Introduction
Inflammation plays a key role in the pathogenesis of
many lung diseases, such as chronic obstructive pulmonary disease
(COPD) (1). Accumulating evidence
has demonstrated that lung epithelial cells not only form the
epithelium, which acts as a physical barrier between the inhaled
air and internal tissues, but also are actively involved in
antimicrobial defense through pattern recognition receptors (PRRs)
on the cell surface and in the cytoplasm (2–4). Cyclic
guanosine monophosphate (GMP)-adenosine monophosphate (AMP)
synthase (cGAS) is a PRR that can sense cytoplasmic DNA and
synthesize cyclic GMP-AMP (cGAMP) (5), which activates the stimulator of
interferon genes/serine/threonine-protein kinase 1 TBK1
(TBK1)/interferon regulatory factor (IRF) 3 axis and eventually
triggers the expression of type I interferons (IFN-I) (5). Therefore, cGAS has crucial functions in
DNA-induced innate immune responses in immune cells (6). As cGAS binds to cytosolic DNA
irrespective of DNA sequence, it can be activated by DNA from
invading pathogens or from the cell itself (7). In macrophages, cGAS can directly
interact with the autophagy protein Beclin-1 to mediate DNA-induced
autophagy (8). However, it remains
unknown whether cGAS plays a role in regulating autophagy in lung
epithelial cells.
Toll-like receptor 4 (TLR4) is a type of PRR that
has crucial functions in the pulmonary immune response against
bacterial infection (9). TLR4 can
recognize and be activated by LPS, an important component of the
outer membrane of Gram-negative bacteria (10). On the one hand, TLR4 activation
stimulates the production of proinflammatory cytokines through the
myeloid differentiation primary response gene 88 (MyD88)-dependent
pathway, in which MyD88, interleukin-1 receptor-associated kinase
1, TNF receptor-associated factor (TRAF) 6, and the transcription
factors nuclear factor (NF)-κB, activator protein 1 (AP-1) and
IRF-5 are key molecules (11). On
the other hand, TLR4 activation also induces the production of
IFN-I, such as IFN-β through the MyD88-independent pathways,
including Toll-interleukin-1 receptor domain-containing
adaptor-inducing interferon-β (TRIF), TRAF3, receptor-interacting
serine/threonine-protein kinase 1 (RIP1), TBK1 and the
transcription factors, NF-κB, AP-1 and IRF3 (11). LPS can induce autophagy in murine and
human macrophages (12), which is
regulated through a TRIF-dependent, MyD88-independent TLR4
signaling pathway (12). RIP1 and
p38 mitogen-activated protein kinase are downstream components of
this pathway (12). LPS also
stimulates cGAS expression in an IFN-I-dependent manner in murine
and human macrophages (13).
However, it remains unknown whether and by what mechanisms LPS
stimulates cGAS expression in lung epithelial cells.
Autophagy is a highly conserved cellular pathway by
which damaged organelles and proteins are delivered to the lysosome
to be degraded by enzymes (14).
Autophagy is also involved in the host defense against microbes by
degradation of microorganisms and delivery of microbial nucleic
acids and antigens for the activation of immune responses (14). Increased levels of autophagy have
been observed in lung tissues from patients with COPD (15), and exposure to cigarette smoke
extract results in increased autophagy in lung epithelial cells
(15). In addition to cigarette
smoke, infectious agents also contribute to COPD initiation and
progression (16). However, whether
and how infectious agents affect autophagy in lung epithelial cells
remains unknown.
In the present study, the effects of DNA on IFN-β
expression and autophagy and the effects of LPS on the expression
and function of cGAS in A549 cells were investigated.
Materials and methods
Antibodies and other reagents
An anti-cGAS (cat. no. 15102; 1:1,000 dilution)
antibody was purchased from Cell Signaling Technology, Inc. An
anti-β-actin antibody (cat. no. TA-09; 1:1,000 dilution) was
obtained from OriGene Technologies, Inc. microtubule-associated
proteins 1A/1B light chain 3B (LC3B; cat. no. NB600-1384; 1:1,000
dilution) and sequestosome-1 (p62) antibodies (cat. no. NBP1-48320;
1:1,000 dilution) were obtained from Novus Biologicals LLC. LPS
[Escherichia coli (E. coli) 055:B5] was purchased from
Sigma-Aldrich (Merck KGaA). The TLR4 inhibitor TAK242 (cat. no.
13871), the TBK1 inhibitor BX795 (cat. no. 14932) and the NF-κB
inhibitor BAY11-7082 (cat. no. 10010266) were obtained from Cayman
Chemical Company. The cGAS inhibitor RU.521 (17) (cat. no. AOB37877) was purchased from
Aobious Inc. E. coli was purchased from the Beijing CWBIO
Company. Recombinant human IFN-β was purchased from Multisciences
Biotech Co., Ltd.
Cell culture, treatments and
transfection
A549 cell line derived from an alveolar cell
carcinoma was used as model of alveolar epithelial cells in the
current study (18,19). A549 cells were obtained from the
Kunming cell bank of the Chinese Academy of Sciences (Kunming,
China). Cells were cultured in RPMI-1640 medium containing 10%
fetal bovine serum purchased from Thermo Fisher Scientific and 1%
penicillin/streptomycin at 37°C in a humidified incubator with 5%
CO2. A549 cells (2×105 cells/well) were
seeded in six-well plates and cultured at 37°C in a 5%
CO2 incubator overnight, followed by further
experimentation. To test the effects of LPS on cGAS expression, LPS
at different concentrations (100, 200, 400 and 800 ng/ml) were used
to treat A549 cells for 4 h, and then cGAS expression was analyzed.
