Introduction
Epidermolysis bullosa simplex (EBS) belongs to a
group of inherited disorders characterized by the high occurrence
of bullous lesions after a certain degree of friction or trauma. It
is most commonly caused by mutations in keratin 5 (KRT5) or
keratin 14 (KRT14) (1,2). The
protein products of these genes, keratin 5 and keratin 14, are
paired intermediate filaments expressed in basal keratinocytes that
contribute to mechanical stability of keratin filament networks
(1,2). EBS is comprised of three major forms:
i) Localized and mild subtype, called EBS, localized (EBS-loc; OMIM
131800) or EBS Weber-Cockayne; ii) generalized and severest
subtype, EBS Dowling-Meara (EBS-DM; OMIM 131760); and iii)
generalized but relatively milder subtype known as EBS, generalized
intermediate (EBS-gen intermed; OMIM 131900) or EBS-Koebner
(3). EBS, generalized severe,
formerly known as EBS Dowling-Meara (EBS-gen sev; OMIM 131760), is
the most severe form of EBS. Its cardinal features include large,
generalized blisters, mucous membrane involvement, and dystrophic
nails. Bullous lesions are usually most severe in the neonatal and
infancy stages, diminishing in severity with age, especially during
later childhood and adulthood (3).
The ultrastructural pathogenesis of EBS-DM includes clumping or
collapsing of keratin filaments in the basal epidermal cells,
leading to basal cell cytolysis and sequent intraepidermal blister
formation (4,5). In most cases, the clinical severity is
related to the location of the mutations. Most of the mutations
responsible for this subtype are located in the highly conserved
α-helical end segments of helix 1A and 2B (including amino acid
residue 125) of KRT5 and KRT14, which are critical
for proper intermediate filament structure (6). The current study reported the distinct
clinical features of three Chinese probands suspected of having EBS
and diagnosed with the EBS-gen sev subtype by molecular
analysis.
Patients and methods
Patients
Three unrelated Chinese families with three probands
clinically suspected of having EBS subtypes were enrolled in the
current study at the outpatient department of Xinhua Hospital
Affiliated to Shanghai Jiaotong University School of Medicine from
October 2016 to October 2018, including two young males (age, 4 and
10 months), and a 19-year-old female.
PCR and Sanger sequencing
Primers flanking all coding exons and intron-exon
boundaries of KRT5 and KRT14 were designed using
Primer Premier 5.0 software (Premier Biosoft International).
Sequences of primers used in the current study are presented in
Table SI. Sanger sequencing was
performed to identify the mutations in all patients and to verify
the sequences of unaffected family members. Peripheral blood
samples of all index patients were collected in EDTA anticoagulant
tubes (Insepack™; Sekisui Medical Co., Ltd.) and frozen at −20°C.
TIANamp Blood DNA kit (Tiangen Biotech Co., Ltd.) was used to
extract genomic DNA from 600-µl blood samples according to the
manufacturer's instructions. Genomic DNA samples were amplified by
PCR using Takara Ex Taq DNA polymerase (Takara Bio,
Inc.). The following thermocycling conditions were used: Initial
denaturation at 94°C for 5 min; 31 cycles of denaturation at 94°C
for 30 sec, annealing (temperature for each primer is listed in
Table SI) for 30 sec, extension at
72°C for 1 min, and a final extension at 72°C for 1 min; and 4°C
for 5 min. PCR products were purified with AxyPrep DNA Gel
Extraction kit (Corning, Inc.) according to the manufacturer's
instructions. The Sequencing Reaction system was based on
BigDye® Terminator v3.1 Cycle Sequencing kit (Thermo
Fisher Scientific, Inc.) according to the manufacturer's
instructions. Purified PCR products were directly sequenced using
an ABI PRISM® 3730 automated sequencer (Applied
Biosystems; Thermo Fisher Scientific, Inc.).
