Introduction
Aortic dissection (AD) is a fatal disease that
accounts for a large proportion of aortic-associated mortalities
(1). Indeed, epidemiological surveys
have demonstrated that the incidence of thoracic AD is 3–4 per
100,000 individuals per year as of 2011 (2,3). AD is
defined as blood flow that enters the aortic media through intimal
tears, followed by a formation of a false lumen addition to the
true lumen (4). The false lumen wall
is composed of the intima and media of the aortic wall, and the
true lumen is the original aortic lumen. Clinically, AD can result
in numerous serious complications, including aortic rupture and
visceral ischemia (4). The majority
of patients with non-operative ascending AD (Stanford type A)
exhibit an overall survival time of <2 weeks (5). Unfortunately, due to its sudden and
unpredictable nature, little is known regarding the pathological
and molecular events underpinning the development or progression of
AD.
Endothelial dysfunction serves an initial role in
the development and pathogenesis of cardiovascular diseases,
including AD (6). Vascular
endothelial cells (VECs) are flat endothelial cells (ECs) in the
inner lining of the major blood vessels. They are important in
regulating blood flow, and thus, are involved in numerous
physiological processes (6).
Inflammation can result in VEC dysfunction and further promote AD
progression (7). Angiotensin II (Ang
II) is the major effector peptide of the renin-angiotensin system
(RAS), which induces vasoconstriction, hypertrophy and
extracellular remodeling via the angiotensin type 1 receptor (AT1R)
(8). Ang II-induced endothelial
dysfunction has been implicated in a variety of cardiovascular
diseases, including atherosclerosis, hypertension, left ventricular
hypertrophy, myocardial infarction and heart failure (9). Additionally, studies have indicated
that Ang II contributes to the development of AD in humans and
animal models (10), and also
promotes the production of reactive oxygen species by inducing
multiple downstream pathways in VECs (11). Angiotensin receptor blocker (ARB) is
an antihypertensive drug that is commonly used to treat patients
with AD by achieving blood pressure control (4). In the present study, the effect of ARB
on AD progression was investigated. Both VEC dysfunction and Ang II
serve an important role in the occurrence of AD and consequently
the association between them was also investigated.
Yes-associated protein (YAP) is a pluripotent
intracellular junction protein that is both a transcriptional
co-activator, and a major effector of the Hippo-YAP signaling
pathway. Nuclear YAP binds to the transcriptional
enhancer-associated domain transcription factors to regulate
transcriptional processes, and ultimately influence cell
proliferation and apoptosis (12).
Mammalian sterile 20-like kinase-1 and −2 are the central mediators
of the Hippo-YAP signaling pathway, which promotes YAP
phosphorylation via signal transduction. Notably, phosphorylated
YAP (p-YAP) cannot enter the nucleus, and remains in the cytoplasm
where it cannot regulate transcription (13). Furthermore, it has been demonstrated
that YAP serves an important role in the mediation of the
proliferation, migration, apoptosis and phenotypic transition of
ECs (14) and vascular smooth muscle
cells (15). Additionally, a
previous study found that the Hippo-YAP signaling pathway is
regulated by Ang II signaling, and that its reactivation induces
apoptosis and proliferation in podocytes (16). To the best of our knowledge, the
influence of the YAP signaling pathway on EC proliferation and
injury, and whether YAP expression in ECs is regulated by AT1R, is
yet to be elucidated. Therefore, the purpose of the present study
was to explore AT1R-mediated regulation of YAP by Ang II in ECs.
Therefore, the present study aimed to identify the mechanisms
underlying the YAP-mediated promotion of proliferation, and
suppression of cell injury, in Ang II-treated HAECs. It was
hypothesized that YAP is a key mediator of VEC Ang II-associated
toxicity.
Materials and methods
Cell culture
Human monocytic HAECs (Shanghai Zhong Qiao Xin Zhou
Biotechnology Co., Ltd.) were cultured in RPMI-1640 medium (Thermo
Fisher Scientific, Inc.) supplemented with 10% fetal bovine serum
(Gibco; Thermo Fisher Scientific, Inc.), 2 mM L-Glutamine and 1%
penicillin, streptomycin and amphotericin-B (Sigma-Aldrich; Merck
KGaA) at 37°C and 5% CO2. When the cells had adhered,
they were transferred to phorbol myristate acetate-free medium
(Sigma-Aldrich; Merck KGaA) to obtain resting HAECs. Cells
(6×104 cells/well) were then seeded into multi-well
plates with a membrane insert of 0.4-µm pore size (Corning Life
Sciences). Different factors were added to the wells after 24 h
incubation as follows: Control group, HAECs only; group 1, HAECs
with Ang II (1 µM) treatment; group 2, HAECs with Ang II (1 µM) and
small interfering RNA (siRNA) negative control (NC) transfection;
group 3, HAECs with Ang II (1 µM) and AT1R siRNA transfection;
group 4, HAECs with Ang II (1 µM) and 1 µl DMSO; and group 5, HAECs
with Ang II (1 µM) and the ARB 1 µl (20 mM) telmisartan treatment.
After adding the various intervention conditions, the cells were
further cultured for 72 h at 37°C and 5% CO2. The total
incubation time of the cells was 96 h.
Cell Counting Kit (CCK)-8 assay
HAECs were inoculated into multi-well plates
(5×103 cells/well). Cells were inoculated with three
replicate sets per experimental group. Subsequently, CCK-8 was used
according to the manufacturer's instructions. Briefly, CCK-8
solution (IS087, USCN Life Sciences, Inc.) was mixed with
serum-free medium (1:10 v/v; DMEM with High Glucose; Gibco; Thermo
Fisher Scientific, Inc.), and 10 µl of the mixture was added to
each well. Cells were then incubated at 37°C with 5% CO2
for 4 h, after which the optical density at 450 nm was
determined.
ELISA
Endothelin (ET)-1 levels in cell culture supernatant
from each treatment group at the end of the 96 h treatment period
were detected using competitive inhibition ELISA with ET-1 Antibody
ELISA kit (USCN Life Sciences, Inc.), and interleukin (IL)-6 and
matrix metallopeptidase (MMP)-9 in the cell solutions were detected
by double antibody ELISA with IL-6 ELISA Kit (USCN Life Sciences,
Inc.) and MMP9 ELISA Kit (USCN Life Sciences, Inc.), respectively.
