Introduction
Non-small cell lung cancer (NSCLC) includes squamous
cell carcinoma, adenocarcinoma and large-cell carcinoma and
accounts for >80% of all lung cancer cases globally (1). Despite various types of treatments
developed for NSCLC in the past decades, the 5-year survival rate
remains unsatisfactory at only ~18% (2). Metastasis remains a leading cause of
NSCLC-associated mortality (3). A
number of metastasis regulators have been identified in NSCLC. For
instance, WNT/TCF signaling was reported to regulate NSCLC
metastasis through lymphoid enhancer binding factor 1 and HOXB9
(4). Jin et al (5) also indicated that LKB1 is involved in
regulating lung cancer metastasis. However, the mechanisms
underlying NSCLC metastasis remain to be further investigated
(6).
TOR signaling pathway regulator (TIPRL), the
mammalian ortholog of the yeast protein Tip41, is a type-2A
phosphatase regulatory protein (7).
Mechanistically, TIPRL may bind to protein phosphatase (PP)2A,
PP4R2, PP4R3 and PP6 (7,8). Previous studies have demonstrated that
through the interaction between TIPRL and the PP4 complex, γ-H2AX
becomes dephosphorylated to promote cell death (7), and that the interaction between TIPRL
and PP2A leads to the activation of mTORC1-signaling activator
(8). γ-H2AX is a DNA damage response
marker that may serve as a prognostic biomarker for cancer
(9,10). mTOR signaling has crucial roles in
cancer progression and has been reported to be involved in
regulating tumor growth (11),
metastasis (12), autophagy
(13), radioresistance (14) and chemoresistance (15). These studies indicated the important
roles of TIPRL in human cancers. A recent study demonstrated that
TIPRL was upregulated in hepatocellular carcinoma, while its
knockdown induced cancer cell apoptosis (16). However, the molecular functions of
TIPRL in NSCLC remain to be further investigated.
The present study focused on investigating the
prognostic value and functional roles of TIPRL in NSCLC. The
expression of TIPRL in NSCLC samples was assessed and the
association between TIPRL expression and survival time was
determined. Loss-of-function assays were performed to investigate
the influence of TIPRL on NSCLC migration and invasion. The present
results suggest that TIPRL may serve as a biomarker for the
prognosis of patients with NSCLC and also as a therapeutic
target.
Materials and methods
Datasets
The present study evaluated the expression levels of
TIPRL in NSCLC samples using The Cancer Genome Atlas (TCGA) dataset
(no. GSE27262), which was downloaded from the TCGA data portal
(https://tcga-data.nci.nih.gov). The TCGA
data subset for lung adenocarcinoma (LUAD) included 59 normal
samples and 517 LUAD samples. Students t-test was used to determine
statistical significance between normal and LUAD samples. P<0.05
was considered to indicate a statistically significant difference.
The clinical information used in the study was downloaded from
cBioPortal database (https://www.cbioportal.org/), which was uploaded as
Table SI.
Cell culture
The NSCLC cell line A549 was purchased from the Cell
Bank of the Type Culture Collection of the Chinese Academy of
Sciences and cultured in RPMI-1640 medium (HyClone; GE Healthcare
Life Sciences) supplemented with 10% fetal bovine serum (FBS;
Gibco; Thermo Fisher Scientific, Inc.), penicillin (100 U/ml) and
streptomycin (100 U/ml). The A549 cells were cultured at 37°C in a
humidified atmosphere of 95% air and 5% CO2.
Construction of TIPRL knockdown
lentivirus
The short hairpin (sh)RNA sequence targeting TIPRL
(5′-CCGGGTGCTGAAGAGTGGCAAGAAACTCGAGTTTCTTGCCACTCTTCAGCACTTTTT-3′)
was obtained from GeneChem, Inc. Recombinant lentiviral vectors
were constructed according to previous studies (17). Concentrated lentiviruses were
transfected at a multiplicity of infection of 40 in serum-free
RPMI-1640 medium. The supernatant was replaced with complete
culture medium (RPMI-1640 medium containing 10% FBS) after 24 h.