Results
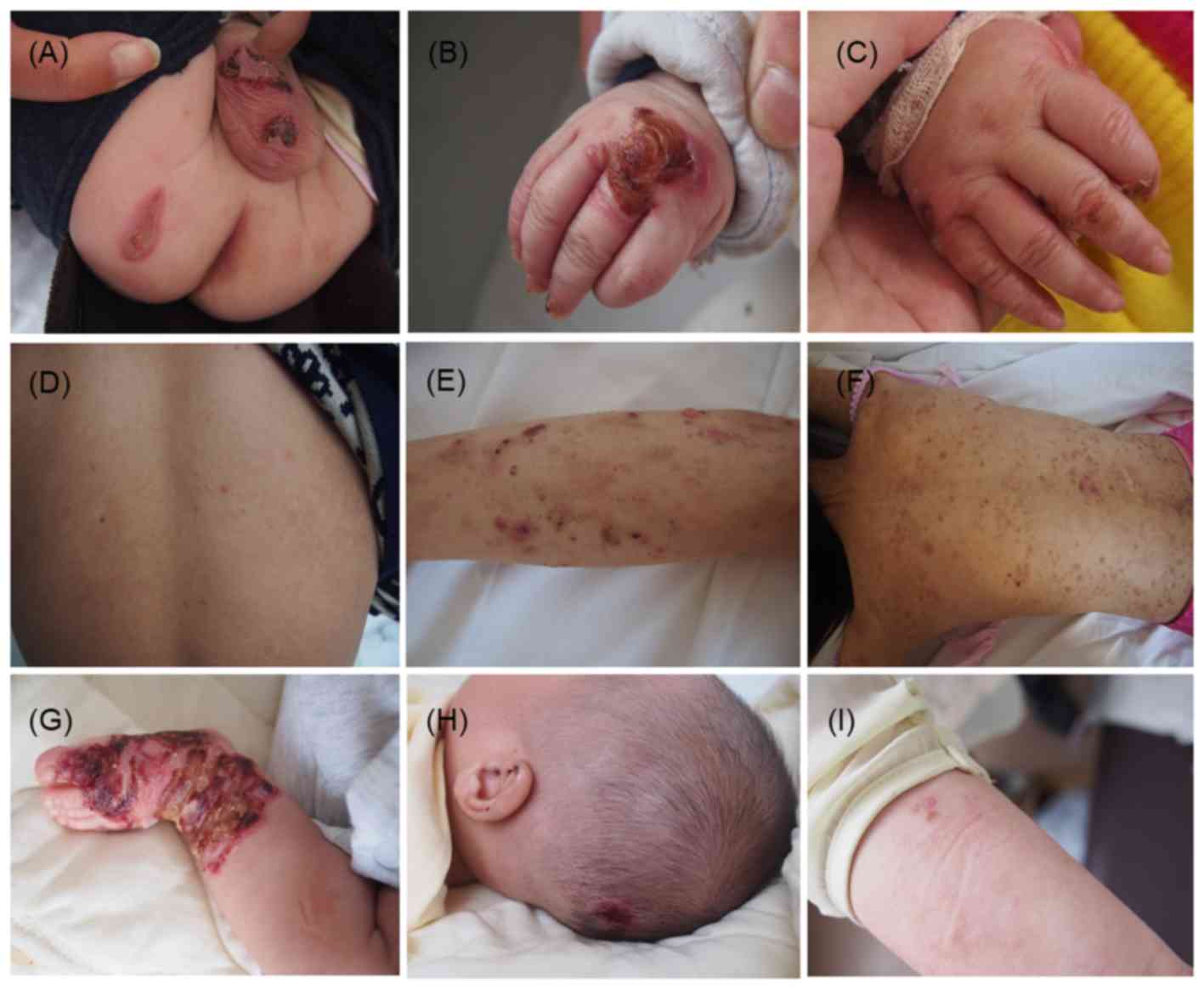
The proband in family 1 was a 10-month-old male. His
mother first came to the Department of Dermatology, Xinhua Hospital
in April 2014 complaining of generalized skin blisters on the
baby's hands and feet since birth (Fig.
1A and B), especially after friction or trauma. Seven months
later, the skin lesions over his extremities were slightly improved
(Fig. 1C). His 26-year-old father
with a similar history of blistering was also present at this
visit. Dermatological investigation showed no scarring, but only
mild, post-inflammatory pigmentation on skin regions previously
covered in blisters (Fig. 1D),
although he presented with widespread blisters as an early
infant.