ELISAs were performed in accordance with the manufacturer's
protocols.
Western blotting for AT1R and YAP
Cell lysates were prepared using RIPA lysis buffer
(P0013B, Beyotime Institute of Biotechnology) and total protein was
extracted and quantified using a bicinchoninic acid protein
concentration kit (cat. no. p0010; Beyotime Institute of
Biotechnology) according to the manufacturer's protocol. Equal
amounts of proteins (20 µg) were separated via SDS-PAGE (15 g SDS,
15.6 ml 2 M Tris pH 6.8, 57.5 g glycerol, 16.6 ml
β-mercaptoethanol) and transferred to PVDF membranes. The membranes
were blocked with 5% BSA (16000-044, Gibco; Thermo Fisher
Scientific, Inc.) in Tris-buffered saline with 20% Tween-20 for 2 h
at room temperature. Membranes were subsequently incubated
overnight at 4°C with the following primary antibodies: Anti-YAP
(cat. no. AF6328; polyclonal, rabbit anti-human and mouse; 1:1,000;
Affinity Biosciences), anti-p-YAP (Ser127) (cat. no. AF3328;
polyclonal, rabbit anti-human and mouse; 1:1,000; Affinity
Biosciences), anti-AT1R (cat. no. DF4910; polyclonal, rabbit
anti-human and mouse; 1:1,000; Affinity Biosciences), anti-GAPDH
(cat. no. ab9485; polyclonal, rabbit anti-human and mouse; 1:2,500;
Abcam). The membranes were washed three times using PBS, after
which anti-rabbit immunoglobulin G (IgG; H+L; cat. no. BA1054;
polyclonal, goat anti-rabbit; 1:5,000; Boster Biological
Technology) was added and the membranes were incubated at 37°C for
1 h. Bands were detected using a Gel Doc systems (Bio-Rad
Laboratories, Inc). The density of the bands were assessed by
quantitative densitometric analysis using ImageJ (V1.8.0, National
Institutes of Health).
AT1R siRNA transfection
AT1R siRNA was designed and synthesized by Shanghai
GenePharma Co., Ltd. as follows: AT1R-165, GCAUUAAUGCCUCCAUUUATT;
AT1R-880, GCUCAAGCCCUGUCAGAUATT; NC, AAUGUACUCACUACGACUGCG. AT1R
siRNAs were transfected into cells with Lipofectamine®
2000 (Thermo Fisher Scientific, Waltham, MA USA) according to the
manufacturer's protocol. HAECs in a logarithmic growth phase with
good growth status were inoculated into a 6-well plate at
5×105 cells/well, and cultured overnight at 37°C in a 5%
CO2 incubator. At 2 h prior to transfection, they were
mixed with serum-free MEMα medium (Gibco; Thermo Fisher Scientific,
Inc.).
Reverse transcription-quantitative PCR
(RT-qPCR)
After siRNA transfection for 48 h, AT1R relative
mRNA was collected from transfected HAECs as described in the
previous section (AT1R siRNA transfection). RevertAid First Strand
cDNA Synthesis kit (cat. no. K1622, Thermo Fisher Scientific, Inc.)
were used according to the manufacturer's protocol in order to
reverse transcribe 2 µg RNA into first-strand cDNA. FastStart
Universal SYBR Green Master (Rox; 04913914001, Hoffmann-La Roche,
Ltd.) was then used to perform quantitative PCR as follows: 2 min
at 50°C, 10 min at 95°C, and then 40 cycles of 15 sec at 95°C and
60 sec at 60°C. Amplification was performed on a PCR System 7300
(Applied Biosystems; Thermo Fisher Scientific, Inc.). The
2−ΔΔCq method was used to analyze the results (17). In addition, β-actin was used as an
internal reference gene to account for unknown sources of
variation. siRNA-165 was used for subsequent experiments following
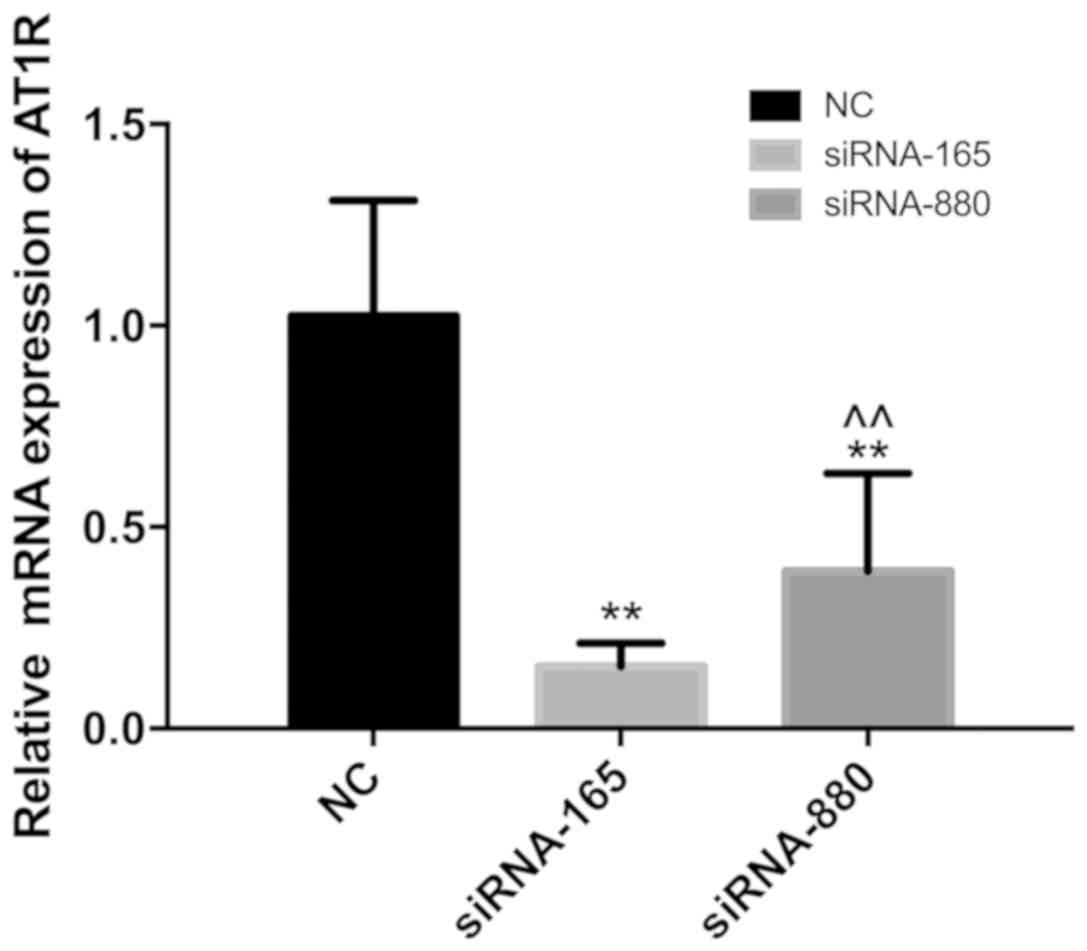
the effectiveness of AT1R siRNA which is exhibited in Fig. 1.