The transfection efficiency of shTIPRL was determined using reverse
transcription-quantitative (RT-q)PCR and western blot analysis
after 72 h.
Cell proliferation and flow cytometric
analysis
For cell proliferation assay, an MTT kit was used to
detect the effect of TIPRL on cell proliferation. To dissolve the
formazan crystals, 100 µl dimethyl sulfoxide was added to each well
and the absorbance at 570 nm was measured using a microplate
reader. Data from three independent experiments were analyzed. For
apoptosis assay, 100,000 cells/well A549 cell were seeded into
6-well plates. A total of 48 h later, these cells were collected
and washed twice using PBS. Then the apoptosis was detected using
an Annexin V-APC Apoptosis Detection kit (eBioscience; Thermo
Fisher Scientific, Inc.) by a flow cytometer (BD Biosciences;
Becton, Dickinson and Company). Cell cycle distribution was
analyzed as a typical DNA content histogram using BD CellQuest™
cell cycle analysis software (version 5.1; BD Biosciences).
Western blot analysis
Protein was extracted and western blot analysis was
performed as described previously (18). The PVDF membranes were blocked with
5% skimmed milk at room temperature for 1 h. The antibodies used in
the experiments were as follows: Anti-TIPRL (1:1,000; cat. no.
ab70795; Abcam), anti-GAPDH (1:1,000; cat. no. sc-32233; Santa Cruz
Biotechnology, Inc.), anti-epithelial (E)-cadherin (1:1,000; cat.
no. 3195; Cell Signaling Technology, Inc.), anti-vimentin (1:1,000;
cat. no. 5741; Cell Signaling Technology, Inc.), anti-twist
(1:1,000; cat. no. ab49254; Abcam) and anti-neural (N)-cadherin
(1:1,000; cat. no. 13116; Cell Signaling Technology, Inc.). Goat
anti-mouse IgG-horseradish peroxidase (1:4,000; cat. no. A4416;
Sigma-Aldrich; Merck KGaA) and goat anti-rabbit IgG-horseradish
peroxidase (1:4,000; cat. no. A0545; Sigma-Aldrich; Merck KGaA)
secondary antibodies were used. ECL chemiluminescence reagent
(Santa Cruz Biotechnology, Inc.) was used to visualize the bands.
The grey values of the protein bands were quantified with ImageJ
software (version 1.8; National Institutes of Health).
RT-qPCR
PCR analysis was performed as described previously
(18). Total RNA was extracted from
cells using TRIzol® reagent (Invitrogen; Thermo Fisher
Scientific, Inc.) and complementary (c)DNA was synthesized using
the RevertAid First Strand cDNA Synthesis kit (Promega Corp.). The
reverse transcription conditions were: 25°C for 10 min, 37°C for
120 min and 85°C for 5 min. The miScriptSYBR GreenPCR kit (Qiagen,
Inc.) was used to perform qPCR. The PCR amplification was performed
at 95°C for 30 sec, followed by 40 cycles of denaturation at 95°C
for 5 sec and annealing/extension at 60°C for 30 sec using an ABI
7300 Thermocycler (Applied Biosystems; Thermo Fisher Scientific,
Inc.). The qPCR primers used in the present study were TIPRL
forward, 5′-GGCGTCCAAGACCCACATC-3′ and reverse,
5′-ACAGGCCACTTTAAGCATTCC-3′; and GAPDH forward,
5′-TGACTTCAACAGCGACACCCA-3′, and reverse,
5′-CACCCTGTTGCTGTAGCCAAA-3′.
Wound healing assay
A549 cells were seeded into a 6-well plate until the
cells were confluent and cultured in serum-free medium for 24 h to
obtain a monolayer. A scratch was made using an Oris™ plate. The
cells were rinsed with PBS and incubated with fresh serum-free
medium at 37°C. Images were captured at 0, 24 and 48 h using an
inverted light microscope (TS100; Nikon Corporation) and analyzed
using ImageJ. Each assay was performed in triplicate.