The second proband, a 19-year-old female, presented
with diffuse pigmentation, scar formation after rupture of the
blisters, and scattered new blistering, resembling dermatitis
herpetiformis (Fig. 1E and F). With
age, the blistering diminished.
The last proband was a 4-month-old infant, with
clinical manifestations similar to those of proband 1 (Fig. 1G and H). His 25-year-old mother had a
similar clinical history since birth, but currently showed only
scattered blisters and mild, post-inflammatory pigmentation
(Fig. 1I).
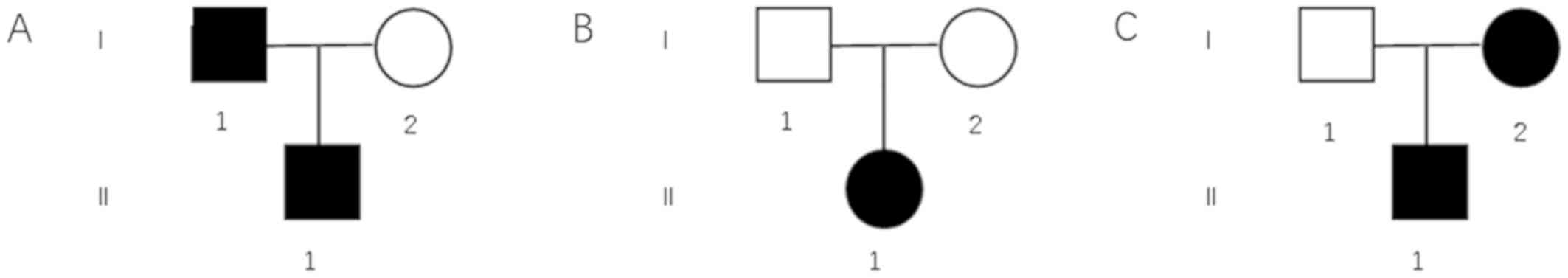
Pedigree charts of these three families are shown in
Fig. 2. Mutation screening of
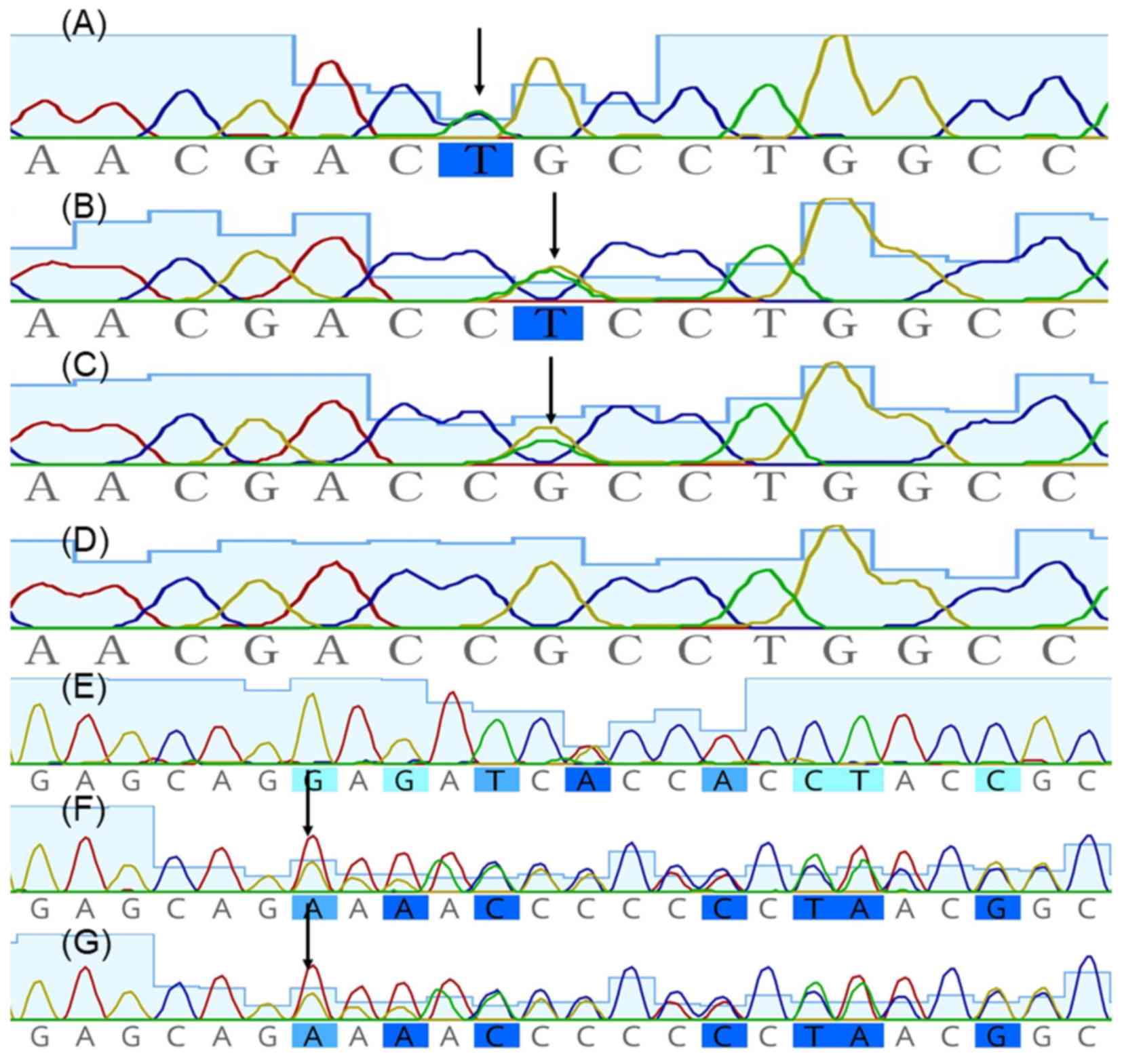
KRT5 was negative, whereas heterozygous missense mutations
c.374G>T (Arg125Leu), c.373C>T (Arg125Cys) and c.1231delG
(p.Glu411Argfs*31) in KRT14 were identified in proband 1,
proband 2 and proband 3, respectively, and were absent in
unaffected family members (Fig. 3).
Mutation delineation was based on comparisons with the reported
cDNA reference sequence (GenBank accession number, NM_ 000526.4 for
KRT14). Sequencing results were analyzed using Geneious,
version 5.6.7 (Biomatters, Ltd.; http://www.geneious.com/).
Discussion
The underlying pathological mechanism of EBS-gen sev
is intraepidermal blister formation via basal cell cytolysis (or,
rarely, acantholysis). Other subtypes can be distinguished by
clumping or significant collapse of keratin filaments in the basal
