Introduction
Subarachnoid hemorrhage (SAH) is associated with a
30-day mortality rate of ~50% and is one of the most
life-threatening cerebrovascular diseases (1). Deaths are mainly due to the initial
hemorrhage and no effective treatment is currently available for
brain injury (2). In recent years,
neuronal death has been implicated in brain injury following
experimental SAH and in a clinical setting, and is considered to
play a crucial role in SAH-related brain injury (1,2).
Brain-derived neurotrophic factor (BDNF) plays a key
role in neuronal survival, synaptogenesis, neuronal differentiation
and synaptic plasticity in the central nervous system (3–5).
Clinical studies demonstrated that the BDNF Val66Met polymorphism
is associated with poor recovery, but not associated with learning
and memory performance following SAH in patients (6,7). Animal
studies demonstrated that the BDNF/tropomyosin-related kinase
receptor B (TrkB) signaling pathway is involved in neuronal
apoptosis following SAH (8,9). Expression of mature BDNF was found to
be decreased at 4, 48 and 72 h, and increased at 5 and 7 days
post-SAH in a blood injection SAH model (10–12).
Furthermore, recombinant human BDNF inhibited hemolysate-induced
neuron death in an in vitro model of SAH (13). However, the role of BDNF in
SAH-induced neuronal apoptosis and neurological deficits is
unclear.
The aim of the present study was to investigate
whether intracerebroventricular (ICV) injection of BDNF can protect
against neuronal apoptosis and neurological deficits following SAH
in a rat model.
Materials and methods
Experimental animals and design
A total of 117 male Sprague-Dawley rats (age,
12-weeks old; weight, 290–330 g) were obtained from the Animal
Center of Xiangyang No. 1 People's Hospital (Xiangyang, China) and
used according to the Guide of Care and Use of Laboratory Animals
of the National Institutes of Health. All the animal experimental
protocols were approved by the Animal Use and Care Committee of
Hubei University of Medicine. The rats were group-housed with
access to food and water ad libitum in a cage under controlled
temperature (25±2°C) and humidity (55±10%) conditions, with a 12
h-light/dark cycle. The experiment consisted of two parts: i) In
order to investigate the time course of BDNF expression following
SAH, 48 rats were subdivided into the sham and SAH groups at 6, 12,
24, 48 and 72 h. Rats were decollated under deep anesthesia with
pentobarbital sodium (40 mg/kg, i.p.) at each time point mentioned
above. Then the basal cortex was collected for ELISA and western
blot analysis (n=6); ii) in order to investigate the role of
exogenous BDNF in neuronal apoptosis after SAH, 69 rats were
subdivided into sham, SAH + vehicle, and SAH + BDNF groups. The
rats received a single ICV injection of BDNF at 30 min after SAH.
Mortality was calculated at 72 h. Following neurological assessment
at 72 h, rats were sacrificed under deep anesthesia with
pentobarbital sodium (40 mg/kg, i.p.), 6 rats from each group were
used for western blot analysis and 6 rats from each group were used
for immunofluorescence staining. A total of 6 rats in each group
were used for adhesive removal task and Frey test at day 14.
SAH model and grading system
The rat SAH model was constructed by the
endovascular perforation method, as described previously (14,15).
Briefly, a 4-cm skin incision in the ventral neck was performed
following anesthesia with pentobarbital sodium (40 mg/kg, i.p.) and
the right common, external and internal carotid arteries were
exposed. Subsequently, the external carotid artery was ligated and
transected, and a blunted 4-0 monofilament nylon suture was
inserted into the internal carotid artery for 18 mm and advanced a
further 3 mm to perforate the wall of the bifurcation with the
middle cerebral artery. The suture was removed after 10 sec,
allowing reperfusion of the internal carotid artery. The
sham-operated rats underwent an identical procedure, apart from the
perforation. After completing the procedure, rats were arranged in
a recovery cage for 30–60 min. After the rats started to eat some
semi-fluid food and move around, they were housed in clean and new
cages.
SAH grade score was obtained at 72 h using an
18-point SAH grading system (14).
Briefly, rats were decollated under anesthesia with pentobarbital
sodium (40 mg/kg, i.p.) and the brains were collected quickly. The
SAH grade score was calculated according to the blood amount of six
segments in the basal cistern. Each segment was scored (0–3) as
follows: 0, no subarachnoid blood; 1, minimal subarachnoid blood;
2, moderate blood clot with visible arteries; and 3, blood clot
covering all arteries. Total score was calculated by adding the
scores from the six segments.
Drug administration
ICV injection was performed on experimental SAH rats
as previously reported (16,17). Briefly, the rats were fixed to the
stereotactic frame after anesthesia with pentobarbital sodium (40
mg/kg, i.p.). Subsequently, a 30-gauge needle of a Hamilton syringe
was inserted into the left lateral ventricle via a burr hole (−1.5
mm posterior, −1.0 mm lateral and −3.5 mm from bregma). The rats
received ICV injection of recombinant rat BDNF protein (10 µl, 0.5
nmol; cat. no. Ab9794; Abcam) or vehicle (10 µl PBS) at 30 min
after SAH. After injection, the needle was kept for 5 min and then
retracted slowly. The burr hole was quickly sealed with bone wax.
Dosage drug administration was based upon prior research using an
intracerebral hemorrhage rat model (18).