Transwell assay
Transwell assays were performed using Transwell
inserts with an 8-μm pore size. The cell invasion assay was
performed in Transwell plates pre-coated with Matrigel Basement
Membrane Matrix at 37°C for 1 h (coating concentration, 1 mg/ml; BD
Biosciences). After transfection, A549 cells were seeded into the
upper well at 5×104 cells/well in RPMI-1640 medium
containing 1% (fetal bovine serum) FBS. Medium with 10% FBS was
added to the bottom wells of the system. After 3 days of
incubation, the cells on the upper and lower sides of the Transwell
membrane were fixed with 4% paraformaldehyde at 37°C for 30 min and
stained with 0.5% crystal violet for 5 min at room temperature. The
cells on the upper side, which had not transgressed through the
membrane, were gently removed with a cotton swab and the numbers of
invasive cells in 5 random fields of view were counted under a
light microscope (magnification, ×100; Olympus Corporation).
Statistical analysis
All experiments were performed as 3 independent
biological replicates and data are presented as the mean ± standard
deviation. All statistical analyses were performed using SPSS
software version 16.0 (SPSS, Inc.). Students t-test was used to
determine statistical significance between two groups. For multiple
groups, one-way analysis of variance followed by the Newman-Keuls
post hoc test was used. P<0.05 was considered to indicate a
statistically significant difference.
Results
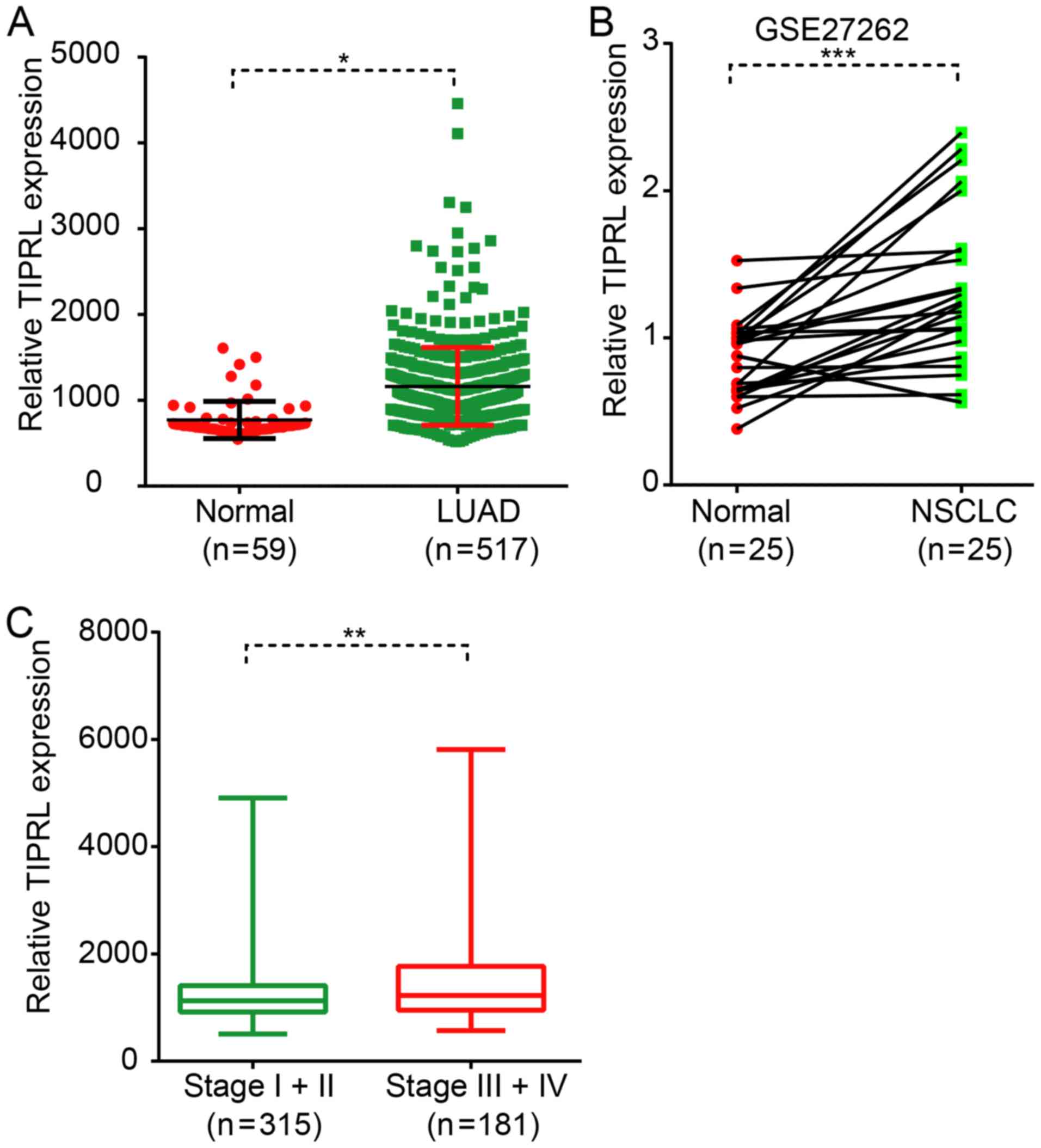
TIPRL is upregulated in NSCLC
The expression pattern of TIPRL in NSCLC has
remained elusive. The present study evaluated the expression levels
of TIPRL in NSCLC and normal lung samples by analyzing a public
dataset, LUAD, which was downloaded from TCGA database. As
presented in Fig. 1, TIPRL was
identified to be significantly overexpressed in LUAD samples
compared with in the matched normal tissues (P<0.05; Fig. 1A). In order to further validate the
present findings, an independent dataset, GSE27262 was analyzed,
which was based on the microarray technology. The results also
showed that TIPRL was significantly overexpressed in NSCLC samples
compared with the normal tissues (P<0.001; Fig. 1B). Of note, higher TIPRL expression
