Introduction
The influenza A virus is a major human pathogen. The
severity of infection ranges from mild to severe and may even lead
to death. Seasonal viruses can cause annual epidemics that result
in 3–5 million cases of severe illness and 290,000–650,000 deaths
(1).
In order to effectively diagnose and treat severe
influenza virus infections, it is important to determine the
infection status and the quality of the host immune response.
Therefore, the identification of specific biomarkers that enable
accurate diagnosis of the disease and have a prognostic value for
predicting disease severity is required (2). Identifying biomarkers of influenza A
infection is challenging, as many viral and cellular factors
influence virulence, host response and pathogenicity of the virus.
Hemagglutinin, neuraminidase, NS and polymerase PB1 and PB2 gene
products serve a central role in determination of virulence
(3–7). Abundant viral replication in the lungs
and dissemination into non-respiratory tract tissues may result in
increased pathogenicity and mortality (7,8). Both
innate and adaptive immune responses are crucial for the control of
influenza infection. The activity of innate and adaptive immune
cells is coordinated by cytokines. However, an overactive or
unbalanced immune response may result in the overproduction of
cytokines that leads to severe inflammation, whereby an excessive
number of neutrophils and mononuclear cells are recruited to the
site of infection (9). Interleukin
(IL)-6 and chemokines, C-C motif chemokine ligand (CCL)2, CCL4,
C-X-C motif chemokine ligand (CXCL)8, CXCL9 and CXCL10, are
associated with the pathogenicity of both avian (H5N1 and H7N9) and
human (pdmH1N1 and H3N2) viruses (10–12).
Chemokines, CCL2, CXCL8, CXCL9 and CXCL10, have been associated
with mortality (13–17).
In present study, the cytokine expression profile in
the lungs of mice infected with a nonlethal dose (LD0) of the
A/PR/8/34 virus (H1N1) was analysed and compared with that of mice
infected with a lethal dose (LD100) of the same virus. The aim of
the present study was to identify the cytokines with altered
expression patterns following infection with LD100 when compared
with LD0. The results provide novel insights into the pathology of
influenza A infection and may have applications for the improvement
of influenza diagnosis and therapy.
Materials and methods
Cells and viruses
MDCK (ATCC CCL-34) cells were grown in Dulbecco's
modified Eagle's medium (Lonza) containing 10% fetal calf serum
(HyClone Laboratories). Influenza virus A/PR/8/34 [H1N1] was
cultured in 10-days-old fertile hen's eggs.
Female BALB/c mice (age, 4 weeks; body weight
approximately 20 g) were purchased from the Faculty of Medicine,
Masaryk University (Czech Republic). A total of 30 mice in two
groups of 15 mice were anesthetized with Zoletil (5 mg/kg)
intraperitoneally and inoculated intranasally with 103
plaque-forming units (PFU; LD100) or 101 PFU (LD0) of
virus (40 µl). To ensure that the mice were fully anesthetized, the
monitoring of rear foot reflexes was made before infection, and
continual observation of respiratory pattern, mucous membrane
colour and responsiveness to manipulations was made throughout the
procedure. The mice were monitored daily and humanely sacrificed at
the experimental endpoint. The criteria for euthanasia were
establish by using the total score for observation of possible
animal distress. Weight loss exceeding 25% of the original body
weight, decrease of appetite, weakness, shivering, depression and
moribund state of animals were monitored twice per day. For the
cytokine assay, all groups of 4 mice were sacrificed by cervical
dislocation and the lungs were aseptically collected at 0, 2 and 4
days post-infection (p.i.). Experiment lasted for 8 days. Organ
homogenates were pooled together, and aliquots were stored at
−80°C.
Determination of virus titers
Cellular debris was removed from lung tissue samples
by centrifuging at 160 × g for 10 min at 4°C and the supernatants
were used for virus quantification. The viral titers are expressed
as PFU/ml of lung homogenate in MDCK cells using a plaque assay as
previously described (18). The
results are expressed as the mean of two independent
experiments.
Semi-quantitative reverse
transcription-polymerase chain reaction (RT-PCR)
Total RNA from the lungs was extracted using the SV
Total RNA Isolation System (Promega). A total of 400 ng/µl RNA was
reverse transcribed using random hexanucleotide primers and the
MuLV reverse transcriptase (Finnzyme; Thermo Fisher Scientific).
Viral transcripts were detected by semi-quantitative RT-PCR as
previously described (18). The
sequences of all additional primers used for PCR were as follows:
Interferon regulatory factor (IRF)3, forward,
5′-GTCCTCAGATCTGGCTATTG-3′ and reverse, 5′-GCTTCAGTGGATTTTCTTGG-3′;
IRF4, forward, 5′-TCACTTGTTCGTGGAGCATC-3′ and reverse,
5′-TCTGGAGTCAGTGCTGATGG-3′; IRF7, forward,
5′-CCACGGAAAATAGGGAAGAA-3′ and reverse, 5′-CATAGGGTTCCTCGTAAACA-3′,
Arginase 1 (Arg1), forward, 5′-GCAGTGGCTTTAACCTTGGC-3′ and reverse,
5′-CTGTGATGCCCCAGATGGTT-3′; CD38, forward,
5′-TGGCCTTGCTGGAATAGGTG-3′ and reverse, 5′-TCATCAAGGTGGGAGCATGG-3′;
early growth response protein 2 (Egr2), forward,
5′-ACCTCCTTCCTACCCATCCC-3′ and reverse, 5′-ACAGGGAAACGGCTTTCGAT-3′;
G-protein coupled receptor 18 (Gpr18), forward
5′-TGGCCATCGTACAGCCAAAA-3′ and reverse, 5′-GTCGGAGATCTTCAGGCAGG-3′.
Formyl peptide receptor 2 (Fpr2) transcripts were detected by using
primers as previously decribed (19).
The band intensity of the PCR products was
determined using Gene Tools image analysis software (Syngene).
β-actin was used as an internal control to normalize the expression
of the mRNA levels between different samples.
Cytokine array
Lung tissue homogenates (100 µl) were lysed and the
protein concentration was determined using the Pierce BCA Protein
assay kit (Thermo Fisher Scientific). Chemokine expression in lung
tissue lysates was assessed using the Proteome Profiler Mouse XL
Cytokine array kit (R&D Systems). Signal intensities on
autoradiography films were quantified using Gene Tools software
(Sygene). The expression levels of cytokines were normalized to the
expression level of reference spots. The assay was performed in
duplicate to ensure reproducibility of the results.
Statistical analysis
Statistical analysis was performed by comparing
control group (noninfected mice) to infected mice or LD0 to LD100,
respectively. Data were analyzed using the unpaired Student's
t-test for data with two groups and ANOVA followed by Tukey's post
hoc test was performed for data with ≥2 groups. P-values <0.05
were considered to be significant. Statistical analysis was
performed using GraphPad Prism software (https://goodcalculators.com/one-way-anova-calculator/
and http://www.graphpad.com/quickcalcs/ttest1.cfm).