Western blot analysis
Western blotting was performed as described
previously (16,17). After sample preparation, 50 µg
protein of each sample was loaded onto an 12% SDS-PAGE gel. After
electrophoresis and transfer of proteins to a nitrocellulose
membrane, the membranes were blocked at room temperature and
incubated for 15 h at 4°C with the following primary antibodies:
Anti-BDNF (1:1,000; cat. no. ab108319), anti-Bcl-2 (1:1,000; cat.
no. ab59348), anti-Bax (1:1,000; cat. no. ab32503) and
anti-activated caspase-3 (1:1,000; cat. no. ab2302), all from
Abcam; anti-activated caspase-9 (1:1,000; cat. no. 9507) and
anti-β-actin (1:1,000; cat. no. 4970), both from Cell Signaling
Technology, Inc. Anti-rabbit or mouse IgG HRP-linked antibodies
(all, 1:4,000; cat. no. 7074 and cat. no. 7076, respectively; Cell
Signaling Technology, Inc.) were selected to incubate with the
membrane at room temperature for 2 h. The blots were visualized
with ECL reagent (cat. no. 32106; Thermo Fisher Scientific, Inc.).
The protein bands were selected to perform densitometry
quantification with ImageJ software (v1.8; National Institutes of
Health).
ELISA
To detect the level of BDNF, ELISA was performed as
described previously (12,19). The concentration of BDNF was measured
with a rat BDNF ELISA kit (cat. no. ERBDNF; Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol and was
quantified as µg/g tissue.
Modified Garcia score, rotarod test and beam balance
test. The neurological deficits were assessed by an observer
blinded to the experiment at 72 h after SAH using the modified
Garcia score, beam balance test and rotarod test, as previously
described (14,15,20,21). The
modified Garcia scoring system consisted of six tests that were
scored as 0, 1, 2 or 3 (no activity, spontaneous activity,
spontaneous movements of all limbs and forelimb outstretching,
respectively), and as 1, 2 or 3 (climbing wall of wire cage, body
proprioception, and response to vibrissae touch, respectively). For
the rotarod test, the rats were trained to stay on an accelerating
rotarod (4–40 rpm until 5 min, increasing by 4 rpm per 30 sec
interval); three trials each day were performed for 1 week prior to
SAH. The observer recorded the mean fall latency of each rat. For
the beam balance test, the observer placed the rat on a 60-cm
square narrow wooden beam (1 cm wide and 50 cm above the floor),
and recorded the duration of the rat remaining on the center of the
beam, until 60 sec elapsed.
Immunohistochemical and Tunel
staining
Activated caspase-3/NeuN immunostaining and Tunel
staining were performed as described previously (15,22).
Briefly, frozen brain sections were fixed in 4% paraformaldehyde
for 20 min at 4°C, washed in PBS and then blocked in 5% normal goat
serum for 30 min at room temperature, and incubated for 15 h at 4°C
with the primary antibodies: Anti-activated caspase-3 (1:200; cat.
no. ab2302; Abcam) and anti-NeuN (1:200; cat. no. ab177487; Abcam).
The sections were washed in PBS and incubated for 2 h at room
temperature with secondary antibodies: Fluorescein
isothiocyanate-conjugated anti-rabbit IgG (1:400; cat. no. F9887;
Sigma-Aldrich; Merck KGaA) and tetramethylrhodamine -conjugated
anti-mouse IgG (1:400; cat. no. T5393; Sigma-Aldrich; Merck KGaA),
then washed with PBS and a coverslip was added. For Tunel staining,
the In situ Cell Death Detection kit with Fluorescein was used
according to the manufacturer's protocol (Roche Diagnostics).
Images were captured using a fluorescence microscope.
Adhesive removal task and Frey test. The long-term
sensorimotor behavior deficit was assessed by an observer blinded
to the experiment at day 14 after SAH using the adhesive removal
task and Frey test, as described previously (23). Briefly, for the adhesive removal
task, the observer placed stickers on the right and left forepaws,
then recorded the mean time of the three stickers' complete
removal. The sticker placement on the forepaw was alternated. For
the Frey test, the observer placed the rat on a wire grid bottom
using the von Frey hairs (bending force 1–15 g). The Frey hair
force was increased or decreased based on the rat response. The
observer recorded mean time of the clear paw withdrawal, licking or
shaking with increase in mechanical sensitivity and calculated the
50% paw withdrawal threshold.
Statistical analysis
All the data were presented as the mean ± standard
deviation obtained from 6 experimental repeats. The significance of
the differences among multiple groups was analyzed by one-way
analysis of variance followed by Dunn's post-hoc test in SPSS 16.0
statistical software (SPSS, Inc.). P<0.05 was considered to
indicate a statistically significant difference.
Results
SAH significantly decreases BDNF levels. The
mortality rates were 0% (0 of 6 rats), 14.3% (1 of 7 rats), 25.0%
(2 of 8 rats), 25.0% (2 of 8 rats), 33.3% (3 of 9 rats) and 40.0%
(4 of 10 rats) in the sham and SAH groups at 6, 12, 24, 48 and 72
h, respectively (Fig. 1A). The SAH
grading scores were 11.8±0.7, 12.3±0.8, 11.9±1.3, 11.8±1.1 and
12.1±1.1 in SAH groups at 6, 12, 24, 48 and 72 h, respectively,
there was no significant difference among the five groups (Fig. 1B). To determine the time course of
BDNF expression following SAH, rat basal cortices were dissected at
6, 12, 24, 48 and 72 h after SAH, and were assayed by western blot
analysis and ELISA. As shown in Fig. 1C
and D, SAH significantly decreased the BDNF level in the basal
cortex compared with the sham group (P<0.05).