Immunocytochemistry to detect YAP
localization
ECs after 96 h culture were fixed with 3%
formaldehyde solution for 10–25 min at room temperature after
washing with PBS. Fixed cells were washed three times with PBS,
then permeabilized with 1% Triton X-100 (Beyotime Institute of
Biotechnology), incubated for 5–10 min at room temperature, and
rewashed three times with PBS. Each sample site and 3% BSA blocking
solution was sealed at room temperature for 30 min. YAP antibody
(AF6328, Affinity Biosciences) was diluted 1:500, and membranes
were blocked overnight in a humid chamber at 4°C and washed three
times with PBS. The fluorescent CY3 goat anti-rabbit IgG
(rhodamine-labeled; cat. no. BA1036; Boster Biological Technology)
and 4′,6-diamidino-2-phenylindole were added to membranes (both
diluted 1:500) for 30–60 min at room temperature in the dark,
followed by washing three times with PBS. Slides were enclosed with
fluorescence-quench mount and observed using a confocal laser
scanning microscope (×1,000 magnification).
Statistical analysis
Statistical analysis was performed using SPSS
software (v19.0; IBM Corp.) to evaluate differences among groups.
Comparisons among >2 groups were performed using one-way ANOVA
followed by Tukey's post-hoc test. Data are presented as mean ±
standard deviation, and P<0.05 was considered to indicate a
statistically significant difference. All experiments were repeated
in triplicates.
Results
Knockdown of siRNA-165 is more
efficient compared with siRNA-880
The knockdown efficiency of siRNA-165 and siRNA-880
was determined using quantitative PCR (Fig. 1). The results indicated that
siRNA-165 knockdown efficiency was more stable and the effect was
more notable, and it was consequently selected for subsequent
experimentation.
Ang II reduces VEC proliferation and
triggers an inflammatory response, which is suppressed by treatment
with ARB and AT1R siRNA
Cell proliferation was investigated using a CCK-8
assay (Fig. 2A). Compared with the
control group, cell proliferation was significantly decreased
following treatment with Ang II (1 µM; 24 h; P<0.05).
Additionally, compared with groups 1 and 2 (Ang II treatment with
or without NC siRNA), cell proliferation was significantly
increased following transfection with AT1R siRNA after culturing
for 48 h (group 3; P<0.05), and also increased following
treatment with telmisartan after culturing for 48 h (group 4;
P<0.05). This meant that the inhibition of the proliferative
activity of ECs by Ang II is achieved by regulating AT1R. Based on
the present results, it was speculated that Ang II induced an
inflammatory reaction in HAECs. Since Ang II is known to upregulate
ET-1, proinflammatory chemokines, such as interleukin-6 (IL-6), and
MMP-9 to promote endothelial inflammation (18,19), the
expression levels of these factors were measured in supernatants
from each group (Fig. 2B-D).
Circulating levels of IL-6 and ET-1 were all significantly higher
in groups 1, 2 and 4 than in the control group and groups 3 and 5
(P<0.05). Group 5 and group 3 had a decrease in MMP 9
circulating levels compared with group 4 and group 2, respectively.
The current findings indicated that Ang II promoted endothelial
inflammation and that transfection with ARB and AT1R siRNA may
alleviate these proinflammatory effects.