epidermal cells, which can be observed with immunofluorescence
mapping (IFM) or electron microscopy (EM) (7). However, these primary methods may lead
to discordance with the actual diagnosis (7). In addition, skin biopsy in infants is
usually regarded as a somewhat unacceptable trauma for parents, and
complementary genetic testing is required.
EBS-gen sev is inherited in an autosomal dominant
pattern. Except for those rare cases caused by truncated mutations,
including nonsense/in-frame deletion/frameshift/splicing mutations,
most EBS-gen sev cases are attributed to missense mutations that
exert a dominant negative effect on functional protein structure by
altering inter-chain interactions (8). There is a close correlation between the
mutational locus and the severity of EBS. Compared with other two
major forms, EBS-loc and EBS-gen, most EBS-gen sev cases were
associated with mutations in the highly conserved end segments of
KRT5 or KRT14 rod domain (6), which highlights the importance of
molecular diagnosis.
The site Arg125 (CGC) of KRT14 contains a CpG
dinucleotide, making it a hotspot for EBS-causing mutations due to
the disposition of a spontaneous mutant. Mutations in Arg125
(including Arg125His, Arg125Cys, Arg125Ser, Arg125Gly and
Arg125Leu) have been shown to be responsible for at least 40% of
EBS-gen sev cases. In particular, Arg125His and Arg125Cys accounted
for the majority of the mutations in Arg125 (1,6,9–14).