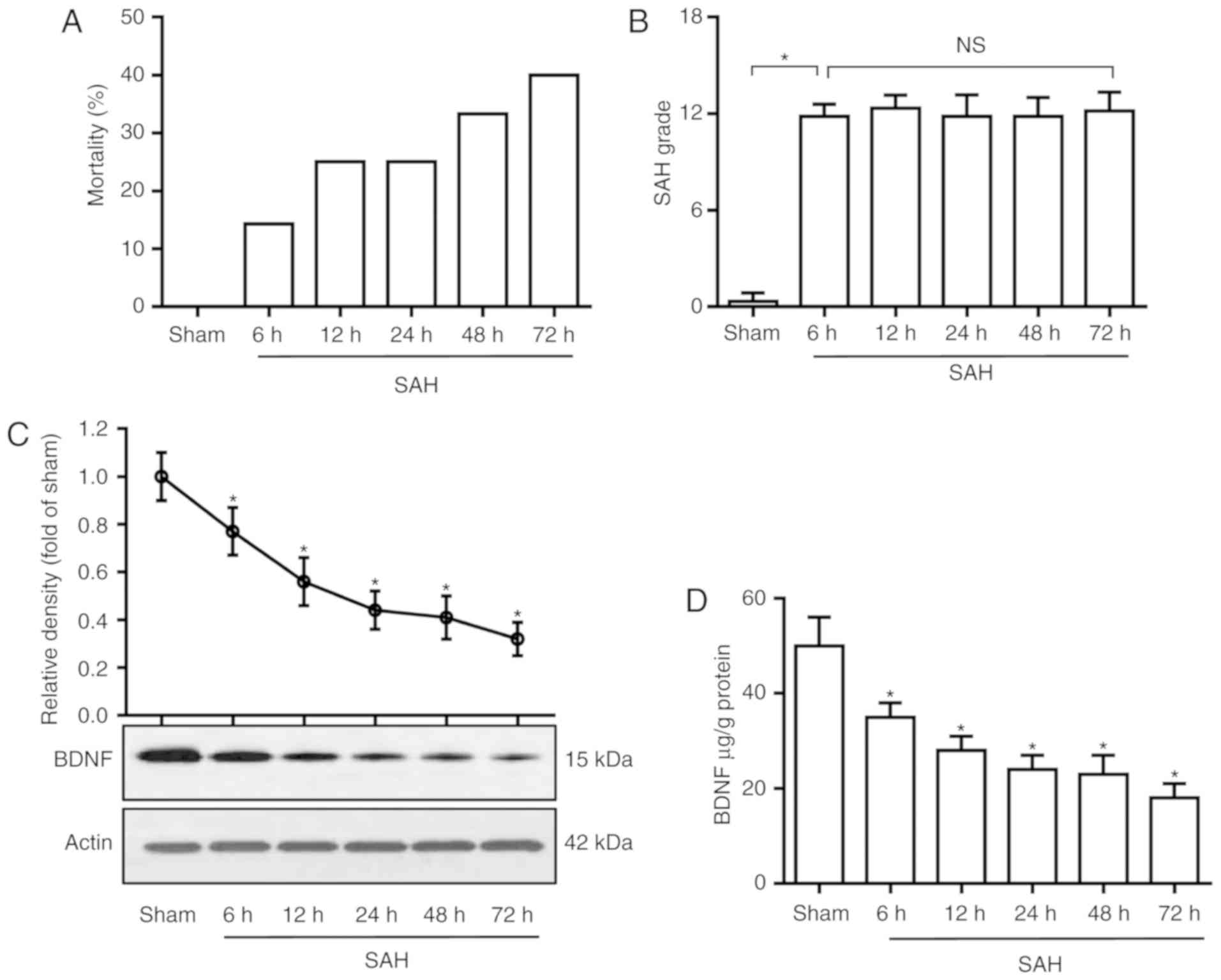 | Figure 1.BDNF level decreases after SAH. (A)
Mortality rate, (B) SAH grade scores, (C) western blot analysis of
BDNF protein in basal cortex and (D) ELISA analysis of BDNF
concentration in the basal cortex, in the sham and SAH groups at 6,
12, 24, 48 and 72 h. (B) P=0.88; (C and D) P=0.01. *P<0.05 vs.
sham; one-way analysis of variance comparison test, n=6 in each
group. BDNF, brain-derived neurotrophic factor; SAH, subarachnoid
hemorrhage; ns, not significant. |
Exogenous BDNF attenuates neurological
deficit at 72 h after SAH
The SAH grading scores were 12.3±1.0 and 12.2±0.8 in
the SAH + vehicle and SAH + BDNF groups, respectively. There was no
significant difference between the two groups (Fig. 2A), indicating that the variation of
SAH size did not affect the result. The mortality rates were 0% (0
of 18 rats), 33.3% (9 of 27 rats) and 25.0% (6 of 24 rats) in the
sham, SAH + vehicle and SAH + BDNF groups, respectively (Fig. 2B). The modified Garcia score and the
time on the rotarod and beam balance significantly decreased at 72
h in the SAH + vehicle group when compared with the sham group
(P<0.05), suggesting that SAH caused neurological deficits and
impaired the animals' ability to remain on the rotarod and beam
(Fig. 2C-E). However, these
neurobehavioral deficits improved with BDNF treatment compared with
the SAH + vehicle group (Fig.