was associated with the advanced stage of NSCLC. The present
results demonstrated that TIPRL was significantly upregulated in
stage-3 and stage-4 LUAD samples compared with that in stage-1 and
stage-2 LUAD samples (P<0.01; Fig.
1C).
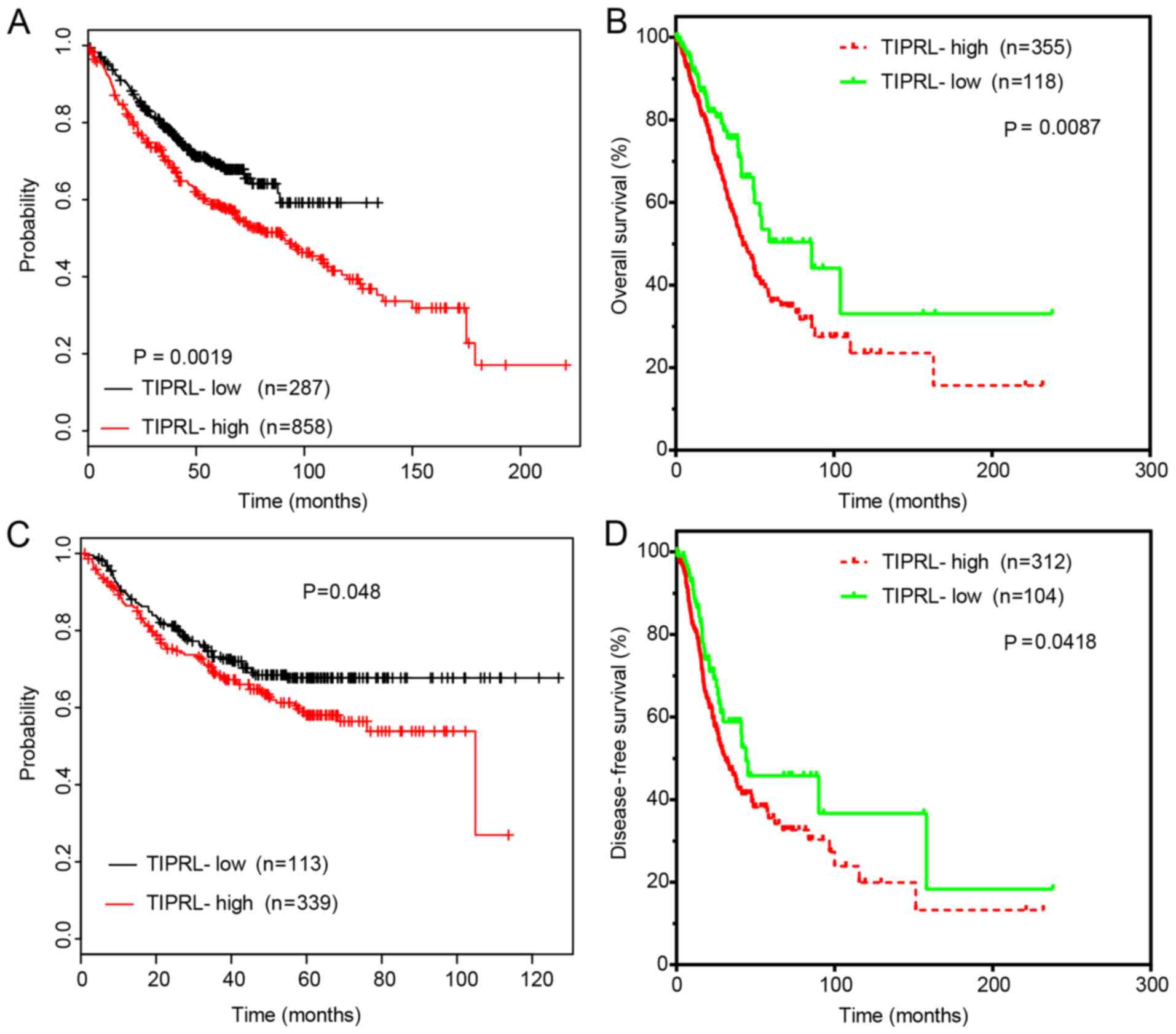
TIPRL is correlated with a shorter
survival time in NSCLC
Kaplan-Meier curve analysis was then performed to
evaluate the prognostic value of TIPRL in NSCLC. The upper quartile
of TIPRL expression in LUAD samples was selected as the cutoff.
First, the Kaplan-Meier Plotter dataset was analyzed. The results
demonstrated that high TIPRL expression in lung cancer samples was
associated with shorter overall survival (OS) (Fig. 2A) and disease-free survival (DFS)
time (Fig. 2C). Analysis of TCGA
datasets indicated that high TIPRL expression levels in LUAD
samples were associated with reduced OS (Fig. 2B) and DFS time (Fig. 2D). These results suggested that
dysregulation of TIPRL may indicate the prognosis of NSCLC.
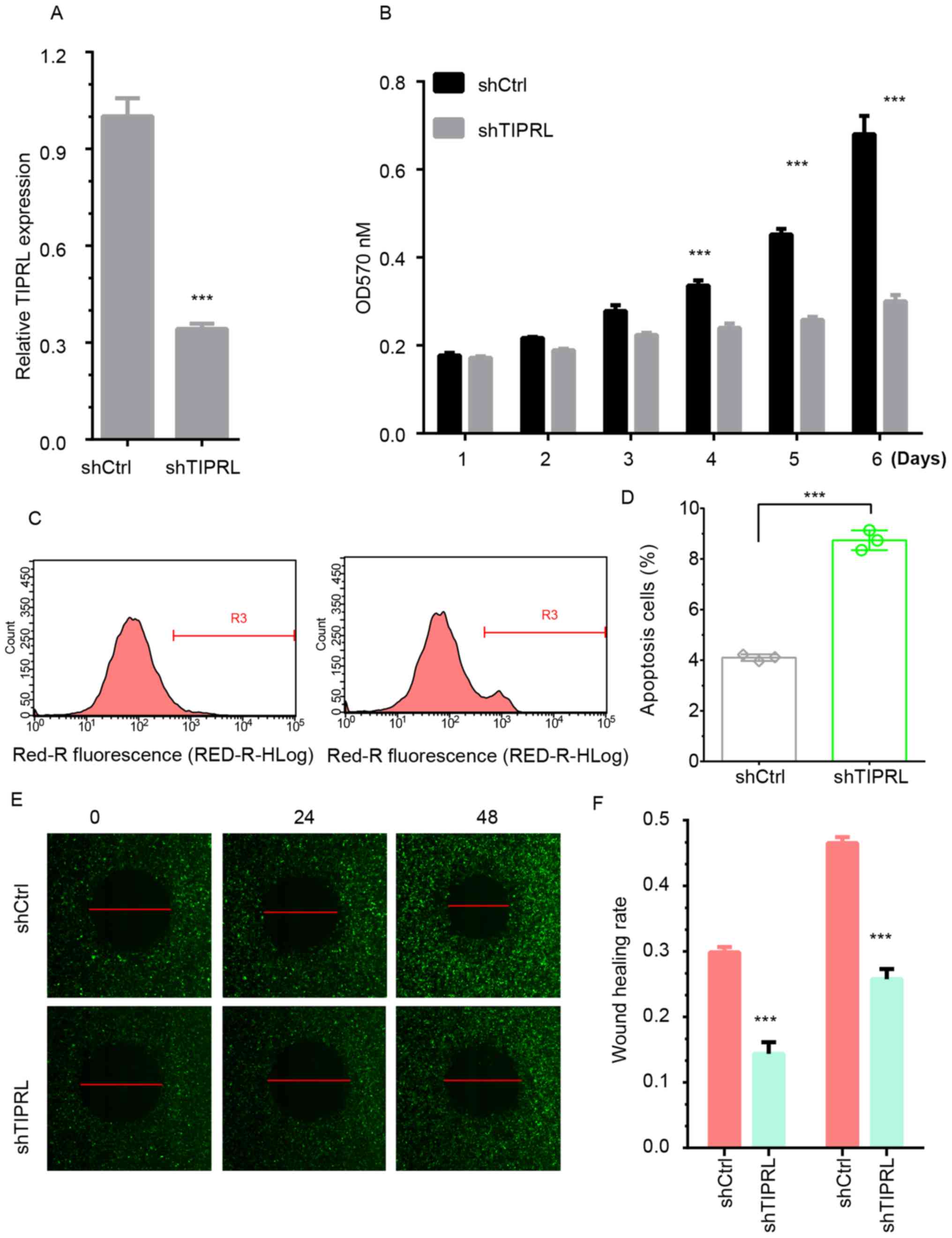
Silencing of TIPRL inhibits NSCLC cell
proliferation
An shRNA was designed to knock down endogenous
expression of TIPRL in NSCLC. As presented in Fig. 3, the mRNA (Fig. 3A) and protein (Fig. 5A) expression of TIPRL was
significantly decreased in A549 cells (P<0.01). By using an MTT
assay, it was found that silencing of TIPRL significantly
suppressed A549 cell proliferation compared with the control group
after 4 days (P<0.001; Fig. 3B).
Flow cytometric analysis showed that silencing of TIPRL induced
A549 cell apoptosis by 2-fold compared with the control group
(Fig. 3C and D). These results
suggested that TIPRL played an important role in regulating cancer
proliferation.
Silencing of TIPRL inhibits NSCLC cell
migration and invasion
A wound healing assay was first performed to detect
the influence of TIPRL on cell metastasis. The repair rate in the
TIPRL knockdown group was significantly suppressed in comparison
with that in the control group (P<0.05). The results
demonstrated that 11 and 23% of the wound area in the TIPRL
knockdown group was repaired by migrating cells after 24 and 48 h,
respectively. However, ~30 and 45% of the wounded area in the
control groups was repaired under the same incubation conditions
(Fig. 3E and F).