For inhibitor, A549 cells were pretreated with TAK242 (10 µM),
BX-795 (10 µM) or BAY11-7082 (20 µM) for 1 h, followed by LPS (400
ng/ml) treatment for 4 h. For transfection experiments, A549 cells
(2×105 cells/well) were seeded in six-well plates
overnight, then transfected with E. coli DNA (2 µg/ml) using
Lipofectamine® 3000 (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's instructions. The
cGAS inhibitor RU.521 was added at the indicated concentrations
(0.5, 1, 1.5 and 2 µM) to cell culture wells concurrently with the
transfection materials. The control group was treated under the
same condition but without DNA and RU.521. A total of 24 h
post-transfection, the cells and cell culture media were harvested
separately for further analysis.
Western blot analysis
Western blot assays were performed as previously
described (20). In brief, A549
cells were collected and lysed with lysis buffer (cat. no. R0020;
Beijing, Solarbio Science and Technology Co., Ltd.) on ice for 10
min. The supernatant was obtained by centrifugation at 13,500 × g
for 20 min at 4°C, and the protein concentration of the supernatant
was measured with a BCA kit (cat. no. P0009; Beyotime Institute of
Biotechnology) according to the manufacturer's instructions. A
total of 20 µg protein was loaded per lane and separated by 12 or
15% SDS-PAGE and then transferred onto polyvinylidene difluoride
membranes. The membranes were blocked with 5% non-fat milk at room
temperature for 1-h, followed by incubation with the primary
antibody (LC3B, p62, β-actin and cGAS) at 4°C overnight. The
membrane was then incubated with a horseradish
peroxidase-conjugated goat anti-rabbit/mouse secondary antibody at
room temperature for 1 h. Blots were developed using an ECL kit
(cat. no. P0018; Beyotime Institute of Biotechnology). The gray
value of the target protein and β-actin were analyzed using Image J
software (version 4.0; National Institutes of Health,
Bethesda).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
RT-qPCR assays were performed as described
previously (21). In brief, total
RNA from A549 cells was isolated using TRIzol® (cat. no.
15596026; Thermo Fisher Scientific, Inc.). cDNA was amplified with
2 µg of total RNA from each sample using the RT kit (cat. no.
KR106; Tiangen Biotech Co., Ltd) according to the manufacturer's
protocol. The FQ-RT primer Mix for cDNA consisted of oligo-dT
primer and Random primers (8N). Reverse transcription conditions
were as follows: 42°C for 15 min, then 95°C for 3 min. RT-qPCR was
performed using SYBR Green I (cat. no. RR820A; Takara Bio Inc.) and
a CFX touch real-time PCR detection system (Bio-Rad Laboratories,
Inc.). GAPDH (forward, 5′-CAGGAGGCATTGCTGATGAT-3′ and reverse,
5′-AAGGCTGGGGCTCATTT-3′) was used as the internal control. Primers
for cGAS (forward, 5′-GTACCCAGAACCCTCAAGACA-3′ and reverse,
5′-GTCCTGAGGCACTGAAGAAAG-3′) for RT-qPCR were obtained from
GeneCopoeia (cat. no. HQP001767). Thermocycling conditions were as
follows: 95°C for 30 sec, 40 cycles of 95°C for 5 sec and 60°C for
30 sec. The relative mRNA expression level of the genes was
determined using the 2−ΔΔCq method (22).
E. coli DNA preparation
An E. coli strain (cat. no. CW0808) was
purchased from Beijing CWBIO Company and DNA was extracted using a
bacterial DNA kit (cat. no. DP302; Tiangen Biotech Co., Ltd)
according to the manufacturer's instructions. The DNA concentration
was measured using a NanoDrop 2000 spectrophotometer (Thermo Fisher
Scientific, Inc.).
ELISA
A549 cells and culture media were collected
separately at the end of the various treatments of inhibitors, LPS
or E. coli DNA alone or combined. Cell lysates were prepared
as previously described by adding 200 µl lysis buffer (21). IFN-β levels in the cell culture media
and in the supernatant of the cell lysates were measured with a
human IFN-β ELISA kit (cat. no. EK1236; Multisciences Biotech Co.,
Ltd.) according to the manufacturer's instructions. IFN-β levels in
the cell culture media and the cell lysates were combined to
determine the total IFN-β level under each condition indicated in
figures.
Statistical analysis
SPSS v20 (IBM, Corp.) and GraphPad Prism v5
(GraphPad Software Inc.) were used to analyze all data. All
experiments were performed independently at least three times, and
the results are shown as the mean ± standard error of the mean.
Comparison between groups was performed using either the Student's
t-test or one-way analysis of variance test followed by the
Student-Newman-Keuls post hoc test. P<0.05 was considered to
indicate a statistically significant difference.