Arg125 is located in a highly conserved region and strongly
perturbs keratin network formation and keratin filament assembly
(15). Despite the fact that other
identical sites of mutations, even in one pedigree, may lead to
distinct subtypes (1,16), Arg125 mutations are confined mainly
to the most severe subtype EBS-gen sev with similar clinical
courses. To the best our knowledge, only three pathogenic mutations
of EBS-gen sev in the Chinese population (Arg165Ser in KRT5,
and Arg125His and Arg125Cys in KRT14) have been reported so
far. Only Arg125Leu has been reported in Korean and Polish
populations. Phenotypes of patients with Arg125Leu and Arg125Cys in
the current study were in accordance with the previously reported
phenotypes (1,6,9–14).
Novel mutation c.1231delG (p.Glu411Argfs*31) located
near the highly conserved α-helical end segments of helix 2B could
cause a stop codon in this highly conserved region, which may
result in a more severe phenotype, such as EBS-gen sev. EBS harbors
risk of death at an early age (17).
Currently, there is no well-established cure, other than general
care to prevent trauma, infection control, and good nutrition
(18).
Gene therapy research of EBS is rare, although gene
therapy and bone marrow transplantation for the relatively severe
dystrophic or junctional epidermolysis bullosa, having the
possibility of cure, have been well investigated (19,20).
These studies highlight that a corrective gene therapy could be an
ideal therapy for EBS, however more studies are required before it
can be developed and used in daily clinical practice. Prenatal or
preimplantation genetic diagnosis is another sensible option for
families at high risk of EBS.
The present study provided valuable information and
assistance in genetic counseling such that the symptoms of EBS-gen
sev patients declined with age. In clinical dermatology, a
diagnosis of EBS-gen sev should be considered for patients with
widespread, herpetiform, clustered blistering, especially in the
neonatal period, diminishing in late childhood. It is generally
challenging to distinguish EBS subtypes clinically in newborns.
Related effective methods such as IFM and EM on freshly induced
blisters combined with molecular genetic testing can be conducted
to make a clear diagnosis.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from the
Shanghai Sailing Program (grant no. 17YF1411900) and from National
Nature Science Foundation of China (grant no. 81903197).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YZ and ZY conceived and designed the present study.
ML collected clinical data. JZ and YD assessed the results and
wrote the paper.
Ethics approval and consent to
participate
The current study was approved by the Institutional
Review Board of Xinhua Hospital, Shanghai JiaoTong University
School of Medicine and was conducted in accordance with the
principles of the Declaration of Helsinki. Ethical approval was
obtained from the Ethics Committee of the Xinhua Hospital
Affiliated to Shanghai Jiao Tong University School of Medicine. All
participants or their legal guardians gave their written informed
consent for participation.
Patient consent for publication
Patients or patients' guardians provided consent for
the publication of images in the present study.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bolling MC, Lemmink HH, Jansen GH and
Jonkman MF: Mutations in KRT5 and KRT14 cause epidermolysis bullosa
simplex in 75% of the patients. Br J Dermatol. 164:637–644.
2011.PubMed/NCBI
|
|
2
|
Fuchs E: The cytoskeleton and disease:
Genetic disorders of intermediate filaments. Annu Rev Genet.
30:197–231. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Fine JD, Bruckner-Tuderman L, Eady RA,
Bauer EA, Bauer JW, Has C, Heagerty A, Hintner H, Hovnanian A,
Jonkman MF, et al: Inherited epidermolysis bullosa: Updated
recommendations on diagnosis and classification. J Am Acad
Dermatol. 70:1103–1126. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Coulombe PA, Hutton ME, Vassar R and Fuchs
E: A function for keratins and a common thread among different
types of epidermolysis bullosa simplex diseases. J Cell Biol.
115:1661–1674. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Coulombe PA, Hutton ME, Letal A, Hebert A,
Paller AS and Fuchs E: Point mutations in human keratin 14 genes of
epidermolysis bullosa simplex patients: Genetic and functional
analyses. Cell. 66:1301–1311. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yasukawa K, Sawamura D, Goto M, Nakamura
H, Jung SY, Kim SC and Shimizu H: Epidermolysis bullosa simplex in
Japanese and Korean patients: Genetic studies in 19 cases. Br J
Dermatol. 155:313–317. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hiremagalore R, Kubba A, Bansel S and
Jerajani H: Immunofluorescence mapping in inherited epidermolysis
bullosa: A study of 86 cases from India. Br J Dermatol.