2C-E).
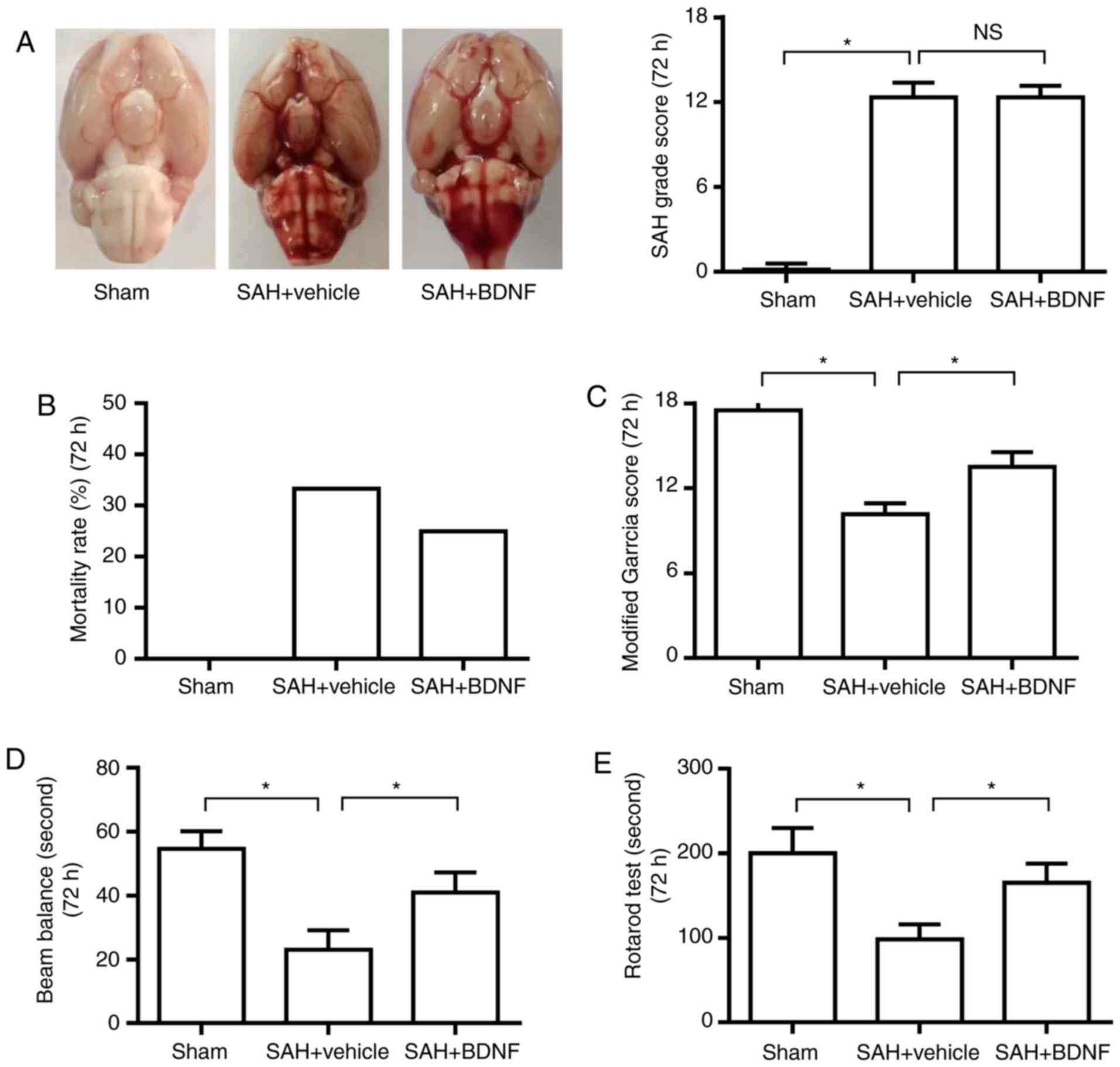 | Figure 2.Exogenous BDNF attenuates neurological
deficit at 72 h after SAH. (A) Representative brain photos of sham,
SAH + vehicle and SAH + BDNF groups (left), SAH grade scores
(right), (B) mortality rate, (C) modified Garcia score, (D) beam
balance test score and (E) rotarod test score in the sham, SAH +
vehicle and SAH + BDNF groups. (n=6, *P<0.05). (A) P=0.73 and
(C-E) P=0.01. *P<0.05; one-way analysis of variance comparison
test, n=6 in each group. BDNF, brain-derived neurotrophic factor;
SAH, subarachnoid hemorrhage. |
Exogenous BDNF attenuates neuronal
apoptosis at 72 h after SAH
Cell apoptosis was assessed at 72 h using western
blotting and immunofluorescence staining. As shown in Fig. 3, SAH caused marked downregulation of
Bcl-2 and upregulation of Bax, activated caspase-9 and activated
caspase-3 (Fig. 3A and B). However,
the expression of Bcl-2 markedly increased, whereas that of Bax,
activated caspase-9 and activated caspase-3 notably decreased in
the SAH + BDNF group compared with the SAH + vehicle group
(Fig. 3A and B). Furthermore,
statistical analysis results revealed that SAH induction
significantly increased the number of activated caspase-3- and
TUNEL-positive neurons in the SAH + vehicle group compared with the
sham group (P<0.001; Fig. 3C and
D), while BDNF treatment significantly reduced this increase
(P<0.001).