Results
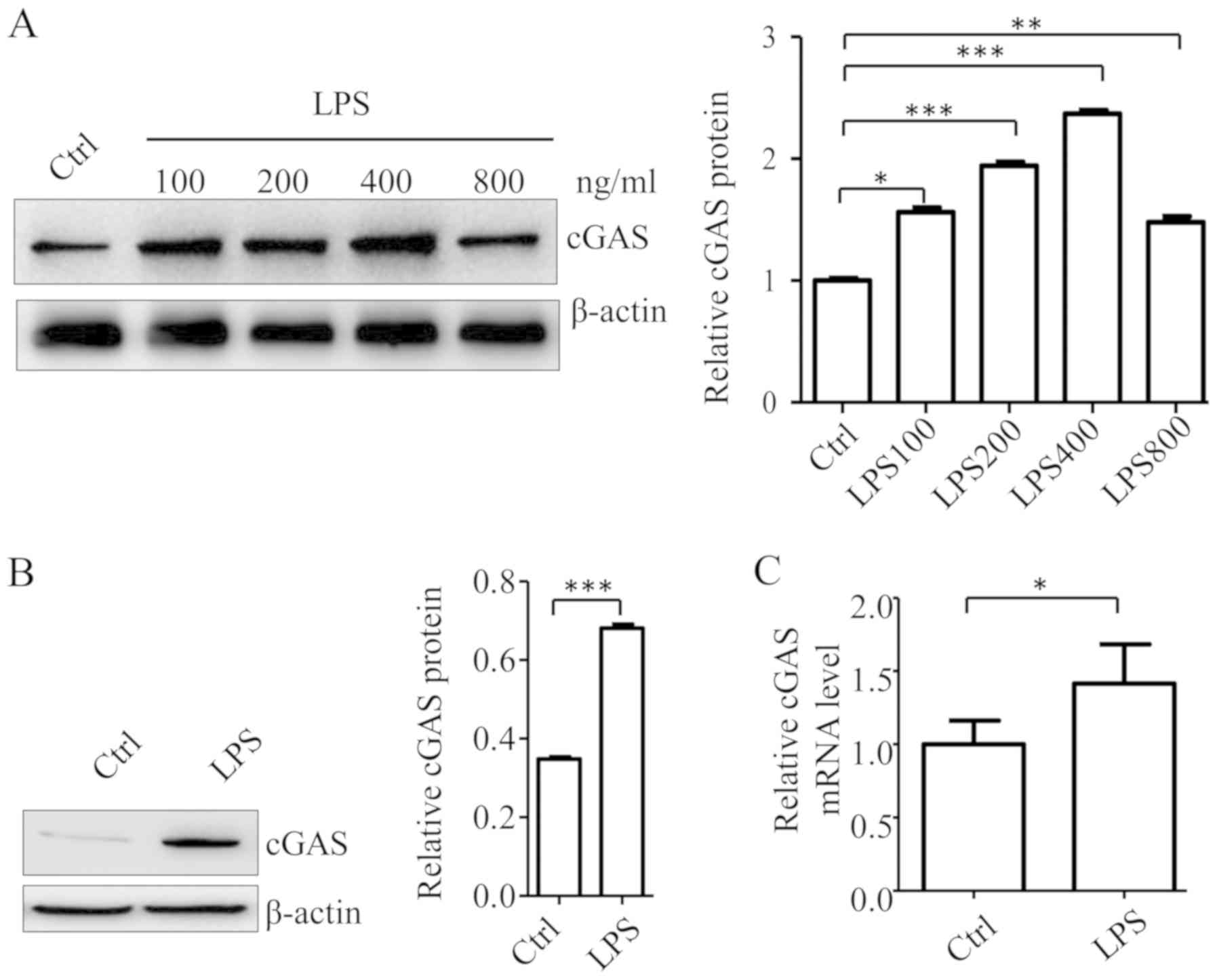
LPS treatment upregulates cGAS
expression in A549 cells
LPS treatment can upregulate cGAS expression in both
murine and human macrophages (13).
To assess whether LPS produces the same effects on cGAS expression
in lung epithelial cells, A549 cells were treated with LPS at
different concentrations (i.e., 100, 200, 400 and 800 ng/ml) for 4
h and cGAS protein levels were then examined. The results showed
that LPS treatment induced cGAS expression in A549 cells, and this
effect was dose-dependent, from 100–400 ng/ml LPS. The cGAS protein
levels in cells treated with 400 ng/ml LPS was higher compared with
that in cells treated with 800 ng/ml LPS (Fig. 1A). As 400 ng/ml LPS treatment had the
strongest effect on cGAS expression, this concentration was
selected for further experiments. To confirm the effect of LPS on
cGAS expression at the mRNA level, A549 cells were treated with LPS
(400 ng/ml) for 4 h and cGAS mRNA levels were determined using
RT-qPCR. The results showed that cGAS mRNA levels in A549 cells
were significantly increased compared with the control group, which
was consistent with the western blot results (Fig. 1B and C). These findings demonstrated
that LPS treatment can induce cGAS expression in A549 cells.
LPS regulates cGAS expression through
the MyD88-independent TLR4 pathway in A549 cells
LPS stimulates cGAS expression in an IFN-I-dependent
manner in immune cells (13), and
LPS binds to TLR4 and further activates the MyD88-independent
pathway to trigger IFN-I production in immune cells (11). TAK242 is a selective TLR4 inhibitor
that interferes with the interaction between TLR4 and its adaptor
molecules (23). To elucidate the
mechanisms by which LPS upregulates cGAS in A549 cells, the cells
were pretreated with TAK242 (10 µM) for 1 h, followed by LPS
treatment for 4 h, and subsequently the cGAS protein and mRNA
levels were determined. The results showed that TLR4 inhibitor
treatment blocked the effect of LPS on cGAS expression at the
protein and mRNA levels (Fig. 2A and
B), indicating that TLR4 is essential for LPS-stimulated cGAS
expression.