172:384–391. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Banerjee S, Wu Q, Yu P, Qi M and Li C: In
silico analysis of all point mutations on the 2B domain of K5/K14
causing epidermolysis bullosa simplex: A genotype-phenotype
correlation. Mol Biosyst. 10:2567–2577. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Arin MJ, Grimberg G, Schumann H, De
Almeida H Jr, Chang YR, Tadini G, Kohlhase J, Krieg T,
Bruckner-Tuderman L and Has C: Identification of novel and known
KRT5 and KRT14 mutations in 53 patients with epidermolysis bullosa
simplex: Correlation between genotype and phenotype. Br J Dermatol.
162:1365–1369. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Garcia M, Santiago JL, Terron A,
Hernández-Martín A, Vicente A, Fortuny C, De Lucas R, López JC,
Cuadrado-Corrales N, Holguín A, et al: Two novel recessive
mutations in KRT14 identified in a cohort of 21 Spanish families
with epidermolysis bullosa simplex. Br J Dermatol. 165:683–692.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kang TW, Lee JS, Kim SE, Oh SW and Kim SC:
Novel and recurrent mutations in Keratin 5 and 14 in Korean
patients with Epidermolysis bullosa simplex. J Dermatol Sci.
57:90–94. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Minakawa S, Nakano H, Nakajima K,
Matsuzaki Y, Takiyoshi N, Akasaka E, Rokunohe D and Sawamura D:
Mutational analysis on 16 Japanese population cases with
epidermolysis bullosa simplex. J Dermatol Sci. 72:330–332. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Müller FB, Küster W, Wodecki K, Almeida H
Jr, Bruckner-Tuderman L, Krieg T, Korge BP and Arin MJ: Novel and
recurrent mutations in keratin KRT5 and KRT14 genes in
epidermolysis bullosa simplex: Implications for disease phenotype
and keratin filament assembly. Hum Mutat. 27:719–720. 2006.
View Article : Google Scholar
|
|
14
|
Pfendner EG, Sadowski SG and Uitto J:
Epidermolysis bullosa simplex: Recurrent and de novo mutations in
the KRT5 and KRT14 genes, phenotype/genotype correlations, and
implications for genetic counseling and prenatal diagnosis. J
Invest Dermatol. 125:239–243. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Letai A, Coulombe PA, McCormick MB, Yu QC,
Hutton E and Fuchs E: Disease severity correlates with position of
keratin point mutations in patients with epidermolysis bullosa
simplex. Proc Natl Acad Sci USA. 90:3197–3201. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jankowski M, Wertheim-Tysarowska K,
Jakubowski R, Sota J, Nowak W and Czajkowski R: Novel KRT14
mutation causing epidermolysis bullosa simplex with variable
phenotype. Exp Dermatol. 23:684–687. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hon KL, Li JJ, Cheng BL, Luk DC, Murrell
DF, Choi PC and Leung AK: Age and etiology of childhood
epidermolysis bullosa mortality. J Dermatolog Treat. 26:178–182.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Langan SM and Williams HC: A systematic
review of randomized controlled trials of treatments for inherited
forms of epidermolysis bullosa. Clin Exp Dermatol. 34:20–25. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Siprashvili Z, Nguyen NT, Bezchinsky MY,
Marinkovich MP, Lane AT and Khavari PA: Long-term type VII collagen
restoration to human epidermolysis bullosa skin tissue. Hum Gene
Ther. 21:1299–1310. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wagner JE, Ishida-Yamamoto A, McGrath JA,
Hordinsky M, Keene DR, Woodley DT, Chen M, Riddle MJ, Osborn MJ,
Lund T, et al: Bone marrow transplantation for recessive dystrophic
epidermolysis bullosa. N Engl J Med. 363:629–639. 2010. View Article : Google Scholar : PubMed/NCBI
|