Results
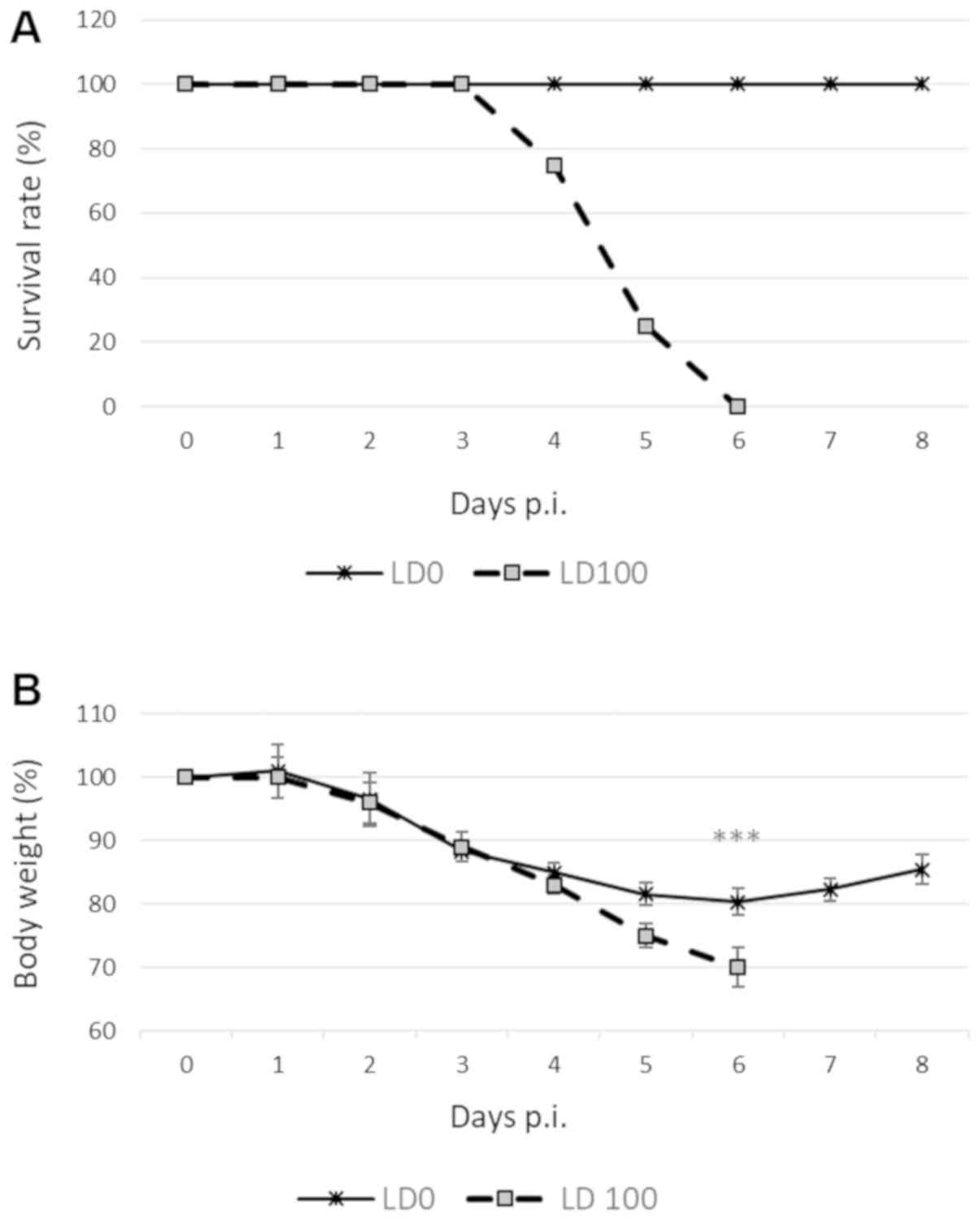
Mortality and virus replication
To determine the impact of viral infection on
influenza-specific mortality, BALB/c mice were inoculated with
either 101 PFU or 103 PFU of H1N1 virus. All
animals rapidly developed severe signs of disease, including
ruffled fur, huddling, lethargy and weight loss. Mice infected with
LD0 exhibited body weight loss of up to 20% within 4 days of
infection, followed by a gradual recovery by the end of the study
(Fig. 1A). By contrast, animals
infected with LD100 exhibited a >25% loss of body weight, and
the aforementioned symptoms were more severe. These mice either
died or were euthanized, and the death was recorded as
infection-associated mortality (Fig.
1B).
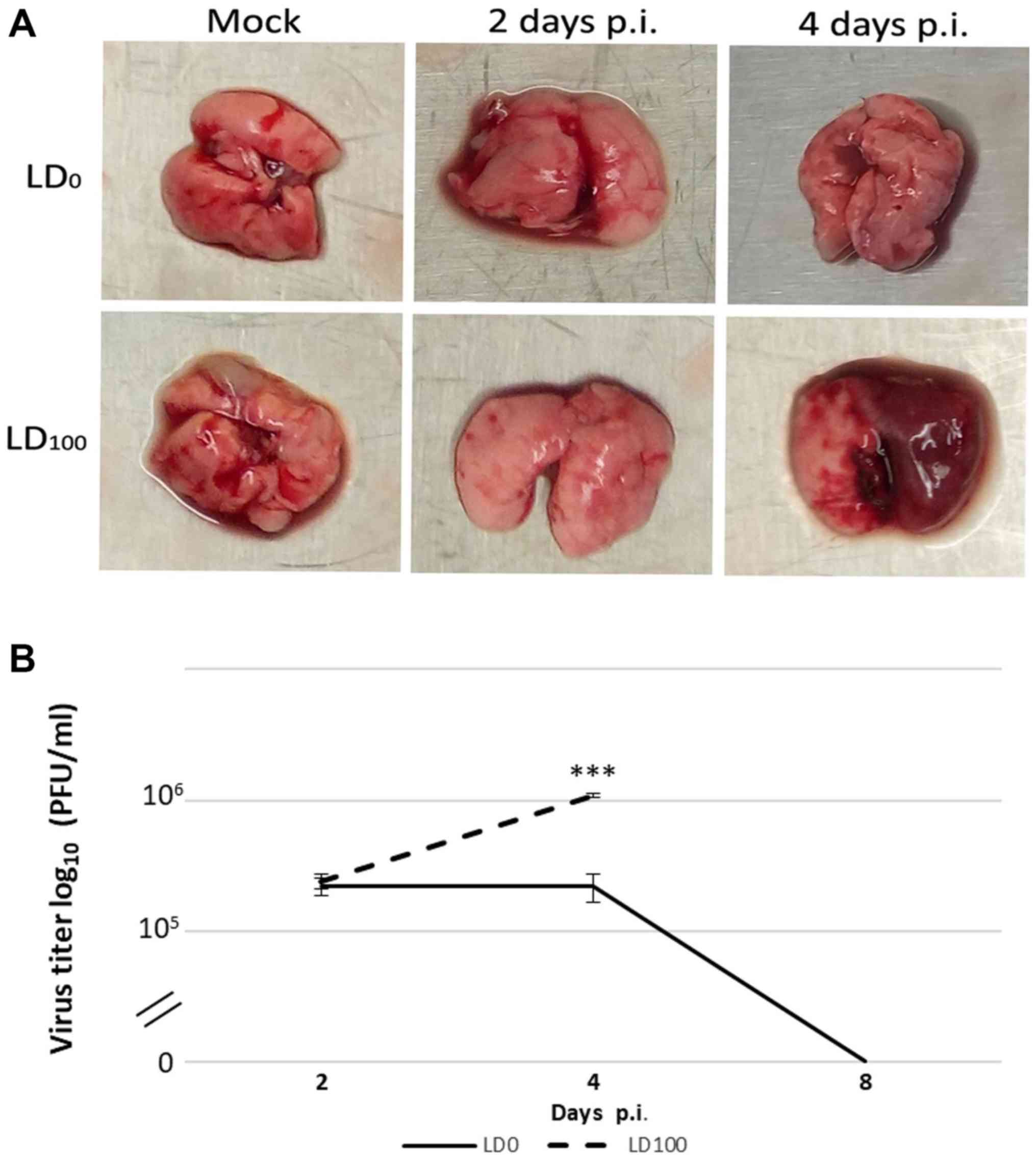
The results demonstrated that macroscopic changes in
the lungs were associated with virus pathogenicity. Naked-eye
observations of the lungs revealed that the most severe signs of
damage were identified in mice infected with LD100 at 4 days p.i.