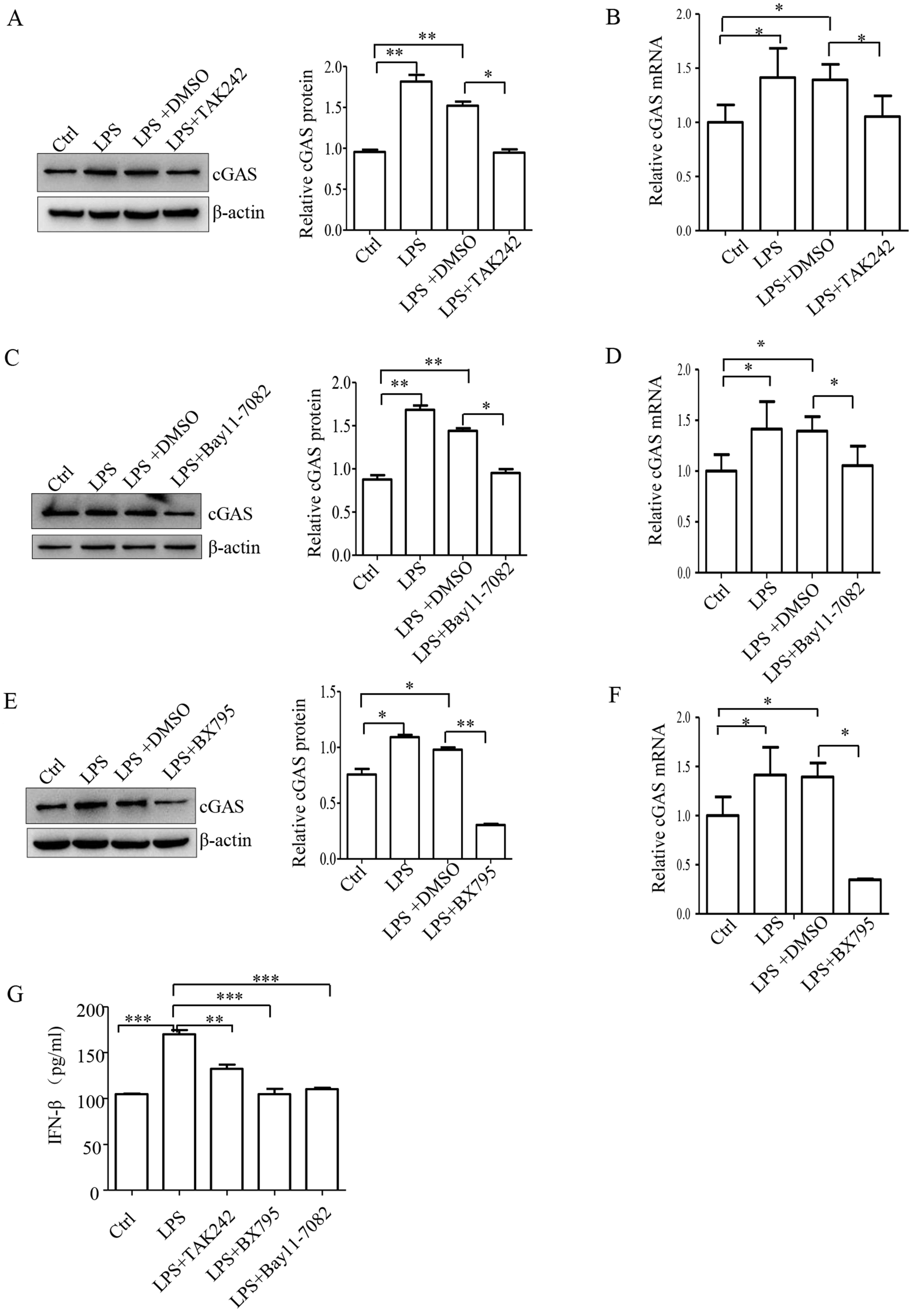 | Figure 2.LPS regulates cGAS expression through
the MyD88-independent TLR4 signaling pathway in A549 cells. The
cells were treated with either (A and B) 10 µM TAK242 TLR4
inhibitor, (C and D) 20 µM Bay11-7082 NF-κB inhibitor or (E and F)
10 µM BX-795 TBK1 inhibitor for 1 h, followed by 400 ng/ml LPS
treatment for 4 h. (A, C and E) cGAS protein levels were determined
using western blot analysis. β-actin was used as a loading control.
(B, D and F) cGAS mRNA levels were determined using reverse
transcriptase-quantitative polymerase chain reaction. (G) TAK242,
Bay11-7082 and BX-795 treatment inhibit IFN-β expression induced by
LPS. The cells were treated with TAK242 (10 µM), BX795 (10 µM),
Bay11-7082 (20 µM) for 1 h, respectively, followed with LPS
treatment for 4 h. IFN-β expression was measured using ELISA.
*P<0.05, **P<0.01 and ***P<0.001 with comparisons
indicated by lines. cGAS, cyclic guanosine monophosphate-adenosine
monophosphate synthase; Ctrl, control; LPS, lipopolysaccharide;
IFN-β, interferon-β. |
BAY 11–7082 is an irreversible inhibitor of IκB,
which inhibits cytokine-induced activation of NF-κB (24) and BX795 is a TBK1 inhibitor that can
selectively block IRF3, but not NF-κB signaling (25). To further reveal the roles of NF-κB
and TBK1, which are two key molecules downstream of TLR4 in the
MyD88-independent pathway, on cGAS expression upon LPS treatment,
A549 cells were pretreated with either Bay11-7082 or BX795 for 1 h,
followed by LPS treatment for 4 h, following which the cGAS protein
and mRNA levels were determined. The results showed that treatment
with both the NF-κB inhibitor and TBK1 inhibitor significantly
reduced LPS-stimulated cGAS expression (Fig. 2C-F), indicating that LPS stimulates
cGAS expression through both NF-κB and TBK1 signaling. The efficacy
of these inhibitors was validated by exposing cells to these
inhibitors for the indicated times, and then assessing the total
IFN-β expression levels using ELISA. The results demonstrated that
all three inhibitors significantly blocked IFN-β expression induced
by LPS (Fig. 2G), indicating they
functioned properly. Taken together, these data suggest that LPS
induces cGAS expression through the MyD88-independent TLR4
signaling pathway in A549 cells.