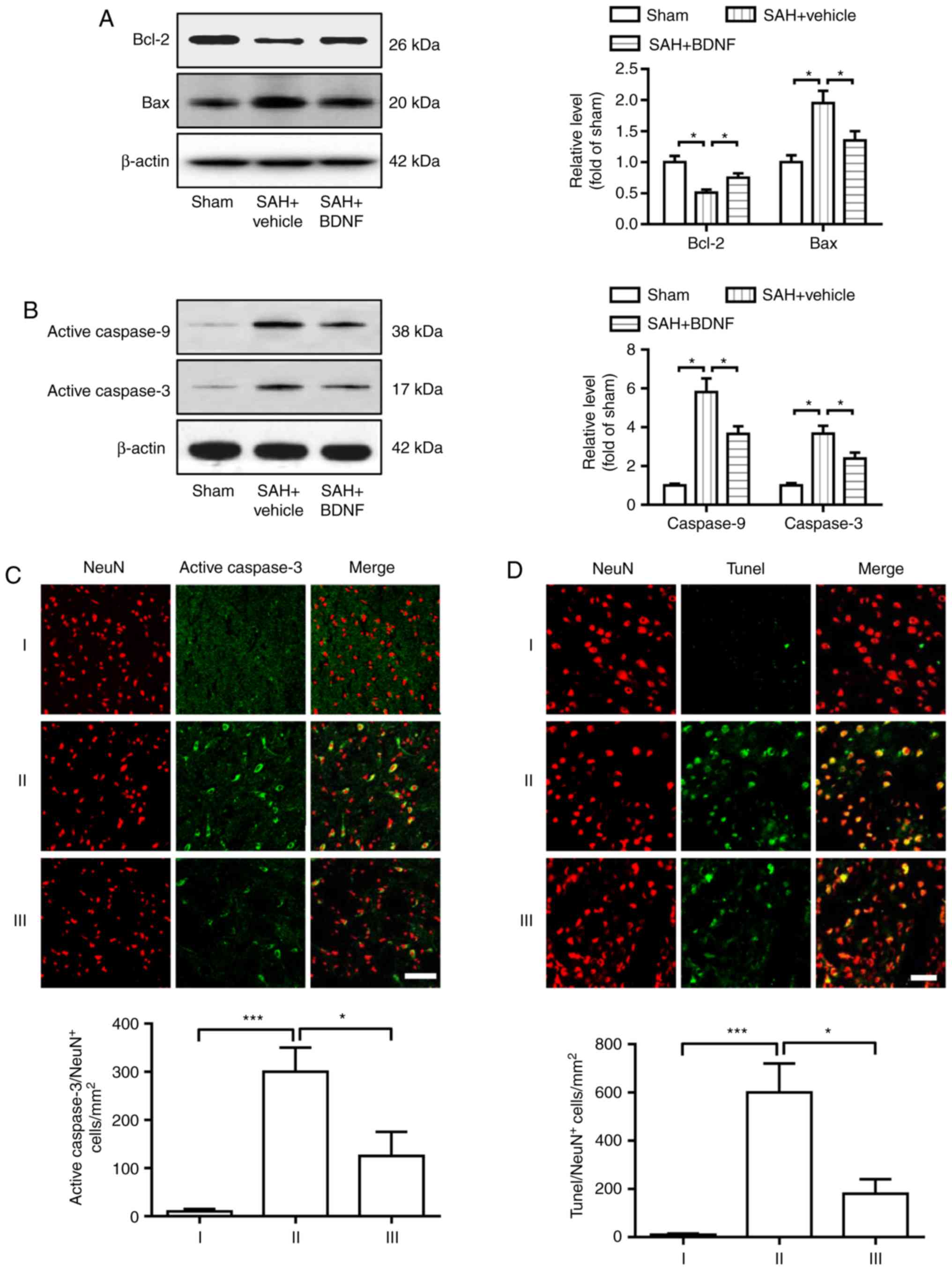 | Figure 3.Exogenous BDNF attenuates neuronal
apoptosis at 72 h after SAH. (A) Expression of Bcl-2 and Bax, (B)
activation of caspase-9 and caspase-3, (C) activated caspase-3/NeuN
staining-positive neurons and (D) Tunel/NeuN staining-positive
neurons, in the basal cortex of the sham (I), SAH + vehicle (II)
and SAH + BDNF (III) groups. (A-D) P=0.01, *P<0.05; (C-D)
P=0.0009, ***P<0.001; one-way analysis of variance comparison
test, n=6 in each group. Scale bar, 50 µm. BDNF, brain-derived
neurotrophic factor; SAH, subarachnoid hemorrhage. |
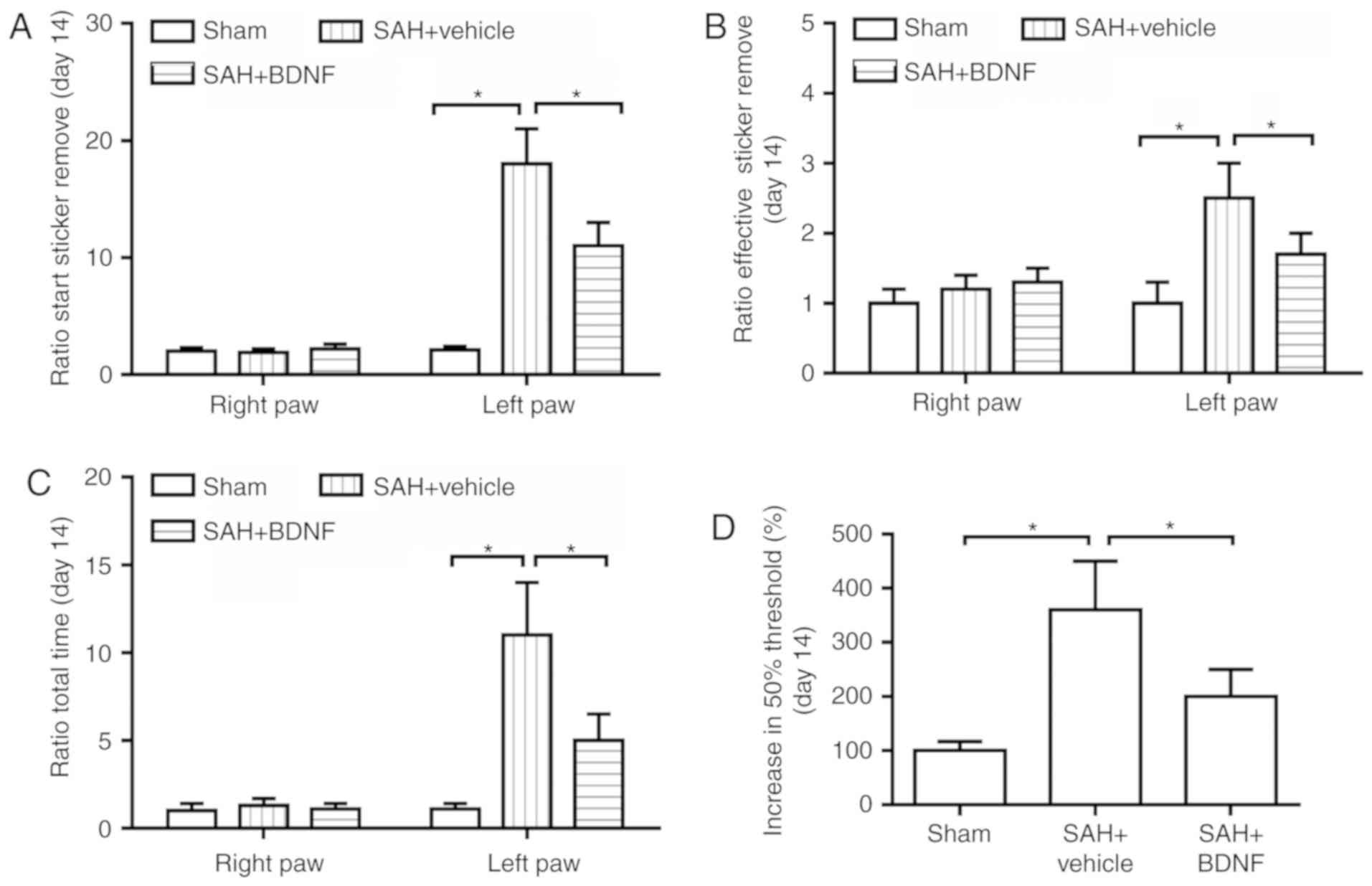
Exogenous BDNF attenuates long-term
sensorimotor behavior deficits after SAH
Next, the long-term sensorimotor behavior deficits
at day 14 after SAH were evaluated using the adhesive removal task
and Frey test. The time for taking on latency to start sticker
removal (sensory function; Fig. 4A),
the time to effective sticker removal (motor function; Fig. 4B) and the time for removing the
adhesive from the left forepaw (impaired; Fig. 4C) were significantly increased in the
SAH + vehicle group when compared with the sham group (P<0.05),
while BDNF treatment significantly attenuated these increases
(Fig. 4A-C). Furthermore, compared
with the sham group, SAH caused impairment in mechanical
sensitivity to innocuous stimuli, while BDNF treatment
significantly attenuated this impairment (P<0.05; Fig. 4D), indicating that BDNF improved
SAH-induced loss of sensory function.
Discussion