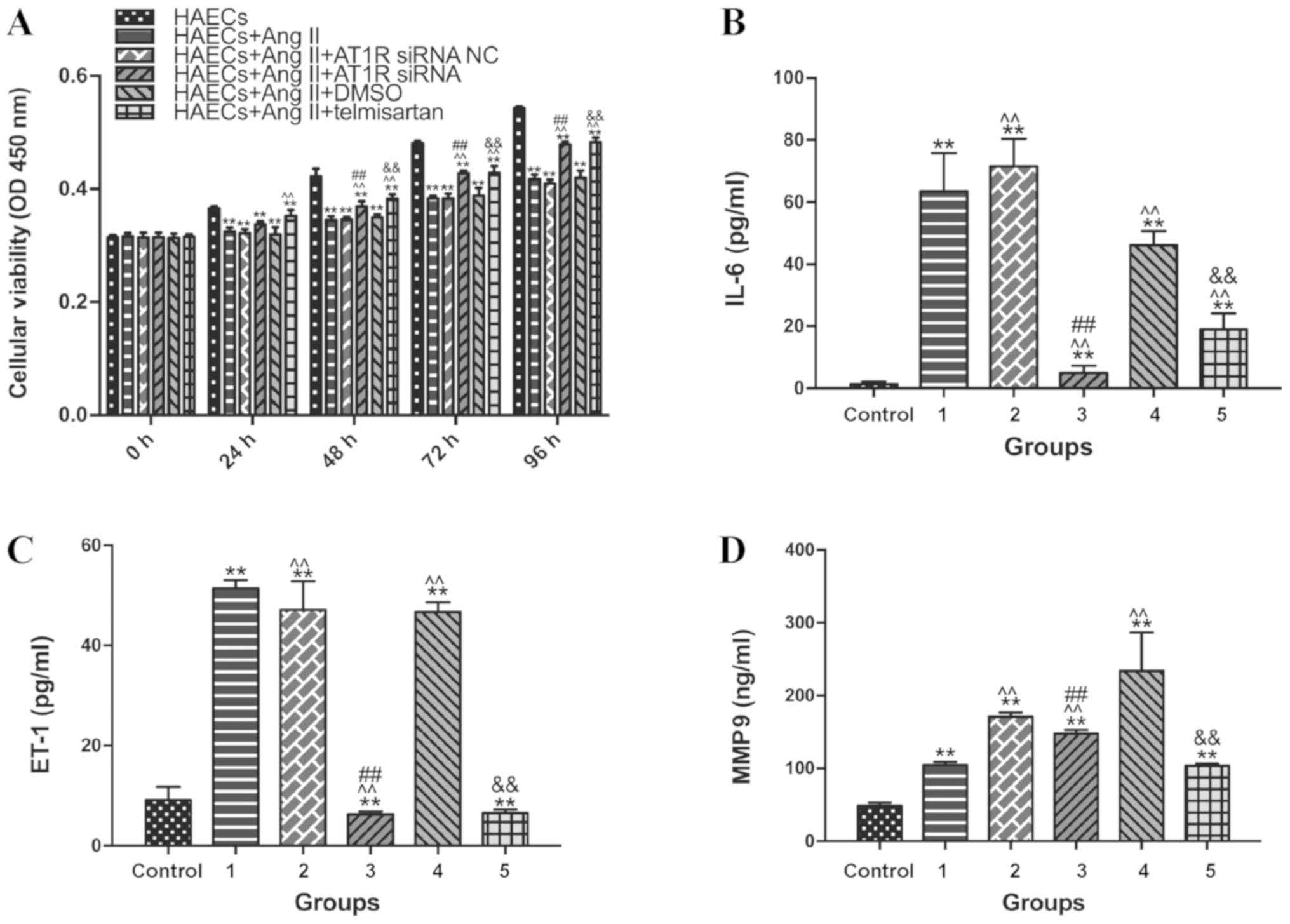 | Figure 2.Ang II inhibits cell proliferation and
promotes inflammation in HAECs, and telmisartan counteracts this
effect. Grouping: Control group, HAECs only; group 1, HAECs with
Ang II (1 µM) treatment; group 2, HAECs with Ang II (1 µM) and
siRNA NC transfection; group 3, HAECs with Ang II (1 µM) and AT1R
siRNA transfection; group 4, HAECs with Ang II (1 µM) and 1 µl
DMSO; and group 5, HAECs with Ang II (1 µM) and the ARB 1 µl (20
mM) telmisartan treatment. (A) CCK-8 detection of cell
proliferation in HAECs at 0, 24, 48, 72 and 96 h. After 96 h, Ang
II treatment (1 µM) significantly decreased the proliferative
activity of HAECs (P<0.05), and this decrease was significantly
reduced by treatment with telmisartan (20 µM). (B) Detection of
IL-6 in supernatants from different samples by ELISA after 96 h
culture. (C) Detection of ET-1 in supernatants from different
samples by ELISA after 96 h culture. (D) Detection of MMP-9 in
supernatants from different samples using ELISA after 96h culture.
Ang II treatment (1 µM) significantly increased the concentrations
of IL-6, ET-1 and MMP9 in the supernatants compared with the
control group (which was not treated with Ang II). IL-6, ET-1 and
MMP-9 levels were significantly lower in the group 5 compared with
the group 4. **P<0.05 compared with the group control;
^^P<0.05 compared with the group 1;
##P<0.05 compared with the group 4;
&&P<0.05 compared with the group 2. siRNA,
small interfering RNA; NC, negative control; AT1R, angiotensin type
1 receptor; Ang II, angiotensin II; HAEC, human aortic endothelial
cell; ET-1, endothelin-1; IL-6, interleukin-6; MMP9, matrix
metalloproteinase 9. |
Ang II decreases the YAP/p-YAP ratio
in VECs; however, treatment with ARB reverses this effect
To identify alterations in YAP and AT1R expression
in each group, western blotting was performed to detect AT1R, YAP
and p-YAP expression in HAECs from each group (Fig. 3A). Compared with the control group,
YAP expression was decreased in HAECs from groups 1, 2 and 4 which
were treated with Ang II (Fig. 3C),
whereas AT1R (Fig. 3B) and p-YAP
(Fig. 3D) expression was
significantly increased in these groups (P<0.05). In groups 3
(transfection with AT1R siRNA) and 5 (treatment with telmisartan),
these expression changes were alleviated to a certain extent
(Fig. 3B-D). This indicated that Ang
II promoted YAP phosphorylation, making it unable to enter the
nucleus.