(Fig. 2A). Minimal pathological
changes were observed in the lungs from mice in the LD0 group. In
the same group, the maximum viral titer was observed at 2 days p.i.
and these levels were not altered over the subsequent 2 days
(Fig. 2B). At 4 days p.i., the viral
titer decreased and was undetectable at 8 days p.i. In
LD100-infected mice, the viral titer increased up to 4 days p.i.
The maximum viral titer in this group of mice was five times higher
than mice in the LD0 group.
Activation of retinoic acid-inducible
gene (RIG)-I-like receptor signaling pathway genes
The mRNA levels of selected genes in the infected
lungs were determined using a semi-quantitative PCR assay at days
0, 2 and 4 days p.i. As demonstrated in Fig. 3A and B, LD0 and LD100 doses of the
virus induced similar expression alterations of RIG-I and Melanoma
differentiation-associated protein 5 (MDA-5) mRNAs. LD100 induced
significantly higher levels of RIG-I mRNA at 4 days p.i. No
signficant difference in IRF3 and NF-ĸB mRNA expression levels was
observed. IRF7 mRNA levels were significantly increased in mice
infected with lethal doses of the virus at 4 days p.i. (P<0.05).
In addition, LD100 induced significant increases in IRF4 mRNA
levels at 2 and 4 days p.i. (P<0.01 and P<0.05). Both viral
doses induced similar expression alterations of interferon (IFN)
mRNA levels, except for IFN-β and IFN-ε. LD0 induced a significant
increase in IFN-β mRNA levels at 4 days p.i. (P<0.05). In
addition, LD100 induced a significant increase in IFN-ε mRNA
expression at 2 days (P<0.05) and 4 days (P<0.001) p.i.
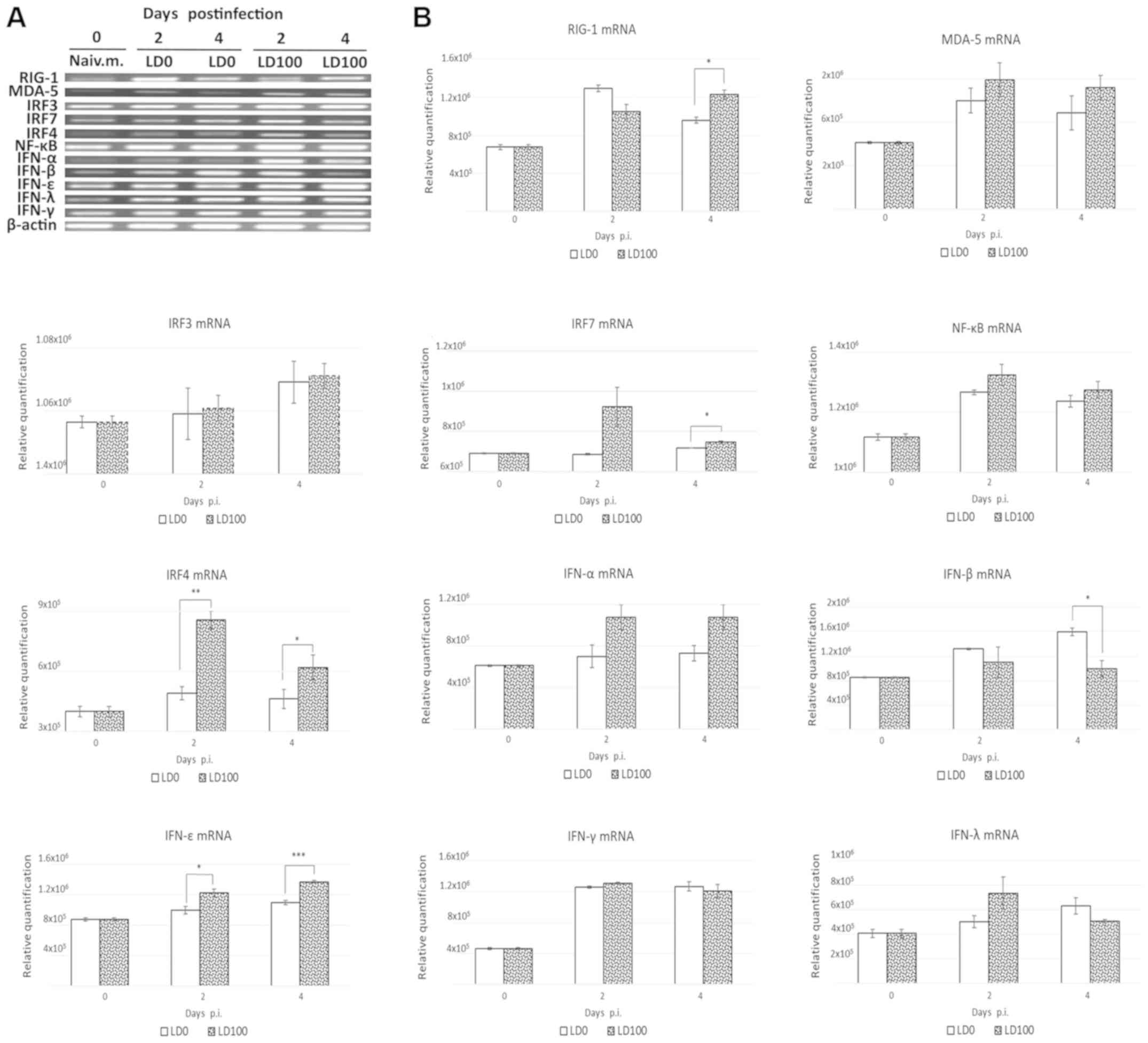 | Figure 3.Induction of RIG-I-like receptor
signalling pathways. BALB/c mice were infected intranasally with
LD0 and LD100 doses of H1N1 virus. The lungs were harvested prior
to infection and at 2 and 4 days p.i., and lung homogenates were
used for assessment of mRNA levels. Representative (A) reverse
transcription PCR blots and (B) relative expression levels of mRNAs
were obtained. The expression values represent the mean of two
separate experiments and are presented as the mean ± standard
deviation. *P<0.05, **P<0.001 and ***P<0.0001, as
indicated. RIG, retinoic acid-inducible gene; LD0, 101
plaque-forming units; LD100, 103 plaque-forming units;
H1N1, A/PR/8/34 virus; IFN, interferon; IRF, interferon regulatory
factor; p.i., post-infection; MDA-5, melanoma