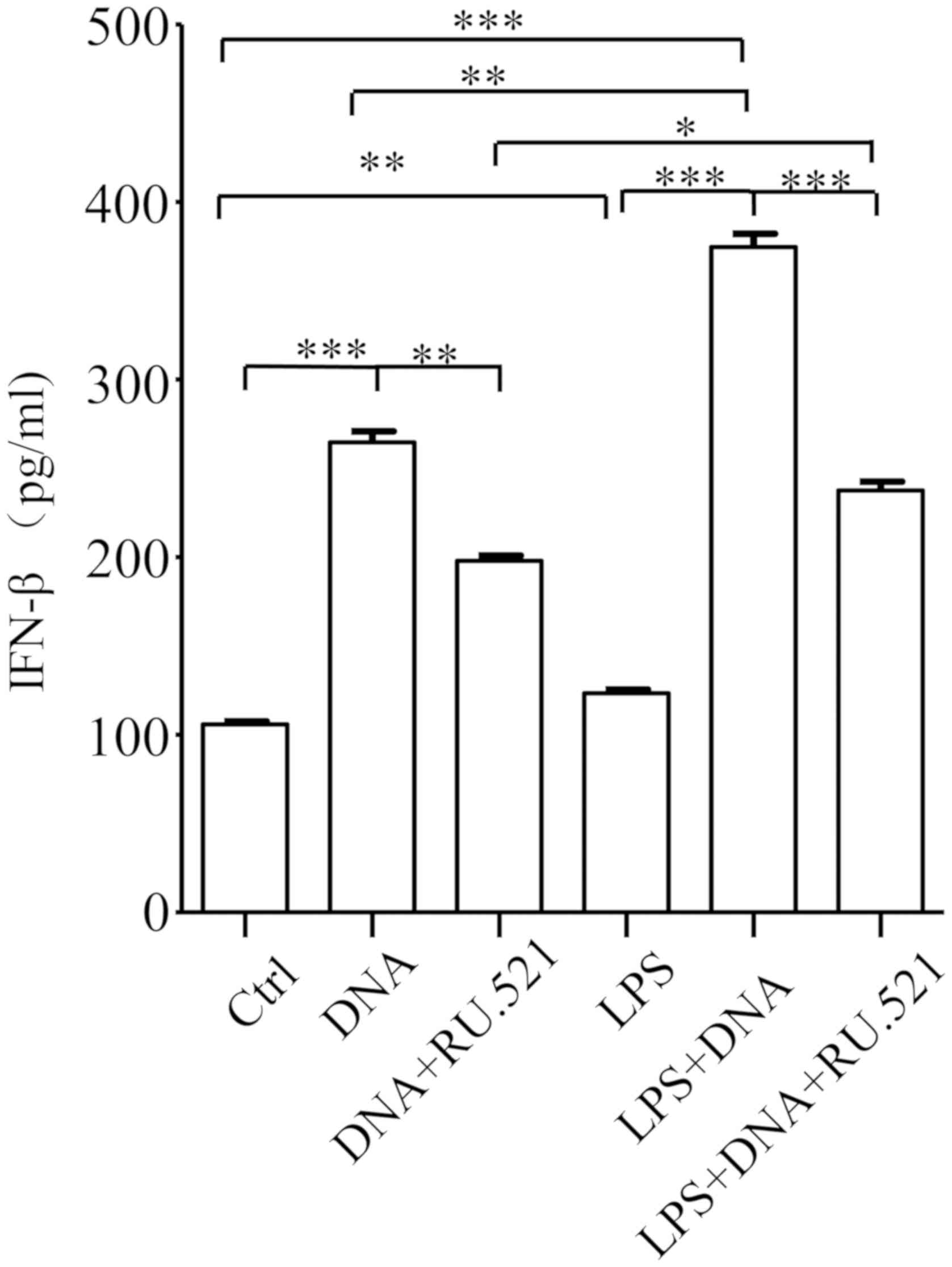
LPS enhances DNA-induced IFN-β
expression in A549 cells
cGAS can sense cytosolic DNA to promote IFN-I
expression in bone marrow-derived macrophage (BMM) cells (13). To determine whether the same function
occurs in A549 cells, cells were transfected with E. coli
DNA, and IFN-β levels in the cell culture media and in cell lysates
were measured using ELISA 24 h post-transfection. The results are
shown in Fig. 3, which indicates
that IFN-β expression in cells transfected with DNA were
significantly increased (over 1.5-fold) compared with that in the
control cells. As LPS could upregulate cGAS expression in A549
cells, the authors hypothesized that LPS might enhance DNA-induced
IFN-β production. To test this hypothesis, A549 cells were
pretreated with LPS for 4 h. Then, LPS was removed, and cells were
transfected with DNA. The total IFN-β levels were measured using
ELISA 24 h post-transfection. The results showed that LPS
pretreatment caused >2.5-fold increase in DNA-induced IFN-β in
A549 cells compared with that in the untreated control cells,
whereas LPS treatment alone caused an ~20% increase in IFN-β
compared with untreated control cells (Fig. 3). These findings indicate that LPS
pretreatment enhances DNA-induced IFN-β production. To evaluate the
role of cGAS in DNA-induced IFN- β production and to assess whether
LPS enhances DNA-induced IFN-β expression through cGAS, the cells
were pretreated with or without either LPS, followed by DNA
transfection and treatment with cGAS inhibitor RU.521 (2 µM)
(17). A total of 24 h after
transfection, IFN-β production was measured using ELISA. The
results demonstrated that RU.521 treatment significantly blocked
the IFN-β increase caused by DNA addition compared with the LPS+DNA
group and LPS alone group, and completely inhibited the enhancing
effect of LPS on DNA-stimulated IFN-β production compared with DNA
alone group (Fig. 3), indicating
that LPS enhances DNA-induced IFN-β production via cGAS.