The present study demonstrated that ICV injection of
BDNF attenuated neurobehavioral deficits and neuronal apoptosis,
reduced the expression of Bax, activated caspase-9 and caspase-3 in
the basal cortex in an endovascular perforation SAH model in rats.
These findings suggest that exogenous BDNF is neuroprotective
against brain injury after SAH, at least in part through an
anti-apoptotic mechanism.
The expression level of BDNF following SAH has been
poorly tested and the reported results are diverse. The
concentration of BDNF in the cerebrospinal fluid was significantly
decreased at day 3, while it increased markedly at days 5 and 7
after SAH (11). The BDNF level in
the cerebral cortex significantly decreased at 4 and 48 h post-SAH
(10,12). There was no significant change in
BDNF expression of the cerebral hemispheres at 4 and 24 h following
SAH (9). In the present study,
western blot analysis and ELISA revealed that the BDNF
concentration of the basal cortex significantly decreased at 6, 12,
24, 48 and 72 h following SAH. In neurons, N-methyl-D-aspartic acid
triggers a Ca2+ signal and induces BDNF release
(24,25). As it has been reported that neuron
apoptosis occurs very early in the basal cortex following SAH
(2), it was hypothesized that the
downregulation of BDNF induced by SAH is possibly caused by
neuronal apoptosis. Further studies are required to investigate the
BDNF expression in short- and long-term brain damage, and elucidate
whether inhibition of neuronal apoptosis enhances BDNF expression
after SAH.