differentiation-associated gene 5. |
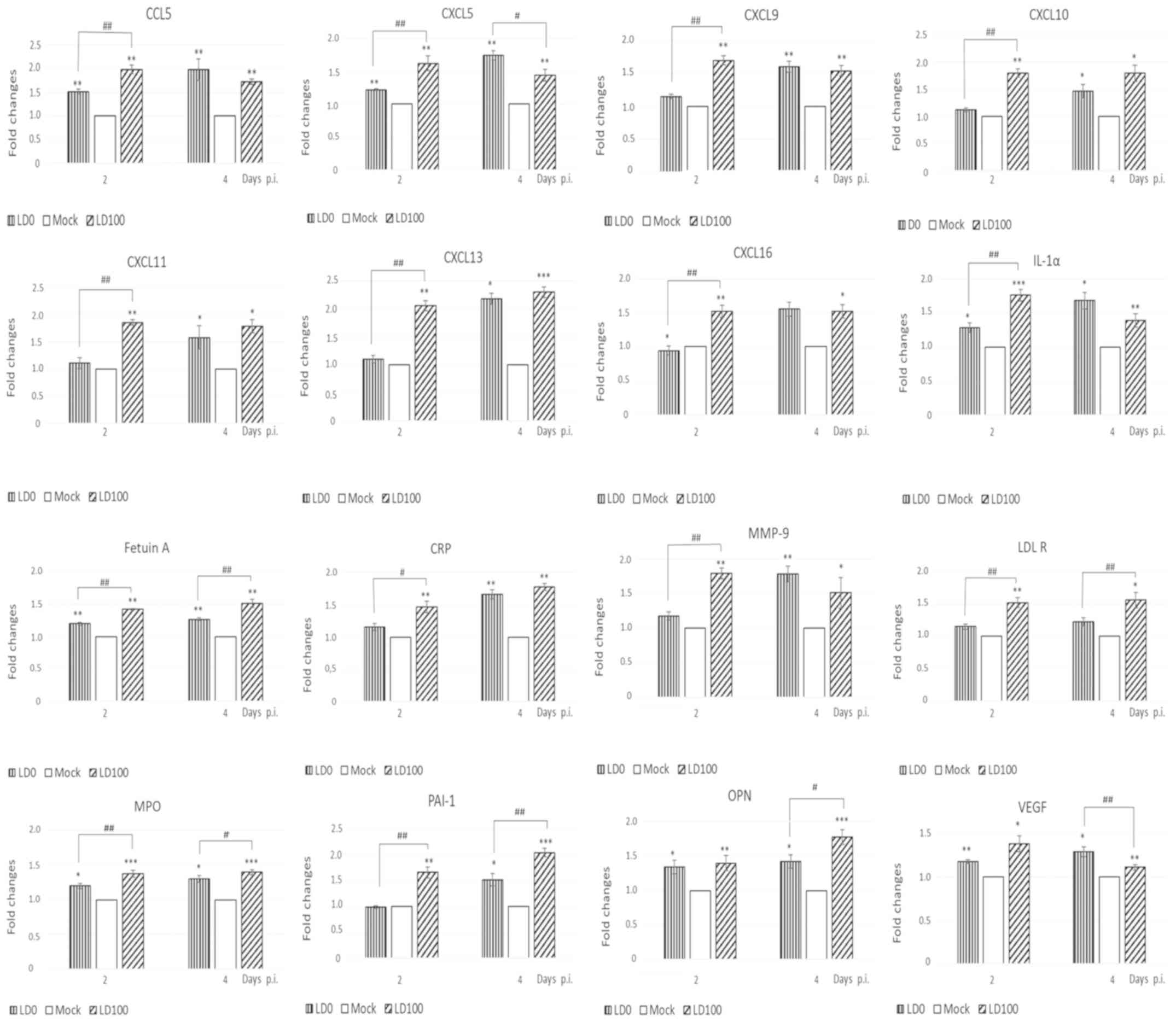
Infection with a lethal dose of virus
significantly increased the expression of cytokines associated with
pathogenicity
Different cytokine expression profiles were observed
following infection with LD0 and LD100. The expression levels of
114 soluble mouse proteins in the lungs of mice infected with
either dose of the virus were compared. Both LD0 and LD100
significantly increased the expression levels of lipocalin 2,
IL-33, CCL6, CCL11 and CCL22. The expression of these cytokines was
not positively correlated with the quantity of virus used for
infection (data not shown).
LD0 and LD100 viral doses increased the expression
of 16 cytokines at 2 and 4 days p.i. (Fig. 4). However, distinct differences in
the protein expression patterns of cytokines induced by LD0 and
LD100 were observed. Cytokines, IL-1α, CCL5, CXCL5, CXCL9, CXCL10,
CXCL11, CXCL13, CXCL16, C-reactive protein (CRP) and matrix
metallopeptidase (MMP)-9 were more abundant in the lungs of mice
infected with LD100, particularly at 2 days p.i. A significant
decrease in the expression of vascular endothelial growth factor
(VEGF) and osteopontin (OPN) was observed at 4 days p.i. An
additional group of the cytokines, including CXCL5, fetuin A,
myeloperoxidase (MPO), plasminogen activator inhibitor type 1
(PAI-1) and low density lipoprotein receptor (LDL-R) were highly
induced by LD100 at 2 and 4 days p.i. The expression of these
cytokines was significantly higher when compared to LD0 at the same
time-points p.i.
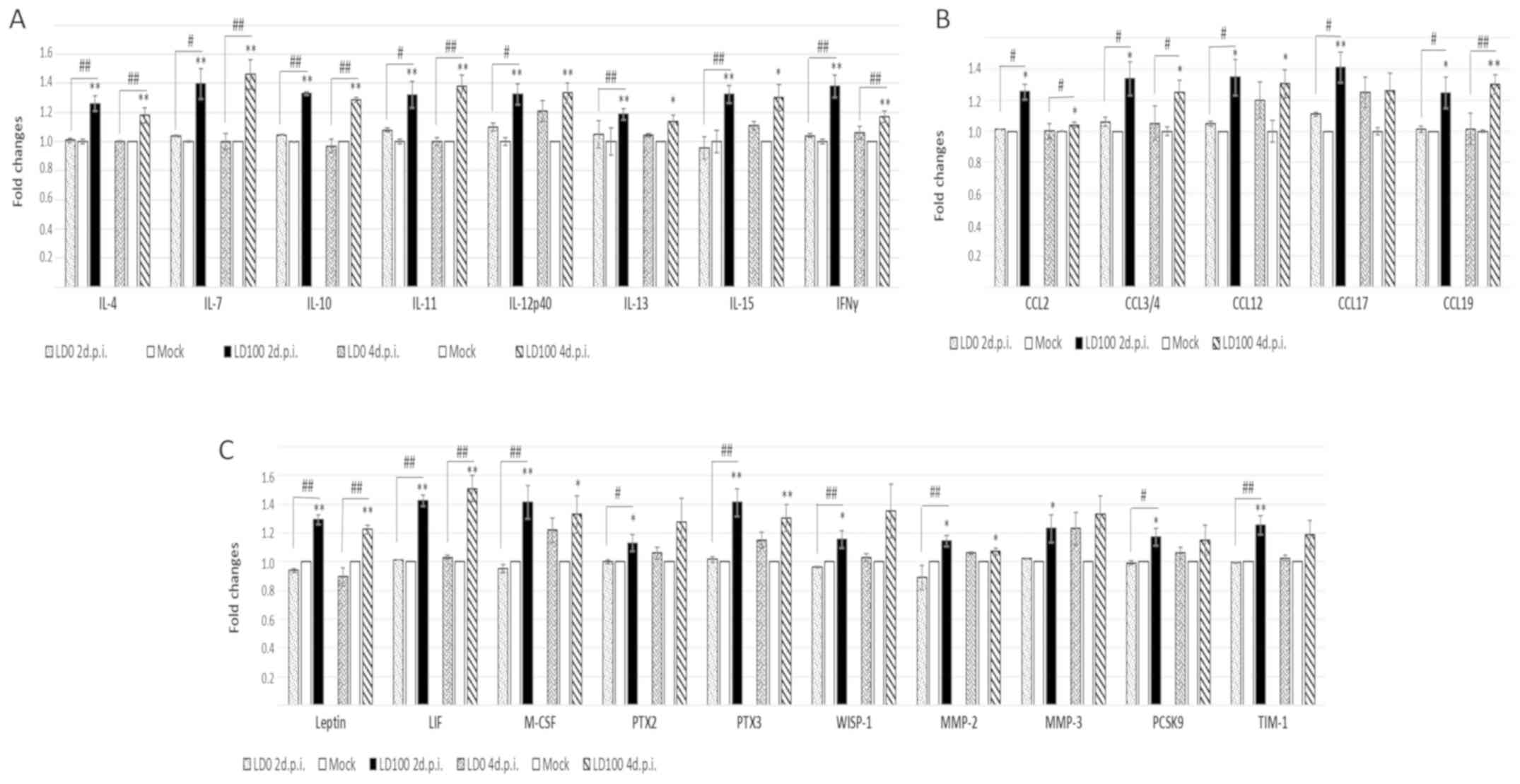
Cytokines induced by the lethal dose
of virus only
The most apparent differences between the cytokine
levels induced by LD0 and LD100 doses of the virus are shown in
Fig. 5. LD0 did not influence the
expression of these cytokines, whereas LD100 increased their
expression at 2 days p.i. These cytokines can be divided into the
following three main groups of proteins: i) IFN-γ; ii) IL-4, IL-7,
IL-10, IL-11, IL-12p40, IL-13, IL-15 and chemokines, CCL2, CCL3/4,
CCL12, CCL17, CCL19; and iii) other immune response proteins,
including leptin, leukaemia inhibitory factor (LIF), macrophage
colony stimulating factor (M-CSF), pentraxin (PTX)2 and 3,
WNT1-inducible-signaling pathway protein 1 (WISP-1), MMP-2, MMP-3,
proprotein convertase subtilisin/kexin type 9 (PCSK9), T cell
immunoglobulin and mucin domain (TIM-1).