LPS enhances cGAS-mediated autophagy
in A549 cells
It is well-known that LC3B conversion (LC3-I to
LC3-II) and lysosomal degradation of LC3-II reflect the progression
of autophagy, and that the p62 protein is degraded by autophagy
(26). Therefore, LC3B and p62 can
both serve as markers of autophagy (26). As a previous study reported that cGAS
mediates DNA-induced autophagy in macrophages (8), whether DNA could induce autophagy in
A549 cells was investigated in the present study by transfecting
with E. coli DNA (2 µg/ml). LC3B and p62 levels were
measured using western blot analysis, 24 h after transfection. The
results showed that DNA treatment led to increased LC3-II
expression, along with a decreased p62 expression (Fig. 4A), implying that DNA can induce
autophagy in A549 cells. To determine the effect of LPS treatment
on DNA-induced autophagy, A549 cells were pretreated with LPS for 4
h and then transfected with E. coli DNA (2 µg/ml). LC3B and
p62 levels were analyzed using a western blot assay, 24 h following
transfection. The results demonstrated that LPS pretreatment
significantly promoted LC3-II and p62 degradation caused by the
addition of the DNA comparing LPS+DNA group with DNA only group
(Fig. 4A), suggesting that LPS
enhances the effects of DNA on autophagy in A549 cells. To evaluate
the role of cGAS in DNA-induced autophagy, cells were treated with
RU.521 at 0.5, 1.0 and 2.0 µM together with E. coli DNA
stimulation, and autophagic marker levels were measured using a
western blot analysis. The results showed that RU.521 significantly
inhibited LC3-I to LC3-II conversion and p62 degradation (Fig. 4B), indicating that DNA-induced
autophagy in A549 cells is mediated by cGAS. To evaluate whether
the effect of LPS on DNA-induced autophagy is mediated by cGAS,
cells were pretreated with LPS, followed by treatment with RU.521
(2.0 µM) and E. coli DNA. The autophagy marker levels were
examined using western blot analysis. The results showed that
RU.521 blocked the effects of LPS on DNA-induced autophagy
(Fig. 4C), suggesting that LPS
promotes DNA-induced autophagy via cGAS. Although there is
difference between LC3-II proteins of DNA+LPS group, the results in
Fig. 4A and C both show increased
autophagy in DNA+LPS group. Lower LC3-II protein in Fig. 4A was due to an intense autophagic
flux that consumes this protein, which is consistent with lower p62
in Fig. 4A.
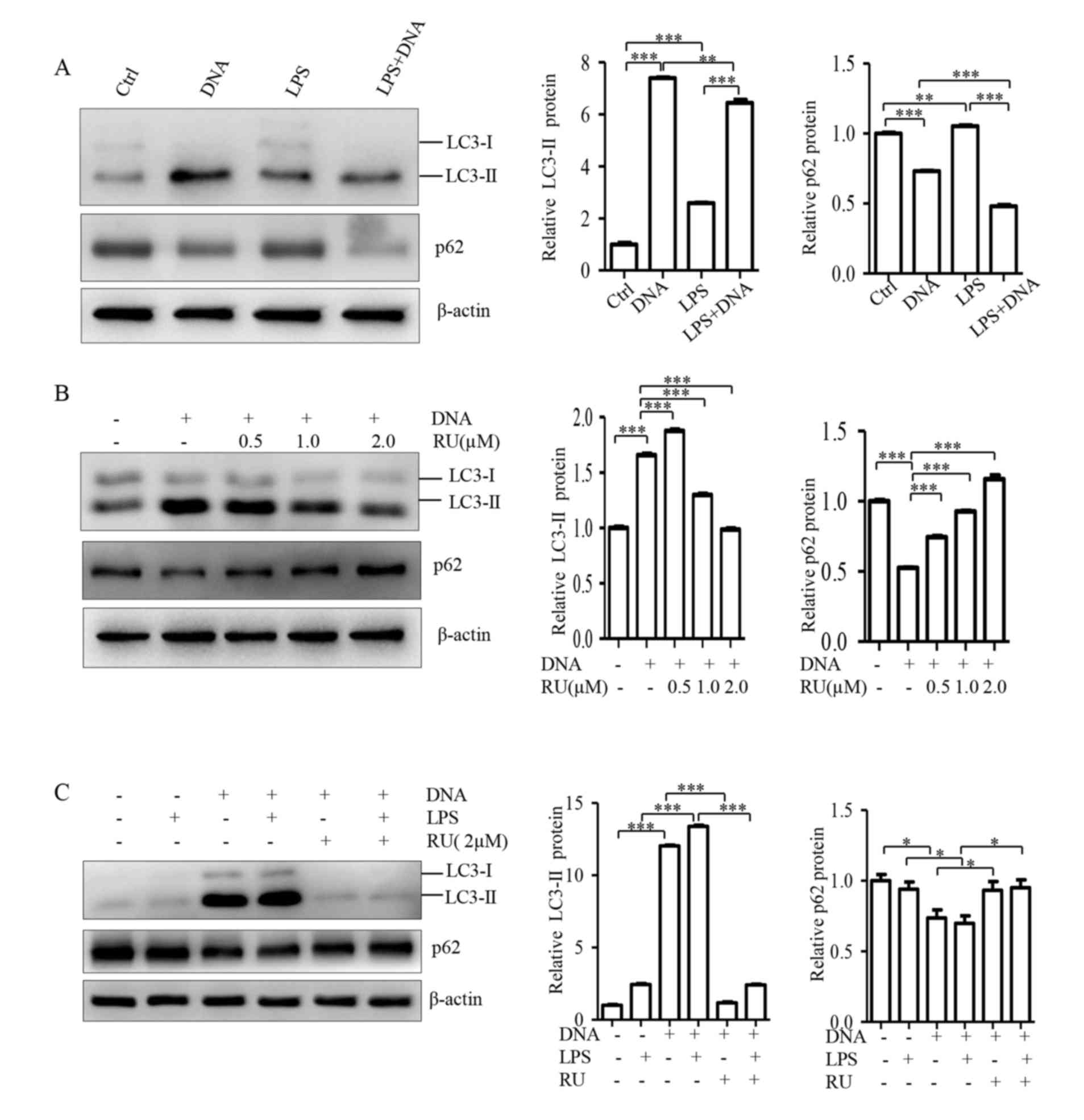 | Figure 4.LPS enhances cGAS-mediated autophagy
in A549 cells. Cells were either (A) pretreated with or without 400
ng/ml LPS for 4 h, followed by 2 µg/ml DNA transfection, (B)
treated with the cGAS inhibitor RU at the indicated concentration
and subsequently transfected with 2 µg/ml DNA or (C) pretreated
with or without 400 ng/ml LPS for 4 h, followed by 2 µM RU
treatment, and transfected with 2 µg/ml DNA. Following this cells
were harvested 24-h post-transfection, and the protein expression
of LC3-II and p62 was analyzed using western blot analysis.
