan KF, George SK, Abdel-Khalek E, El-Shafey EM
and Abo-Zenah H: Proinflammatory cytokines (TNF-alpha and IL-6) in
Egyptian patients with SLE: Its correlation with disease activity.
Cytokine. 35:148–153. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Arora V, Verma J, Marwah V, Kumar A, Anand
D and Das N: Cytokine imbalance in systemic lupus erythematosus: A
study on northern Indian subjects. Lupus. 21:596–603. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Gigante A, Gasperini ML, Afeltra A,
Barbano B, Margiotta D, Cianci R, De Francesco I and Amoroso A:
Cytokines expression in SLE nephritis. Eur Rev Med Pharmacol Sci.
15:15–24. 2011.PubMed/NCBI
|
|
32
|
Robinson ES and Werth VP: The role of
cytokines in the pathogenesis of cutaneous lupus erythematosus.
Cytokine. 73:326–334. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Nahid MA, Satoh M and Chan EK: Mechanistic
role of microRNA-146a in endotoxin-induced differential
cross-regulation of TLR signaling. J Immunol. 186:1723–1734. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Du L, Chen X, Duan Z, Liu C, Zeng R, Chen
Q and Li M: MiR-146a negatively regulatesdectin-1-induced
inflammatory responses. Oncotarget. 8:37355–37366. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Mookherjee N and El-Gabalawy HS: High
degree of correlation between whole blood and PBMC expression
levels of miR-155 and miR-146a in healthy controls and rheumatoid
arthritis patients. J Immunol Methods. 400-401:106–110. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Sheppard HM, Verdon D, Brooks AE, Feisst
V, Ho YY, Lorenz N, Fan V, Birch NP, Didsbury A and Dunbar PR:
MicroRNA regulation in human CD8+ T cell subsets-cytokine exposure
alone drives miR-146a expression. J Transl Med. 12:2922014.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Cho S, Lee HM, Yu IS, Choi YS, Huang HY,
Hashemifar SS, Lin LL, Chen MC, Afanasiev ND, Khan AA, et al:
Differential cell-intrinsic regulations of germinal center B and T
cells by miR-146a and miR-146b. Nat Commun. 9:27572018. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Liu AM, Wang W and Luk JM: miRNAs: New
tools for molecular classification, diagnosis and prognosis of
hepatocellular carcinoma. Hepat Oncol. 1:323–329. 2014. View Article : Google Scholar : PubMed/NCBI
|