Infection with a lethal dose of virus
significantly increased the expression of M2 macrophage
markers
Different transcriptional mRNA profiling in murine
macrophages were observed in the lungs infected with with LD0 and
LD100. Classically activated macrophages (M1) can be distinquished
from alternatively activated macrophages (M2) by their relative
expression of CD38, Gpr18, Fpr2 and Arg1, Egr2, respectivelly. As
demonstrated in Fig. 6, no
signficant difference in CD38, Gpr18, and Fpr2 mRNA expression
levels was observed at 2 and 4 days p.i. Arg1 and Egr2 mRNA levels
were significantly increased in mice infected with lethal doses of
the virus at 4 days p.i. These data indicate that infection with
LD100 influences macrophage polarization in the lungs.
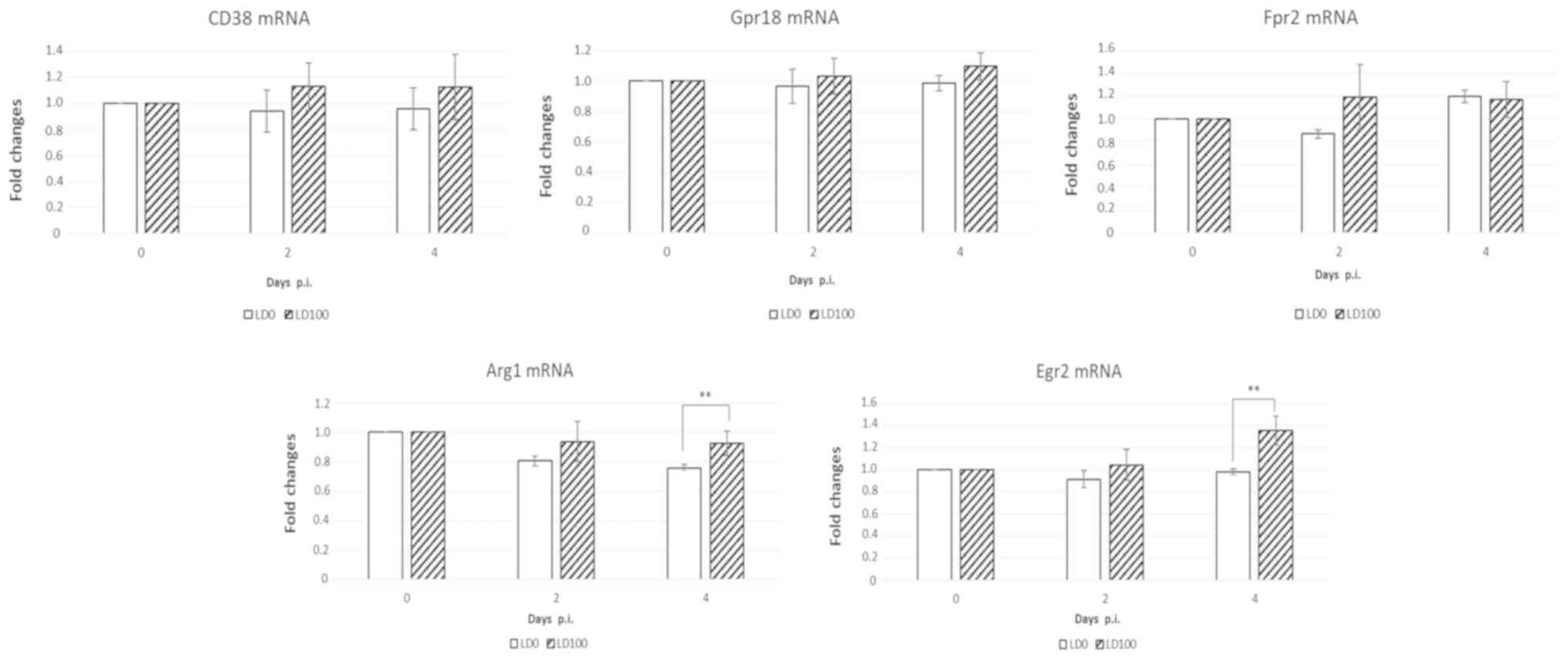 | Figure 6.Induction of M21 and M2 markers in
the lungs of mice infected with LD0 and LD100 doses of H1N1. The
lungs were harvested prior to infection and at 2 and 4 days p.i.,
and lung homogenates were used for assessment of mRNA levels.
Expression values represent the mean of two separate experiments
and are expressed as the mean ± standard deviation. **P<0.001,
as indicated. Gpr18, G-protein coupled receptor 18; Fpr2, formyl
peptide receptor 2; Arg1, arginase-1; Egr2, early growth response
protein 2; LD0, 101 plaque-forming units; LD100,
103 plaque-forming units; H1N1, A/PR/8/34 virus; p.i.,
post-infection. |
Discussion
Cytokines serve an important role in modulating the
host immune response, clearing the virus, and healing any injury
caused by the virus. Numerous factors influence the pathogenicity
of the virus; thus, it is difficult to identify objective markers
that can be used as an effective diagnostic tool and predict
disease severity. It is hypothesized that virus pathogenicity is
directly associated with the replication potential of the virus
(7–11). Comparing the expression of cytokines
induced by nonlethal and lethal doses of the virus may therefore
identify the specific cytokines associated with high pathogenicity,
which could be used as suitable biomarkers.
In the present study, the virus titer in mice
infected with LD0 and LD100 was similar at 2 days p.i.; however,
the cytokine expression profiles induced were different. We
repeated this experiment several times and we obtained the same
results. As it was expected, 100% mortality was observed within
animals infected with LD100. In LD100-infected mice, the virus
titer increased up to 4 days p.i. and none of these mice survived
until 8 day p.i. Many factors as virus input, immune response etc.
influenced the virus replication in the lungs. The most severe sign
of damage was observed in the lungs of mice infected with LD100 at
4 days p.i. Unfortunately, we did not examine histological changes
in the lungs after influenza infection. We would like to do it in
the future.