*P<0.05, **P<0.01 and ***P<0.001 with comparisons
indicated by lines. RU, RU.521; ctrl, control; LPS,
lipopolysaccharide; LC3, microtubule-associated proteins 1A/1B
light chain 3B. |
Discussion
Lung epithelial cells play a key role in immune
responses during the pathogenesis of many chronic pulmonary
diseases, but the underlying mechanisms of their involvement remain
unclear (27). A useful model of
type II alveolar epithelial cells are A549 cells, although it is a
cancerous cell line derived from an alveolar cell carcinoma
(18,19). In the present study, cGAS mediates
A549 cellular responses against cytosolic DNA, providing evidence
that cGAS are active in A549 cells. Although DNA is absent in the
cytoplasm of lung epithelial cells under normal conditions, it may
be released into the cytoplasm to cause immune responses under
certain abnormal conditions, such as infection (28). When a patient is infected with
bacteria or DNA viruses, the pathogen's genomic DNA may enter the
cytoplasm of lung epithelial cells through endocytosis (29,30).
Under these circumstances, cGAS can be activated to stimulate the
expression of IFN-I, which in turn activates host immune responses
through IFN signaling pathway (31).
Therefore, the findings from the present study suggest potential
mechanisms by which lung epithelial cells react against invading
bacteria and DNA viruses. As viral and bacterial infection is a
common cause of the exacerbation of COPD (16), these findings may also provide clues
to understanding the pathophysiology of COPD.
Enhanced levels of autophagy have been observed in
COPD-affected lung tissues (15),
suggesting that autophagy may play an important role in COPD.
However, how autophagy is regulated in COPD remains unclear. Since
cGAS is required for DNA-induced autophagy in BMMs (8), the effects of DNA on autophagy
stimulation in lung epithelial cells was evaluated. The results
show that DNA transfection can cause autophagy in lung epithelial
cells and that cGAS plays a vital role during this process,
suggesting a potential mechanism underlying how autophagy is
regulated in COPD-affected lung epithelia.
Given that cGAS plays such an important role in lung
epithelial cells, how cGAS expression is modulated in these cells
was investigated. A previous study reported that in BMMs LPS could
stimulate cGAS expression through the MyD88-independent TLR4
signaling pathway (13). Therefore,
whether cGAS expression is regulated by the same mechanism in lung
epithelial cells was analyzed. In A549 cells, LPS could stimulate
cGAS expression in a dose-dependent manner, from 100 to 400 ng/ml.
Interestingly, 400 ng/ml LPS treatment had a stronger effect on
cGAS expression compared with 800 ng/ml LPS, indicating there might
be a negative control mechanism to regulate cGAS expression, which
might be triggered by high concentrations of LPS, such as 800
ng/ml. Further experiments are required to address this question in
the future. Treatment with the TLR4 inhibitor TAK242, the NF-κB
inhibitor Bay11-7082 or the TBK1 inhibitor BX795 could completely
block the effects of LPS treatment on cGAS expression, indicating
that TLR4, NF-κB and TBK1, which are all key components of the
MyD88-independent TLR4 signaling pathway, are required for
LPS-induced cGAS expression. Therefore, LPS regulates cGAS
expression through the MyD88-independent TLR4 signaling pathway in
A549 cells.
In summary, the present study demonstrates that LPS
can enhance DNA-induced IFN-β production and autophagy by
upregulating cGAS expression through the MyD88-independent TLR4
signaling pathway in A549 cells. This suggests that there is
crosstalk between the TLR4 signaling pathway and the cGAS signaling
pathway in lung epithelial cells.
Acknowledgements
Not applicable.
Funding
The present study was supported in part by the
National Natural Science Foundation of China (grant nos. 81560453
and 81660008), the Natural Science Foundation of Guangxi (grant
nos. 2015GXNSFAA139178 and 2015GXNSFBA139168), the Guangxi Health
and Family Planning Commission (grant no. S2015-34), and the
Lijiang Scholar Award. GH was supported by the Hundred Talents
Program of Guangxi.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
RW, WW, AL performed experiments. RW, YW and GH
analyzed and interpreted the data. RW, GH, JJ and ZH designed the
experiments and drafted the manuscript. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bagdonas E, Raudoniute J, Bruzauskaite I
and Aldonyte R: Novel aspects of pathogenesis and regeneration
mechanisms in COPD. Int J Chron Obstruct Pulmon Dis. 10:995–1013.
2015.PubMed/NCBI
|
|
2
|
Leiva-Juárez MM, Kolls JK and Evans SE:
Lung epithelial cells: Therapeutically inducible effectors of
antimicrobial defense. Mucosal Immunol. 11:21–34. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Dudek M, Puttur F, Arnold-Schrauf C, Kühl
AA, Holzmann B, Henriques-Normark B, Berod L and Sparwasser T: Lung
epithelium and myeloid cells cooperate to clear acute pneumococcal
infection. Mucosal Immunol. 9:1288–1302. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Brazee P, Dada LA and Sznajder JI: Role of
linear ubiquitination in health and disease. Am J Respir Cell Mol
Biol. 54:761–768. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sun L, Wu J, Du F, Chen X and Chen ZJ:
Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates
the type I interferon pathway. Science. 339:786–791. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Li T and Chen ZJ: The cGAS-cGAMP-STING
pathway connects DNA damage to inflammation, senescence, and
cancer. J Exp Med. 215:1287–1299. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cai X, Chiu YH and Chen ZJ: The
cGAS-cGAMP-STING pathway of cytosolic DNA sensing and signaling.