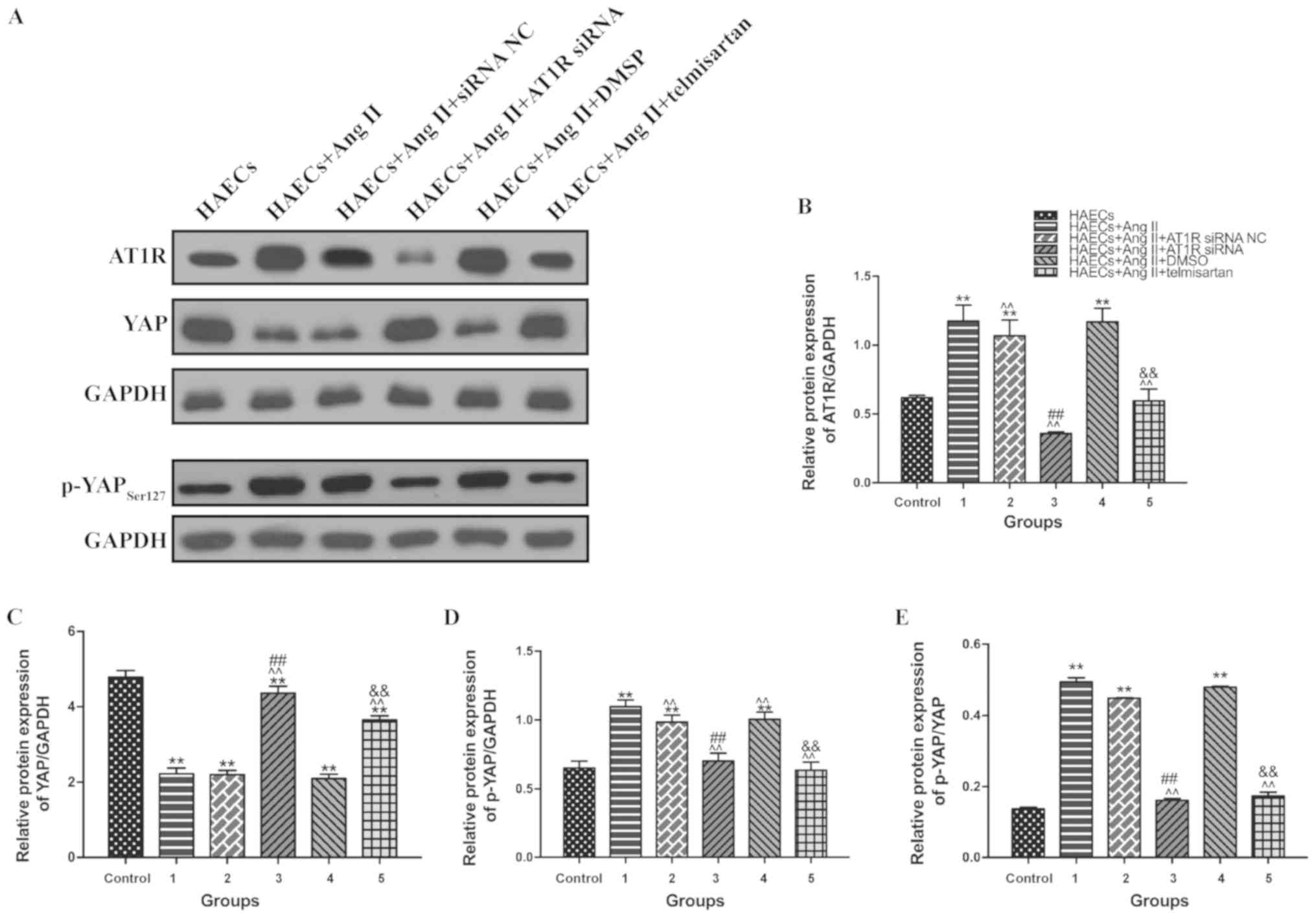 | Figure 3.Ang II promotes AT1R expression and
YAP phosphorylation (Ser127), and treatment with telmisartan or
transfection with AT1R siRNA reverses this effect. Grouping:
Control group, HAECs only; group 1, HAECs with Ang II (1 µM)
treatment; group 2, HAECs with Ang II (1 µM) and siRNA NC
transfection; group 3, HAECs with Ang II (1 µM) and AT1R siRNA
transfection; group 4, HAECs with Ang II (1 µM) and 1 µl DMSO; and
group 5, HAECs with Ang II (1 µM) and the ARB 1 µl (20 mM)
telmisartan treatment. (A) Western blot analysis of sets of six
independent lysates from HAECs that were untreated, treated with
Ang II, treated with Ang II and siRNA NC, treated with Ang II and
AT1R siRNA, treated with Ang II and DMSO, or treated with Ang II
and telmisartan. Treatment with Ang II upregulated AT1R and p-YAP,
and this effect was alleviated by transfection with an AT1R siRNA
or treatment with telmisartan. The ratio between (B) AT1R and
GAPDH; (C) YAP and GAPDH; (D) p-YAP and GAPDH; and (E) p-YAP and
YAP expression is presented. Mean ± SD. **P<0.05 vs. group
control; ^^P<0.05 vs. group 1; ##P<0.05
vs. group 4; &&P<0.05 vs. HAECs + group 2.
siRNA, small interfering RNA; NC, negative control; AT1R,
angiotensin type 1 receptor; Ang II, angiotensin II; HAEC, human
aortic endothelial cell; ET-1, endothelin-1; IL-6, interleukin-6;
MMP9, matrix metalloproteinase 9; YAP, yes-associated protein;
p-YAP, phosphorylated YAP. |
Ang II prevents YAP from entering the
nucleus by binding to AT1R
To elucidate the intracellular localization of YAP,
immunocytochemistry was performed for each treatment group and the
control (Fig. 4A). Following
incubation of HAECs for 72 h under different experimental
conditions, levels of YAP in the nuclei of cells from groups 1, 2,
and 4 were significantly reduced due to the influence of Ang II
(P<0.05). This means that Ang II can prevent YAP from entering
the nucleus. Transfection of AT1R siRNA and treatment with
telmisartan for 72 h effectively alleviated the cytoplasmic
retention of YAP caused by Ang II (Fig.
4B and C; P<0.05). The current immunostaining data supported
the results of western blotting, further indicating that Ang II
promoted YAP phosphorylation via binding to AT1R, thereby
preventing YAP from entering the nucleus.