No significant difference in the activation of
RIG-I-like receptor signaling pathways was observed in the lungs of
mice infected with nonlethal and lethal doses of influenza virus.
Nevertheless, infection with LD100 induced the expression of a
greater abundance and more diverse array of cytokines.
The cytokine profile induced by LD100 was similar to
the profiles previously observed in sera from patients with severe
influenza infection and influenza virus-associated encephalopathy
(11,12,20).
IFN-γ, IL-4, IL-7, IL-10, IL-11, IL-12p40, IL-13 and IL-15 levels
were increased only in the lungs of mice infected with a lethal
dose of influenza. An interesting observation in our study was a
switching in macrophage polarization from M1 to M2, evidenced by
the increase in the M2 markers, Arg1 and Egr2, in the lungs of mice
infected with LD100. IL-12 and IFN-γ are the main T helper (Th)1
cytokines. Th1 cells emerge in the presence of IL-12 and mediate
their function by secreting IFN-γ (21,22). Th1
cytokines promote M1 macrophage differentiation, which serves
important roles in the eradication of viral infection. M1
macrophages secrete chemokines, such as CXCL5, CXCL9 and CXCL10
(23). However, excessive
pro-inflammatory responses can lead to uncontrolled tissue damage.
Anti-inflammatory cytokines, such as IL-4, IL-10 and IL-13 are the
main Th2 cytokines. In addition, increased IRF4 and IL-4 levels
promote the differentiation of naïve CD4+ T cells into
Th2 cells (24–28). Th2 lymphocytes release
anti-inflammatory cytokines, IL-4, IL-5, IL-6, IL-10 and IL-13
(29). In the murine system, IL-10
downregulates the Th1 response (30). Subsequent activation of the
complement system leads to neutrophil or eosinophil influx by IL-4
and IL-5 (31,32). In addition, Th2 cytokines, such IL-4
and IL-13, promote differentiation of M2 macrophages. M2
macrophages which produce high levels of IL-10 are generally
thought to function as anti-inflammatory macrophages and serve key
roles in the suppression of Th1 cell responses, wound healing and
tissue repair (33–35). Overexpression of chemokines and the
Th2 phenotype serve central roles in influenza pathogenesis
(36). The results of the present
study therefore indicate that infection with LD100 invokes a mixed
Th1/Th2 response.
In the current study, infection with LD100 induced
overexpression of specific C-C motif chemokine ligand chemokines,
which may contribute to disease pathogenicity and even death of the
mice. Overexpression of chemokines CCL2, CCL17 and CCL19 is
associated with the activity of M2 macrophages, and serves an
important role in protective immune responses (37–39).
CCL2 and CCL4 chemokines exert potent effects on the recruitment
and degranulation of eosinophils and basophils, which may provide
valuable insights into the immunopathogenesis of respiratory viral
infections (40). CCL2 and CCL12
mediate acute lung injury induced by lethal influenza infection, as
well as by the γ-herpesvirus or fungal (Asperigillus
fumigatus) infections (41–45).
However, the precise role of CCL19 in infuenza infection remains
unknown. CXCL2 and CCL3 chemokines mediate neutrophil infiltration
during the early phase of infection and can serve an important role
in the pathogenesis of the disease in the lungs (46,47).
In the current study, increased expression of CRP,
LDL-R, MMP-9, MPO, PAI-1, OPN and VEGF correlated with increased
inflammatory cytokine expression and lung tissue damage. These
proteins are normally expressed during tissue damage or injury and
serve important roles in tissue repair and lung immunity. Increased
expression of these cytokines has been associated with the
pathogenicity of virus infection. MPO-derived oxidants are
pathogenic and can promote inflammation and tissue damage (48). Increased MMP-9 expression is
associated with organ and tissue damage, and serves an essential
role in infection and in the host response to infection (49). The MMP-9 cycle is an important
mechanism underlying multiple organ failure during severe influenza
infection. A previous study in mouse models demonstrated that H1N1
infection increased the levels of CCL2, MMP-9 and trypsin in serum
and/or the lungs and heart (50).
In the present study, infection of mice with a
lethal dose of the virus induced the expression of cytokines
associated with severe lung injury and pathology. Increased
expression of cytokines, including leptin, LIF, M-CSF, PTX2, PTX3,
WISP-1, MMP-2, MMP-3, PCSK9 and TIM-1, correlated with extensive
lung damage following lethal influenza infection. These proteins
modulate infection response, inflammation and tissue repair. A
previous study demonstrated that PTX2 reduces the severity of acute
lung injury in an animal model and in humans and might be useful
for a variety of disease therapy (51). In addition, the IL-10/CREB/WISP-1
signaling pathway has been shown to link innate immune activation
to mucosal wound repair (52). MMP-2
and MMP-9 serve a protective role through pathogen clearance
(53). However, increased expression
of PCSK9 exacerbates multi-organ damage, and MMP3 contributes to
the pathogenesis of acute respiratory distress syndrome (54,55).
M-CSF promotes the differentiation and survival of macrophages, and
preferentially induces anti-inflammatory M2 rather than
pro-inflammatory M1 macrophages. Increased levels of C-MSF promote
the development of a mixed Th1/Th2 immune response (56). However, it will be necessary to
clarify the function of leptin, LIF, M-CSF, PTX2, PTX3, WISP-1,
MMP-2, MMP-3, PCSK9 and TIM-1 in influenza pathogenicity. It is
possible that these factors are released due to extensive tissue
damage caused by viral replication, the increased expression of
other cytokines or due to induction of a mixed Th1/Th2 immune
response.
The balance between pro- and anti-inflammatory
cytokines is essential for maintaining homeostasis in the
respiratory system, and an imbalance has been implicated in the
pathogenesis of granulomatous diseases and pulmonary fibrosis
(57,58). By comparing the cytokine profiles
obtained from the lungs of mice infected with nonlethal and lethal
influenza doses, the authors of the present study hypothesize that
lethal infection may induce a mixed Th1/Th2 response. A previous
study demonstrated that Th2-dominated immune responses to influenza
virus infection exacerbate lung tissue damage and delay viral
clearance (59). In addition, the
results of the current study indicate that immune cells, such as
macrophages, eosinophils, neutrophils, monocytes, NK cells and
basophils may serve a greater role in the pathogenesis of influenza
infection than previously thought. However, the mechanisms by which
Th1 and Th2 cells influence the inflammatory response during
influenza virus infection require further investigation in future
studies.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Slovak
Research and Development Agency (grant nos. APVV-0676-12 and VEGA
2/0014/16).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
TB conceived and designed the experiments, performed
experiments, analyzed data and wrote the manuscript. LT and VL
performed experiments and analyzed the data. DS performed
experiments. AK analyzed data and performed the literature
search.