Mol Cell. 54:289–296. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Liang Q, Seo GJ, Choi YJ, Kwak MJ, Ge J,
Rodgers MA, Shi M, Leslie BJ, Hopfner KP, Ha T, et al: Crosstalk
between the cGAS DNA sensor and beclin-1 autophagy protein shapes
innate antimicrobial immune responses. Cell Host Microbe.
15:228–238. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sender V and Stamme C: Lung cell-specific
modulation of LPS-induced TLR4 receptor and adaptor localization.
Commun Integr Biol. 7:e290532014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Qiao S, Luo Q, Zhao Y, Zhang XC and Huang
Y: Structural basis for lipopolysaccharide insertion in the
bacterial outer membrane. Nature. 511:108–111. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lu YC, Yeh WC and Ohashi PS: LPS/TLR4
signal transduction pathway. Cytokine. 42:145–151. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Xu Y, Jagannath C, Liu XD, Sharafkhaneh A,
Kolodziejska KE and Eissa NT: Toll-like receptor 4 is a sensor for
autophagy associated with innate immunity. Immunity. 27:135–144.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ma F, Li B, Liu SY, Iyerz SS, Yu Y, Wu A
and Cheng G: Positive feedback regulation of type I IFN production
by the IFN-inducible DNA sensor cGAS. J Immunol. 194:1545–1554.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Levine B and Kroemer G: Autophagy in the
pathogenesis of disease. Cell. 132:27–42. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chen ZH, Kim HP, Sciurba FC, Lee SJ,
Feghali-Bostwick C, Stolz DB, Dhir R, Landreneau RJ, Schuchert MJ,
Yousem SA, et al: Egr-1 regulates autophagy in cigarette
smoke-induced chronic obstructive pulmonary disease. PLoS One.
3:e33162008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tuder RM and Petrache I: Pathogenesis of
chronic obstructive pulmonary disease. J Clin Invest.
122:2749–2755. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Vincent J, Adura C, Gao P, Luz A, Lama L,
Asano Y, Okamoto R, Imaeda T, Aida J, Rothamel K, et al: Small
molecule inhibition of cGAS reduces interferon expression in
primary macrophages from autoimmune mice. Nat Commun. 8:7502017.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lieber M, Smith B, Szakal A, Nelson-Rees W
and Todaro G: A continuous tumor-cell line from a human lung
carcinoma with properties of type II alveolar epithelial cells. Int
J Cancer. 17:62–70. 1976. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Foster KA, Oster CG, Mayer MM, Avery ML
and Audus KL: Characterization of the A549 cell line as a type II
pulmonary epithelial cell model for drug metabolism. Exp Cell Res.
243:359–366. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang Q, Li A, Jin J and Huang G: Targeted
interfering DEP domain containing 1 protein induces apoptosis in
A549 lung adenocarcinoma cells through the NF-κB signaling pathway.
Onco Targets Ther. 10:4443–4454. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li A, Wang Q, He G, Jin J and Huang G: DEP
domain containing 1 suppresses apoptosis via inhibition of A20
expression, which activates the nuclear factor κB signaling pathway
in HepG2 cells. Oncol Let. 16:949–955. 2018.
|
|
22
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Matsunaga N, Tsuchimori N, Matsumoto T and
Ii M: TAK-242 (resatorvid), a small-molecule inhibitor of toll-like
receptor (TLR) 4 signaling, binds selectively to TLR4 and
interferes with interactions between TLR4 and its adaptor
molecules. Mol Pharmacol. 79:34–41. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Pierce JW, Schoenleber R, Jesmok G, Best
J, Moore SA, Collins T and Gerritsen ME: Novel inhibitors of
cytokine-induced IkappaBalpha hosphorylation and endothelial cell
adhesion molecule expression show anti-inflammatory effects in
vivo. J Biol Chem. 272:21096–21103. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Clark K, Plater L, Peggie M and Cohen P:
Use of the pharmacological inhibitor BX795 to study the regulation
and physiological roles of TBK1 and IkappaB Kinase epsilon: A
distinct upstream kinase mediates Ser-172 phosphorylationand
activation. J Biol Chem. 284:14136–14146. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Klionsky DJ, Abdelmohsen K, Abe A, Abedin
MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD,
Adeli K, et al: Guidelines for the use and interpretation of assays
for monitoring autophagy (3rd edition). Autophagy. 12:1–222. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Biswal S, Thimmulappa RK and Harvey CJ:
Experimental therapeutics of Nrf2 as a target for prevention of
bacterial exacerbations in COPD. Proc Am Thorac Soc. 9:47–51. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kawasaki T and Kawai T: Discrimination
between self and non-self-nucleic acids by the innate immune
system. Int Rev Cell Mol Biol. 344:1–30. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lu C, Zhang X, Ma C, Xu W, Gan L, Cui J,
Yin Y and Wang H: Nontypeable haemophilus influenzae DNA stimulates
type I interferon expression via STING signaling pathway. Biochim
Biophys Acta Mol Cell Res. 1865:665–673. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lam E and Falck-Pedersen E: Unabated
adenovirus replication following activation of the
cGAS/STING-dependent antiviral response in human cells. J Virol.
88:14426–14439. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ivashkiv LB and Donlin LT: Regulation of
type I interferon responses. Nat Rev Immunol. 14:36–49. 2014.
View Article : Google Scholar : PubMed/NCBI
|