In line with other studies using the modified Garcia
score, beam balance test and rotarod test for evaluation of
neurological score after SAH (14,15,20,21), the
present study observed neurological deficits following SAH, which
improved by ICV injection of BDNF. Moreover, the adhesive removal
task and Frey test were used to assess the long-term sensorimotor
behavior deficit at day 14 after SAH (23). The results of the present study
revealed that SAH rats exhibited impairment in tactile sensitivity
and mechanical sensitivity. However, BDNF treatment restored these
functions. Neuronal apoptosis is considered to play a key role in
brain injury following SAH and is associated with neurological
deterioration and poor outcome (2).
The results of the present study demonstrated that ICV injection of
BDNF reduced the SAH-related increased number of activated
caspase-3- or TUNEL-positive neurons in the basal cortex. Although
not tested in the present study, the anti-apoptotic mechanism of
BDNF may be similar to that reported by previous studies (8,9), where
BDNF suppressed neuronal apoptosis through activation of the
BDNF/TrkB signaling pathway after SAH. Accumulating evidence
indicates that the balance between the anti-apoptotic protein Bcl-2
and the pro-apoptotic protein Bax is closely associated with cell
apoptosis and survival (26,27). The increased Bax level leads to
mitochondrial membrane permeabilization, which induces the
mitochondrial release of pro-apoptotic factors, then promotes the
initiator of caspase-9 activation and consequently the effector of
activated caspase-3 activation (28,29). In
the present study, decreased expression of Bcl-2 and increased
expression of Bax after SAH was observed, but exogenous BDNF
reversed these changes. The results of the present study also found
that SAH induces activation of caspase-9 and caspase-3, which is
attenuated by ICV injection of BDNF. Therefore, exogenous BDNF
appears to reduce neuronal apoptosis through regulation of Bcl-2,
Bax and caspase-3 expression.
In conclusion, the present study demonstrated that
exogenous BDNF attenuated neuronal apoptosis through inhibiting
caspase-9 and caspase-3 activation, and improved the neurological
deficits and long-term behavioral deficits after SAH.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Science Foundation of China (grant no. 81170095).
Authors' contributions
HW designed experiments and wrote the manuscript.
HC, YD, XL and JR performed the experiments and analyzed the
results. All authors read and approved the final manuscript.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Ethics approval and consent to
participate
Animal procedures were approved by the Animal Use
and Care Committee of Hubei University of Medicine.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Nieuwkamp DJ, Setz LE, Algra A, Linn FH,
de Rooij NK and Rinkel GJ: Changes in case fatality of aneurysmal
subarachnoid haemorrhage over time, according to age, sex, and
region: A meta-analysis. Lancet Neurol. 8:635–642. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hasegawa Y, Suzuki H, Sozen T, Altay O and
Zhang JH: Apoptotic mechanisms for neuronal cells in early brain
injury after subarachnoid hemorrhage. Acta Neurochir Suppl.
110:43–48. 2011.PubMed/NCBI
|
|
3
|
Baker-Herman TL, Fuller DD, Bavis RW,
Zabka AG, Golder FJ, Doperalski NJ, Johnson RA, Watters JJ and
Mitchell GS: BDNF is necessary and sufficient for spinal
respiratory plasticity following intermittent hypoxia. Nat
Neurosci. 7:48–55. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bramham CR and Messaoudi E: BDNF function
in adult synaptic plasticity: The synaptic consolidation
hypothesis. Prog Neurobiol. 76:99–125. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kowiański P, Lietzau G, Czuba E, Waśkow M,
Steliga A and Moryś J: BDNF: A key factor with multipotent impact
on brain signaling and synaptic plasticity. Cell Mol Neurobiol.
38:579–593. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Siironen J, Juvela S, Kanarek K, Vilkki J,
Hernesniemi J and Lappalainen J: The Met allele of the BDNF
Val66Met polymorphism predicts poor outcome among survivors of
aneurysmal subarachnoid hemorrhage. Stroke. 38:2858–2860. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Vilkki J, Lappalainen J, Juvela S, Kanarek
K, Hernesniemi JA and Siironen J: Relationship of the Met allele of
the brain-derived neurotrophic factor Val66Met polymorphism to
memory after aneurysmal subarachnoid hemorrhage. Neurosurgery.
63:198–203; discussion 203. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tang J, Hu Q, Chen Y, Liu F, Zheng Y, Tang
J, Zhang J and Zhang JH: Neuroprotective role of an N-acetyl
serotonin derivative via activation of tropomyosin-related kinase
receptor B after subarachnoid hemorrhage in a rat model. Neurobiol
Dis. 78:126–133. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hasegawa Y, Suzuki H, Altay O and Zhang
JH: Preservation of tropomyosin-related kinase B (TrkB) signaling
by sodium orthovanadate attenuates early brain injury after
subarachnoid hemorrhage in rats. Stroke. 42:477–483. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Jiang Y, Liu DW, Han XY, Dong YN, Gao J,
Du B, Meng L and Shi JG: Neuroprotective effects of anti-tumor
necrosis factor-alpha antibody on apoptosis following subarachnoid
hemorrhage in a rat model. J Clin Neurosci. 19:866–872. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lee WD, Wang KC, Tsai YF, Chou PC, Tsai LK
and Chien CL: Subarachnoid hemorrhage promotes proliferation,
differentiation, and migration of neural stem cells via BDNF
upregulation. PLoS One. 11:e01654602016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhang ZY, Yang MF, Wang T, Li DW, Liu YL,
Zhang JH and Sun BL: Cysteamine alleviates early brain injury via
reducing oxidative stress and apoptosis in a rat experimental
subarachnoid hemorrhage model. Cell Mol Neurobiol. 35:543–553.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li M, Wang Y, Wang W, Zou C, Wang X and
Chen Q: Recombinant human brain-derived neurotrophic factor
prevents neuronal apoptosis in a novel in vitro model of
subarachnoid hemorrhage. Neuropsychiatr Dis Treat. 13:1013–1021.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sugawara T, Ayer R, Jadhav V and Zhang JH:
A new grading system evaluating bleeding scale in filament
perforation subarachnoid hemorrhage rat model. J Neurosci Methods.