Ethics approval and consent to
participate
All animal experiments were approved by the
Institutional Animal Care and Use Committee (IACUC) of the
Institute of Virology. The animals were treated according to the
European Union standards and the fundamental ethical principles,
including animal welfare requirements, were respected. All of the
animal experiments were evaluated and approved by the State
Veterinary and Food Administration of the Slovak Republic (approval
nos. 4370/13-221 and 1204/11-221).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Kalarikkal SM and Jaishankar GB: Influenza
VaccineStatPearls [Internet]. StatPearls Publishing; Treasure
Island, FL: 2019, https://www.ncbi.nlm.nih.gov/books/NBK537197/
|
|
2
|
Kollmus H, Pilzner C, Leist SR, Heise M,
Geffers R and Schughart K: Of mice and men: The host response to
influenza virus infection. Mamm Genome. 29:446–470. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Baskin CR, Bielefeldt-Ohmann H, Tumpey TM,
Sabourin PJ, Long JP, García-Sastre A, Tolnay AE, Albrecht R, Pyles
JA, Olson PH, et al: Early and sustained innate immune response
defines pathology and death in nonhuman primates infected by highly
pathogenic influenza virus. Proc Natl Acad Sci USA. 106:3455–3460.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lycett SJ, Ward MJ, Lewis FI, Poon AF,
Kosakovsky Pond SL and Brown AJ: Detection of mammalian virulence
determinants in highly pathogenic avian influenza H5N1 viruses:
Multivariate analysis of published data. J Virol. 83:9901–9910.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
de Wit E, Kawaoka Y, de Jong MD and
Fouchier RA: Pathogenicity of highly pathogenic avian influenza
virus in mammals. Vaccine. 26 (Suppl 4):D54–D58. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ping J, Keleta L, Forbes NE, Dankar S,
Stecho W, Tyler S, Zhou Y, Babiuk L, Weingartl H, Halpin RA, et al:
Genomic and protein structural maps of adaptive evolution of human
influenza A virus to increased virulence in the mouse. PLoS One.
6:e217402011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tumpey TM, García-Sastre A, Taubenberger
JK, Palese P, Swayne DE, Pantin-Jackwood MJ, Schultz-Cherry S,
Solórzano A, Van Rooijen N, Katz JM and Basler CF: Pathogenicity of
influenza viruses with genes from the 1918 pandemic virus:
Functional roles of alveolar macrophages and neutrophils in
limiting virus replication and mortality in mice. J Virol.
79:14933–14944. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
de Jong RM, Stockhofe-Zurwieden N, Verheij
ES, de Boer-Luijtze EA, Ruiter SJ, de Leeuw OS and Cornelissen LA:
Rapid emergence of a virulent PB2 E627K variant during adaptation
of highly pathogenic avian influenza H7N7 virus to mice. Virol J.
10:2762013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Betakova T, Kostrabova A, Lachova V and
Turianova L: Cytokines induced during influenza virus infection.
Curr Pharm Des. 23:2616–2622. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chi Y, Zhu Y, Wen T, Cui L, Ge Y, Jiao Y,
Wu T, Ge A, Ji H, Xu K, et al: Cytokine and chemokine levels in
patients infected with the novel avian influenza A (H7N9) virus in
China. J Infect Dis. 208:1962–1967. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bradley-Stewart A, Jolly L, Adamson W,
Gunson R, Frew-Gillespie C, Templeton K, Aitken C, Carman W,
Cameron S and McSharry C: Cytokine responses in patients with mild
or severe influenza A(H1N1)pdm09. J Clin Virol. 58:100–107. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sun G, Ota C, Kitaoka S, Chiba Y,
Takayanagi M, Kitamura T, Yamamoto K, Fujie H, Mikami H, Uematsu M,
et al: Elevated serum levels of neutrophil elastase in patients
with influenza virus-associated encephalopathy. J Neurol Sci.
349:190–195. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ahn MY, Zhang ZG, Tsang W and Chopp M:
Endogenous plasminogen activator expression after embolic focal
cerebral ischemia in mice. Brain Res. 837:169–176. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Peiris JS, Yu WC, Leung CW, Cheung CY, Ng
WF, Nicholls JM, Ng TK, Chan KH, Lai ST, Lim WL, et al:
Re-emergence of fatal human influenza A subtype H5N1 disease.
Lancet. 363:617–619. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chan MC, Cheung CY, Chui WH, Tsao SW,
Nicholls JM, Chan YO, Chan RW, Long HT, Poon LL, Guan Y and Peiris
JS: Proinflammatory cytokine responses induced by influenza A
(H5N1) viruses in primary human alveolar and bronchial epithelial
cells. Respir Res. 6:1352005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Walsh KB, Teijaro JR, Rosen H and Oldstone
MB: Quelling the storm: Utilization of sphingosine-1-phosphate
receptor signaling to ameliorate influenza virus-induced cytokine
storm. Immunol Res. 51:15–25. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Vogel AJ, Harris S, Marsteller N, Condon
SA and Brown DM: Early cytokine dysregulation and viral replication
are associated with mortality during lethal influenza infection.
Viral Immunol. 27:214–224. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Svancarova P, Svetlikova D and Betakova T:
Synergic and antagonistic effect of small hairpin RNAs targeting
the NS gene of the influenza A virus in cells and mice. Virus Res.
195:100–111. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jablonski KA, Amici SA, Webb LM,
Ruiz-Rosado Jde D, Popovich PG, Partida-Sanchez S and
Guerau-de-Arellano M: Novel markers to delineate murine M1 and M2
macrophages. PLoS One. 10:e01453422015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Davey RT Jr, Lynfield R, Dwyer DE, Losso
MH, Cozzi-Lepri A, Wentworth D, Lane HC, Dewar R, Rupert A, Metcalf
JA, et al: The association between serum biomarkers and disease
outcome in influenza A(H1N1)pdm09 virus infection: Results of two
international observational cohort studies. PLoS One. 8:e571212013.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hsieh CS, Macatonia SE, Tripp CS, Wolf SF,
O'Garra A and Murphy KM: Development of TH1 CD4+ T cells
through IL-12 produced by Listeria-induced macrophages. Science.
260:547–549. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Macatonia SE, Hosken NA, Litton M, Vieira
P, Hsieh CS, Culpepper JA, Wysocka M, Trinchieri G, Murphy KM and
O'Garra A: Dendritic cells produce IL-12 and direct the development
of Th1 cells from naive CD4+ T cells. J Immunol.
154:5071–5079. 1995.PubMed/NCBI
|
|
23
|
Suzuki K, Meguro K, Nakagomi D and
Nakajima H: Roles of alternatively activated M2 macrophages in
allergic contact dermatitis. Allergol Int. 66:392–397. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lohoff M, Mittrücker HW, Prechtl S,
Bischof S, Sommer F, Kock S, Ferrick DA, Duncan GS, Gessner A and
Mak TW: Dysregulated T helper cell differentiation in the absence
of interferon regulatory factor 4. Proc Natl Acad Sci USA.
99:11808–11812. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Rengarajan J, Mowen KA, McBride KD, Smith
ED, Singh H and Glimcher LH: Interferon regulatory factor 4 (IRF4)
interacts with NFATc2 to modulate interleukin 4 gene expression. J
Exp Med. 195:1003–1012. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Staudt V, Bothur E, Klein M, Lingnau K,
Reuter S, Grebe N, Gerlitzki B, Hoffmann M, Ulges A, Taube C, et
al: Interferon-regulatory factor 4 is essential for the
developmental program of T helper 9 cells. Immunity. 33:192–202.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Huber M and Lohoff M: IRF4 at the
crossroads of effector T-cell fate decision. Eur J Immunol.
44:1886–1895. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhang Y, Zhang Y, Gu W and Sun B: TH1/TH2
cell differentiation and molecular signals. Adv Exp Med Biol.
841:15–44. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Rossi D and Zlotnik A: The biology of
chemokines and their receptors. Annu Rev Immunol. 18:217–242. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Del Prete G, De Carli M, Almerigogna F,
Giudizi MG, Biagiotti R and Romagnani S: Human IL-10 is produced by
both type 1 helper (Th1) and type 2 helper (Th2) T cell clones and
inhibits their antigen-specific proliferation and cytokine
production. J Immunol. 150:353–360. 1993.PubMed/NCBI
|
|
31
|
Kobayashi K, Kaneda K and Kasama T:
Immunopathogenesis of delayed-type hypersensitivity. Microsc Res
Tech. 53:241–245. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Mukhopadhyay S and Gal AA: Granulomatous
lung disease: An approach to the differential diagnosis. Arch
Pathol Lab Med. 134:667–690. 2010.PubMed/NCBI
|
|
33
|
Mosser DM and Edwards JP: Exploring the
full spectrum of macrophage activation. Nat Rev Immunol. 8:958–969.