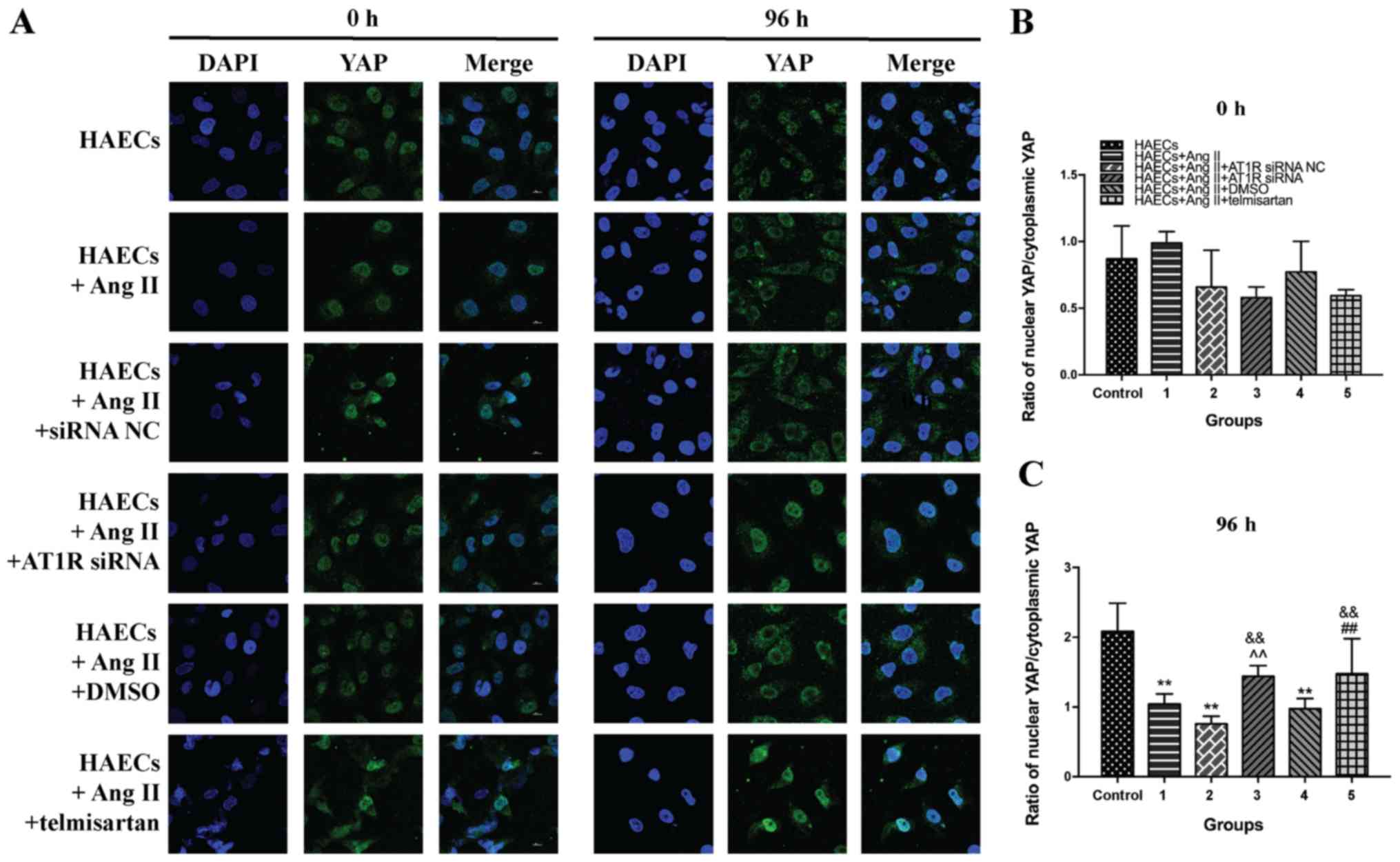 | Figure 4.Ang II prevents YAP from entering the
nucleus by binding to AT1R. Telmisartan and AT1R siRNA attenuated
these effects and promoted YAP into the nucleus. Grouping: Control
group, HAECs only; group 1, HAECs with Ang II (1 µM) treatment;
group 2, HAECs with Ang II (1 µM) and siRNA NC transfection; group
3, HAECs with Ang II (1 µM) and AT1R siRNA transfection; group 4,
HAECs with Ang II (1 µM) and 1 µl DMSO; and group 5, HAECs with Ang
II (1 µM) and the ARB 1 µl (20 mM) telmisartan treatment. (A)
Fluorescence staining of YAP (green) intracellular location of
different groups. Nuclei were labeled with DAPI (blue), and after
96 h of incubation, YAP was concentrated in the cytoplasm under the
influence of Ang II. Telmisartan treatment and AT1R siRNA
transfection caused a large amount of YAP to enter the nucleus. (B)
Ratio between the amount of YAP in the nucleus and the total amount
of intracellular YAP at 0 h. (C) Ratio between the amount of YAP in
the nucleus and the total amount of intracellular YAP at 96 h. Mean
± SD. **P<0.05 vs. the group control; ^^P<0.05 vs. the group
1; ##P<0.05 vs. the group 4;
&&P<0.05 vs. the group 2. siRNA, small
interfering RNA; NC, negative control; AT1R, angiotensin type 1
receptor; Ang II, angiotensin II; HAEC, human aortic endothelial
cell; YAP, yes-associated protein; p-YAP, phosphorylated YAP. |
Discussion
The present study demonstrated that Ang II binding
to AT1R promoted YAP phosphorylation, upregulated YAP cytoplasmic
retention and decreased HAEC proliferation. After quantifying ET1,
MMP-9 and IL-6 expression levels, which are markers of inflammation
and EC injury (20,21), it was revealed that Ang II may
trigger EC inflammation via upregulation of YAP phosphorylation.
Furthermore, it was demonstrated that blocking AT1R reduced the Ang
II-mediated inhibition of proliferation of VECs, downregulating YAP
phosphorylation. YAP is a well-characterized regulator of cell
proliferation (12); hence, the
present results demonstrated that Ang II binding with AT1R promoted
YAP phosphorylation, which resulted in VEC inflammation and
inhibited proliferation. This indicated that the AT1R-YAP signaling
pathway serves an important role in Ang II-mediated intimal
inflammation. However, the specific signaling pathway underlying
this mechanism is yet to be elucidated.
Notably, the present study revealed that the
aforementioned Ang II/AT1R-mediated YAP phosphorylation was
inhibited following treatment with the specific AT1R inhibitor
telmisartan or transfection with an AT1R siRNA, which initiated a
change in YAP intracellular localization from the nucleus to the
cytoplasm. G protein-coupled receptor binding to the G protein
subclass of receptors (Gαq/11) typically activates YAP
(22) and AT1R activates YAP in
HEK293 cells (16); hence, it was
expected that AT1R may have the same effect in VECs. However,
stimulation of AT1R with Ang II resulted in the upregulation of
p-YAP/YAP ratio in VECs. Recent studies have revealed that Ang II
increases EC YAP phosphorylation (23), and that angiotensin-converting enzyme
2 activation attenuates pulmonary vascular remodeling via the
induction of pulmonary arterial cell apoptosis by upregulating YAP
phosphorylation (24). The present
results were consistent with these recent studies because they
indicated that Ang II increased YAP phosphorylation and regulated
VEC activity.