167:327–334. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang Z, Liu J, Fan C, Mao L, Xie R, Wang
S, Yang M, Yuan H, Yang X, Sun J, et al: The GluN1/GluN2B NMDA
receptor and metabotropic glutamate receptor 1 negative allosteric
modulator has enhanced neuroprotection in a rat subarachnoid
hemorrhage model. Exp Neurol. 301:13–25. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang ZY, Sun BL, Liu JK, Yang MF, Li DW,
Fang J, Zhang S, Yuan QL and Huang SL: Activation of mGluR5
attenuates microglial activation and neuronal apoptosis in early
brain injury after experimental subarachnoid hemorrhage in rats.
Neurochem Res. 40:1121–1132. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhu Q, Enkhjargal B, Huang L, Zhang T, Sun
C, Xie Z, Wu P, Mo J, Tang J, Xie Z and Zhang JH: Aggf1 attenuates
neuroinflammation and BBB disruption via PI3K/Akt/NF-κB pathway
after subarachnoid hemorrhage in rats. J Neuroinflammation.
15:1782018. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Guan J, Zhang B, Zhang J, Ding W, Xiao Z,
Zhu Z, Han Q, Wu C, Sun Y, Tong W, et al: Nerve regeneration and
functional recovery by collagen-binding brain-derived neurotrophic
factor in an intracerebral hemorrhage model. Tissue Eng Part A.
21:62–74. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Li H, Yu JS, Zhang DD, Yang YQ, Huang LT,
Yu Z, Chen RD, Yang HK and Hang CH: Inhibition of the receptor for
advanced glycation end-products (RAGE) attenuates neuroinflammation
while sensitizing cortical neurons towards death in experimental
subarachnoid hemorrhage. Mol Neurobiol. 54:755–767. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Huang CY, Wang LC, Wang HK, Pan CH, Cheng
YY, Shan YS, Chio CC and Tsai KJ: Memantine alleviates brain injury
and neurobehavioral deficits after experimental subarachnoid
hemorrhage. Mol Neurobiol. 51:1038–1052. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhou YD and Cai L: Calpeptin reduces
neurobehavioral deficits and neuronal apoptosis following
subarachnoid hemorrhage in rats. J Stroke Cerebrovasc Dis.
28:125–132. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Friedrich V, Flores R and Sehba FA: Cell
death starts early after subarachnoid hemorrhage. Neurosci Lett.
512:6–11. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kooijman E, Nijboer CH, van Velthoven CT,
Mol W, Dijkhuizen RM, Kesecioglu J and Heijnen CJ: Long-term
functional consequences and ongoing cerebral inflammation after
subarachnoid hemorrhage in the rat. PLoS One. 9:e905842014.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Jiang X, Zhu D, Okagaki P, Lipsky R, Wu X,
Banaudha K, Mearow K, Strauss KI and Marini AM:
N-methyl-D-aspartate and TrkB receptor activation in cerebellar
granule cells: An in vitro model of preconditioning to stimulate
intrinsic survival pathways in neurons. Ann N Y Acad Sci.
993:134–145; discussion 159–160. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhu D, Wu X, Strauss KI, Lipsky RH,
Qureshi Z, Terhakopian A, Novelli A, Banaudha K and Marini AM:
N-methyl-D-aspartate and TrkB receptors protect neurons against
glutamate excitotoxicity through an extracellular signal-regulated
kinase pathway. J Neurosci Res. 80:104–113. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang X, He X, Hu S, Sun A and Lu C:
Involvement of Bim in Photofrin-mediated photodynamically induced
apoptosis. Cell Physiol Biochem. 35:1527–1536. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Willis S, Day CL, Hinds MG and Huang DC:
The Bcl-2-regulated apoptotic pathway. J Cell Sci. 116:4053–4056.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hong Y, Shao A, Wang J, Chen S, Wu H,
McBride DW, Wu Q, Sun X and Zhang J: Neuroprotective effect of
hydrogen-rich saline against neurologic damage and apoptosis in
early brain injury following subarachnoid hemorrhage: Possible role
of the Akt/GSK3β signaling pathway. PLoS One. 9:e962122014.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Xie Z, Enkhjargal B, Wu L, Zhou K, Sun C,
Hu X, Gospodarev V, Tang J, You C and Zhang JH: Exendin-4
attenuates neuronal death via GLP-1R/PI3K/Akt pathway in early
brain injury after subarachnoid hemorrhage in rats.
Neuropharmacology. 128:142–151. 2018. View Article : Google Scholar : PubMed/NCBI
|