2008. View
Article : Google Scholar : PubMed/NCBI
|
|
34
|
Mantovani A, Biswas SK, Galdiero MR, Sica
A and Locati M: Macrophage plasticity and polarization in tissue
repair and remodelling. J Pathol. 229:176–185. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Mokarram N and Bellamkonda RV: A
perspective on immunomodulation and tissue repair. Ann Biomed Eng.
42:338–351. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Belperio JA, Dy M, Murray L, Burdick MD,
Xue YY, Strieter RM and Keane MP: The role of the Th2 CC chemokine
ligand CCL17 in pulmonary fibrosis. J Immunol. 173:4692–4698. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Rangel-Moreno J, Moyron-Quiroz JE, Hartson
L, Kusser K and Randall TD: Pulmonary expression of CXC chemokine
ligand 13, CC chemokine ligand 19, and CC chemokine ligand 21 is
essential for local immunity to influenza. Proc Natl Acad Sci USA.
104:10577–10582. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Huang SS, Banner D, Degousee N, Leon AJ,
Xu L, Paquette SG, Kanagasabai T, Fang Y, Rubino S, Rubin B, et al:
Differential pathological and immune responses in newly weaned
ferrets are associated with a mild clinical outcome of pandemic
2009 H1N1 infection. J Virol. 86:13187–13201. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Hsu AT, Lupancu TJ, Lee MC, Fleetwood AJ,
Cook AD, Hamilton JA and Achuthan A: Epigenetic and transcriptional
regulation of IL4-induced CCL17 production in human monocytes and
murine macrophages. J Biol Chem. 293:11415–11423. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Bonville CA, Rosenberg HF and Domachowske
JB: Macrophage inflammatory protein-1alpha and RANTES are present
in nasal secretions during ongoing upper respiratory tract
infection. Pediatr Allergy Immunol. 10:39–44. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Blease K, Mehrad B, Lukacs NW, Kunkel SL,
Standiford TJ and Hogaboam CM: Antifungal and airway remodeling
roles for murine monocyte chemoattractant protein-1/CCL2 during
pulmonary exposure to Asperigillus fumigatus conidia. J Immunol.
166:1832–1842. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Vannella KM, Luckhardt TR, Wilke CA, van
Dyk LF, Toews GB and Moore BB: Latent herpesvirus infection
augments experimental pulmonary fibrosis. Am J Respir Crit Care
Med. 181:465–477. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Stoolman JS, Vannella KM, Coomes SM, Wilke
CA, Sisson TH, Toews GB and Moore BB: Latent infection by
γherpesvirus stimulates profibrotic mediator release from multiple
cell types. Am J Physiol Lung Cell Mol Physiol. 300:L274–L285.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Lai C, Wang K, Zhao Z, Zhang L, Gu H, Yang
P and Wang X: C-C motif chemokine ligand 2 (CCL2) mediates acute
lung injury induced by lethal influenza H7N9 Virus. Front
Microbiol. 8:5872017. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Wolf S, Johnson S, Perwitasari O,
Mahalingam S and Tripp RA: Targeting the pro-inflammatory factor
CCL2 (MCP-1) with bindarit for influenza A (H7N9) treatment. Clin
Transl Immunology. 6:e1352017. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Sakai S, Kawamata H, Mantani N, Kogure T,
Shimada Y, Terasawa K, Sakai T, Imanishi N and Ochiai H:
Therapeutic effect of anti-macrophage inflammatory protein 2
antibody on influenza virus-induced pneumonia in mice. J Virol.
74:2472–2476. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Camp JV, Bagci U, Chu YK, Squier B, Fraig
M, Uriarte SM, Guo H, Mollura DJ and Jonsson CB: Lower respiratory
tract infection of the ferret by 2009 H1N1 pandemic influenza A
Virus triggers biphasic, systemic, and local recruitment of
neutrophils. J Virol. 89:8733–8748. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Strzepa A, Pritchard KA and Dittel BN:
Myeloperoxidase: A new player in autoimmunity. Cell Immunol.
317:1–8. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Luplertlop N, Missé D, Bray D, Deleuze V,
Gonzalez JP, Leardkamolkarn V, Yssel H and Veas F:
Dengue-virus-infected dendritic cells trigger vascular leakage
through metalloproteinase overproduction. EMBO Rep. 7:1176–1181.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Takahashi E, Indalao IL, Sawabuchi T,
Mizuno K, Sakai S, Kimoto T, Kim H and Kido H: Clarithromycin
suppresses induction of monocyte chemoattractant protein-1 and
matrix metalloproteinase-9 and improves pathological changes in the
lungs and heart of mice infected with influenza A virus. Comp
Immunol Microbiol Infect Dis. 56:6–13. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Pilling D and Gomer RH: The development of
serum amyloid P as a possible therapeutic. Front Immunol.
9:23282018. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Quiros M, Nishio H, Neumann PA, Siuda D,
Brazil JC, Azcutia V, Hilgarth R, O'Leary MN, Garcia-Hernandez V,
Leoni G, et al: Macrophage-derived IL-10 mediates mucosal repair by
epithelial WISP-1 signaling. J Clin Invest. 127:3510–3520. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Hendrix AY and Kheradmand F: The role of
matrix metalloproteinases in development, repair, and destruction
of the lungs. Prog Mol Biol Transl Sci. 148:1–29. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Dwivedi DJ, Grin PM, Khan M, Prat A, Zhou
J, Fox-Robichaud AE, Seidah NG and Liaw PC: Differential expression
of PCSK9 modulates infection, inflammation, and coagulation in a
murine model of sepsis. Shock. 46:672–680. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Puntorieri V, McCaig LA, Howlett CJ, Yao
LJ, Lewis JF, Yamashita CM and Veldhuizen RA: Lack of matrix
metalloproteinase 3 in mouse models of lung injury ameliorates the
pulmonary inflammatory response in female but not in male mice. Exp
Lung Res. 42:365–379. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Fleetwood AJ, Lawrence T, Hamilton JA and
Cook AD: Granulocyte-macrophage colony-stimulating factor (CSF) and
macrophage CSF-dependent macrophage phenotypes display differences
in cytokine profiles and transcription factor activities:
Implications for CSF blockade in inflammation. J Immunol.
178:5245–5252. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
da Costa Souza P, Dondo PS, Souza G, Lopes
D, Moscardi M, de Miranda Martinho V, de Mattos Lourenço RD, Prieto
T, Balancin ML, Assato AK, et al: Comprehensive analysis of immune,
extracellular matrices and pathogens profile in lung granulomatosis
of unexplained etiology. Hum Pathol. 75:104–115. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Wu C, Luo Z, Pang B, Wang W, Deng M, Jin
R, Muhataer X, Li Y, Li Q and Yang X: Associations of pulmonary
fibrosis with peripheral blood Th1/Th2 cell imbalance and EBF3 gene
methylation in uygur pigeon breeder's lung patients. Cell Physiol
Biochem. 47:1141–1151. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Graham MB, Braciale VL and Braciale TJ:
Influenza virus-specific CD4+ T helper type 2 T
lymphocytes do not promote recovery from experimental virus
infection. J Exp Med. 180:1273–1282. 1994. View Article : Google Scholar : PubMed/NCBI
|