The vascular endothelium has wide-ranging functions
that maintain internal homeostasis. VECs form a monolayer that
serves as a barrier and separates the basal laminae from the
circulation, preventing thrombosis or hemorrhage (6). Additionally, VECs produce and release
vasoactive factors, both relaxative and constrictive, to regulate
vascular tone and blood pressure (25). Elevation of Ang II is associated with
numerous cardiovascular diseases, including pathological
hypertrophy, atherosclerosis, aortic aneurysm, heart failure and
hypertension (26). In the current
study, YAP was revealed to influence the proliferation and
inflammation of Ang II in VECs, which should be accounted for in
future investigations regarding endothelial dysfunction.
It was revealed that Ang II promoted YAP
phosphorylation in VECs via binding to AT1R, leading to the
downregulation of VEC proliferation and VEC-mediated inflammation.
Therefore, it may be the case that Ang II damages ECs by regulating
YAP. An investigation into the hypothesis that enhancing YAP
dephosphorylation in VECs will help prevent vascular intimal injury
in vivo, should be conducted.
AD is one of the most severe cardiovascular diseases
(27), but its pathogenesis remains
to be elucidated. Medical treatment for AD primarily consists of
controlling blood pressure, but there are no clear guidelines for
clinical drug use (4). Therefore,
investigation of AD pathogenesis and the generation of clinical
recommendations may confer great benefit on patients. In the
present study, it was discovered that ARB binds to AT1R on the
surface of VECs, thereby regulating downstream YAP and reducing the
proinflammatory effects of VECs. The present results suggest that
treatment with ARB may reduce VEC inflammation and increase their
proliferation. Notably, the effect of Ang II on endothelial cells
may contribute to AD pathogenesis because the primary difference
between AD and aortic aneurysm is the formation of intimal tears.
Multiple studies have combined AD and aortic aneurysm when studying
disease mechanism (28,29); however, the results of the present
study indicate that this may be inappropriate. Perhaps the two
diseases share certain pathological mechanisms, but understanding
the mechanism underlying intimal ruptures is key to elucidating the
pathogenesis of AD. Therefore, in the present study, certain
preliminary explorations into the effect of Ang II on endometrial
cells were conducted; in order provide guidance for future animal
experiments on the formation of intimal tears in patients with
AD.
The present study had some limitations. First, all
experiments were conducted in vitro, so in vivo
studies are required to further validate the findings. Second, the
effects of YAP overexpression and inhibition on VEC proliferation
and inflammation were not investigated due to technical reasons and
financial constraints. Further experiments to verify the regulatory
role of YAP in VEC proliferation and inflammation should be
conducted.
Overall, the present study revealed the integrated
roles of AT1R and YAP in regulating VEC proliferation and
inflammation in vascular intimal injury and provided an important
reference for future research concerning AD formation. The role of
YAP in VECs is an important focus for further study, and may
represent a promising target for future pharmacological
intervention in vascular intimal injury.
Acknowledgements
The authors would like to thank Dr Sarah Williams
for editing the English text for a draft of this manuscript.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
XW, HZ and WG conceived and designed the study. YG,
JL and DR analyzed and interpreted the data. XW drafted the
manuscript. WG critically revised the manuscript. XW, LC, YH, GS
and SJ performed cell culture and experimental tests. All authors
read and approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
AD
|
aortic dissection
|
|
Ang II
|
angiotensin II
|
|
AT1R
|
angiotensin type 1 receptor
|
|
YAP
|
yes-associated protein
|
|
EC
|
endothelial cell
|
|
ARB
|
angiotensin receptor blocker
|
|
HAEC
|
human aortic endothelial cell
|
|
CCK-8
|
Cell Counting Kit-8
|
|
ET-1
|
endothelin-1
|
|
IL-6
|
interleukin-6
|
|
MMP9
|
matrix metalloproteinase 9
|
References
|
1
|
Hiratzka LF, Bakris GL, Beckman JA, Bersin
RM, Carr VF, Casey DE Jr, Eagle KA, Hermann LK, Isselbacher EM,
Kazerooni EA, et al: 2010 2010
ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM guidelines for the
diagnosis and management of patients with thoracic aortic disease.
A report of the American college of cardiology foundation/American
heart association task force on practice guidelines, American
association for thoracic surgery, American college of radiology,
American stroke association, society of cardiovascular
anesthesiologists, society for cardiovascular angiography and
interventions, society of interventional radiology, society of
thoracic surgeons, and society for vascular medicine. J Am Coll
Cardiol. 55:e27–e129. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kochanek KD, Xu J, Murphy SL, Miniño AM
and Kung HC: Deaths: Final data for 2009. Natl Vital Stat Rep.
60:1–116. 2011.PubMed/NCBI
|
|
3
|
Olsson C, Thelin S, Ståhle E, Ekbom A and
Granath F: Thoracic aortic aneurysm and dissection: Increasing
prevalence and improved outcomes reported in a nationwide
population-based study of more than 14,000 cases from 1987 to 2002.
Circulation. 114:2611–2618. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Erbel R, Aboyans V, Boileau C, Bossone E,
Bartolomeo RD, Eggebrecht H, Evangelista A, Falk V, Frank H,
Gaemperli O, et al: 2014 ESC guidelines on the diagnosis and
treatment of aortic diseases: Document covering acute and chronic
aortic diseases of the thoracic and abdominal aorta of the adult.
The task force for the diagnosis and treatment of aortic diseases
of the European society of cardiology (ESC). Eur Heart J.
35:2873–926. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chen K, Varon J, Wenker OC, Judge DK,
Fromm RE Jr and Sternbach G: Acute thoracic aortic dissection: The
basics. J Emerg Med. 15:859–867. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Davignon J and Ganz P: Role of endothelial
dysfunction in atherosclerosis. Circulation. 109 (Suppl
1):III27–III32. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Han L, Dai L, Zhao YF, Li HY, Liu O, Lan
F, Jiang WJ and Zhang HJ: CD40L promotes development of acute
aortic dissection via induction of inflammation and impairment of
endothelial cell function. Aging (Albany NY). 10:371–385. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Schlüter KD and Wenzel S: Angiotensin II:
A hormone involved in and contributing to pro-hypertrophic cardiac
networks and target of anti-hypertrophic cross-talks. Pharmacol
Ther. 119:311–325. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Dimmeler S, Rippmann V, Weiland U,
Haendeler J and Zeiher AM: Angiotensin II induces apoptosis of
human endothelial cells. Protective effect of nitric oxide. Circ
Res. 81:970–976. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Daugherty A, Manning MW and Cassis LA:
Angiotensin II promotes atherosclerotic lesions and aneurysms in
apolipoprotein E-deficient mice. J Clin Invest. 105:1605–1612.
2000. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sata M and Fukuda D: Crucial role of
renin-angiotensin system in the pathogenesis of atherosclerosis. J
Med Invest. 57:12–25. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Valis K, Prochazka L, Boura E, Chladova J,
Obsil T, Rohlena J, Truksa J, Dong LF, Ralph SJ and Neuzil J:
Hippo/Mst1 stimulates transcription of the proapoptotic mediator
NOXA in a FoxO1-dependent manner. Cancer Res. 71:946–954. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ren A, Yan G, You B and Sun J:
Down-regulation of mammalian sterile 20-like kinase 1 by heat shock
protein 70 mediates cisplatin resistance in prostate cancer cells.
Cancer Res. 68:2266–2274. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hu J, Liu T, Zhang Z, Xu Y and Zhu F:
Oxidized low-density lipoprotein promotes vascular endothelial cell
dysfunction by stimulating miR-496 expression and inhibiting the
Hippo pathway effector YAP. Cell Biol Int. 43:528–538. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Feng X, Liu P, Zhou X, Li MT, Li FL, Wang
Z, Meng Z, Sun YP, Yu Y, Xiong Y, et al: Thromboxane A2 activates
YAP/TAZ protein to induce vascular smooth muscle cell proliferation
and migration. J Biol Chem. 291:18947–18958. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wennmann DO, Vollenbröker B, Eckart AK,
Bonse J, Erdmann F, Wolters DA, Schenk LK, Schulze U, Kremerskothen
J, Weide T and Pavenstädt H: The Hippo pathway is controlled by
Angiotensin II signaling and its reactivation induces apoptosis in
podocytes. Cell Death Dis. 5:e15192014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Pacurari M, Kafoury R, Tchounwou PB and
Ndebele K: The Renin-Angiotensin-aldosterone system in vascular
inflammation and remodeling. Int J Inflam. 2014:6893602014.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Nurden AT: Platelets, inflammation and
tissue regeneration. Thromb Haemost. 105 (Suppl 1):S13–S33. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wen D, Zhou XL, Li JJ and Hui RT:
Biomarkers in aortic dissection. Clin Chim Acta. 412:688–695. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tsai MH, Wu CH, Lin WN, Cheng CY, Chuang
CC, Chang KT, Jiang RS, Hsu JF and Lee IT: Infection with
Staphylococcus aureus elicits COX-2/PGE2/IL-6/MMP-9-dependent aorta
inflammation via the inhibition of intracellular ROS production.
Biomed Pharmacother. 107:889–900. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yu FX, Zhao B, Panupinthu N, Jewell JL,
Lian I, Wang LH, Zhao J, Yuan H, Tumaneng K, Li H, et al:
Regulation of the Hippo-YAP pathway by G-protein-coupled receptor
signaling. Cell. 150:780–791. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Fu Y, Sun S, Sun H, Peng J, Ma X, Bao L,
Ji R, Luo C, Gao C, Zhang X and Jin Y: Scutellarin exerts
protective effects against atherosclerosis in rats by regulating
the Hippo-FOXO3A and PI3K/AKT signaling pathways. J Cell Physiol.
234:18131–18145. 2019.PubMed/NCBI
|
|
24
|
Yan D, Li G, Zhang Y and Liu Y:
Angiotensin-converting enzyme 2 activation suppresses pulmonary
vascular remodeling by inducing apoptosis through the Hippo
signaling pathway in rats with pulmonary arterial hypertension.
Clin Exp Hypertens. 41:589–598. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Shao J, Weng X, Zhuo L, Yu L, Li Z, Shen
K, Xu W, Fang M and Xu Y: Angiotensin II induced CSF1 transcription
is mediated by a crosstalk between different epigenetic factors in
vascular endothelial cells. Biochim Biophys Acta Gene Regul Mech.
1862:1–11. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Saito Y and Berk BC: Transactivation: A
novel signaling pathway from angiotensin II to tyrosine kinase
receptors. J Mol Cell Cardiol. 33:3–7. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Nienaber CA, Rousseau H, Eggebrecht H,
Kische S, Fattori R, Rehders TC, Kundt G, Scheinert D, Czerny M,
Kleinfeldt T, et al: Randomized comparison of strategies for type B
aortic dissection: The iNvestigation of STEnt grafts in aortic
dissection INSTEAD) trial. Circulation. 120:2519–2528. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang S, Liu Y, Zhao G, He L, Fu Y, Yu C,
Wang Z, Zhao T, Cao F, Gao Y, et al: Postnatal deficiency of
ADAMTS1 ameliorates thoracic aortic aneurysm and dissection in
mice. Exp Physiol. 103:1717–1731. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wang Y, Yin P, Chen YH, Yu YS, Ye WX,
Huang HY, Ji ZC and Shen ZY: A functional variant of SMAD4 enhances
macrophage recruitment and inflammatory response via TGF-β signal
activation in Thoracic aortic aneurysm and dissection. Aging
(Albany NY). 10:3683–3701. 2018. View Article : Google Scholar : PubMed/NCBI
|