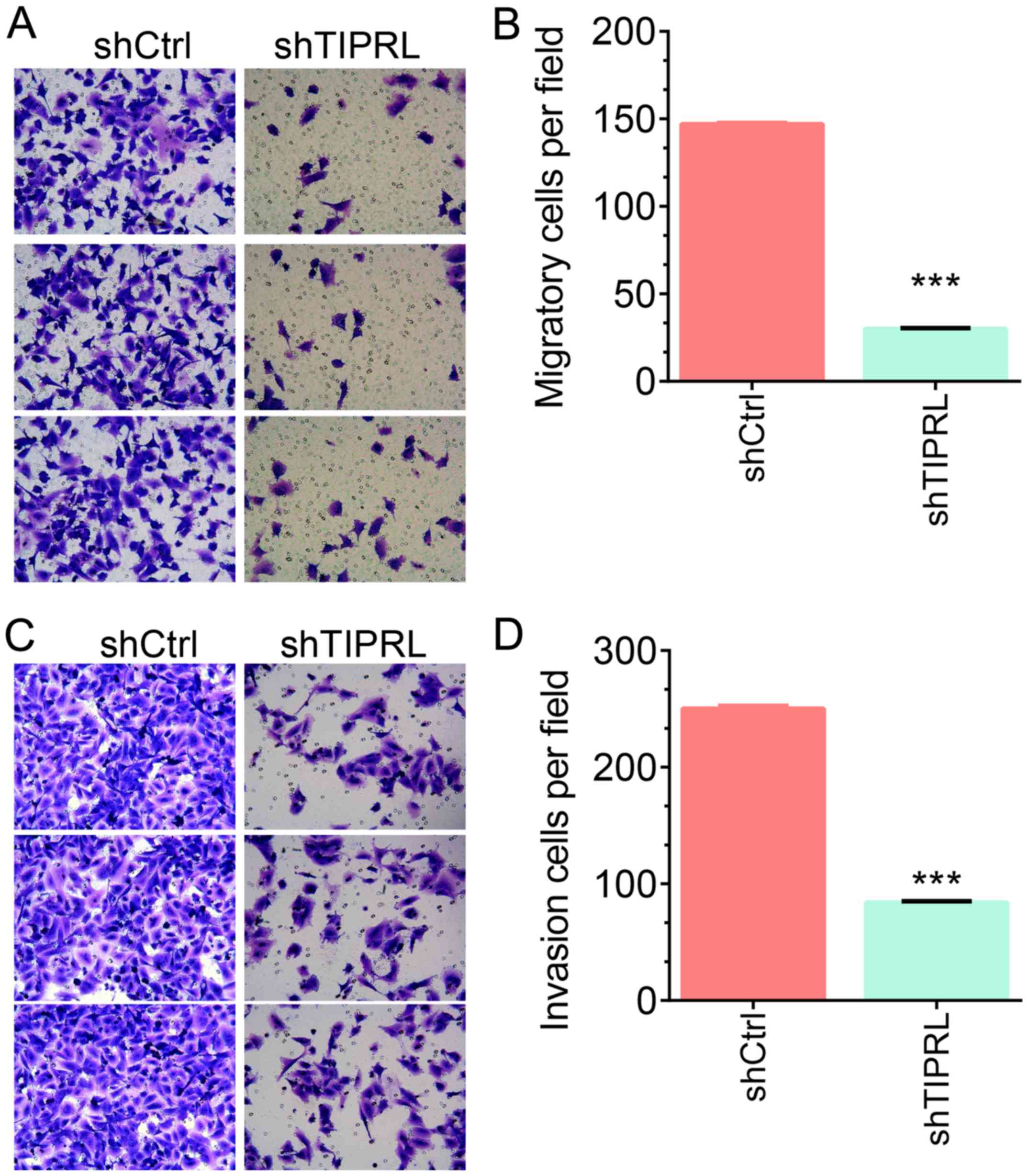
Next, Transwell assays were
performed
It was demonstrated that the numbers of migrating
A549 cells were significantly decreased by 80% in the TIPRL
knockdown group compared with those in the control group
(P<0.001; Fig. 4A and B). In an
invasion assay using Matrigel, it was observed that silencing of
TIPRL significantly inhibited the invasion ability of A549 cells by
70% compared with that in the control groups (P<0.001; Fig. 4C and D). These results indicated that
TIPRL acts as a negative regulator of metastasis in NSCLC.
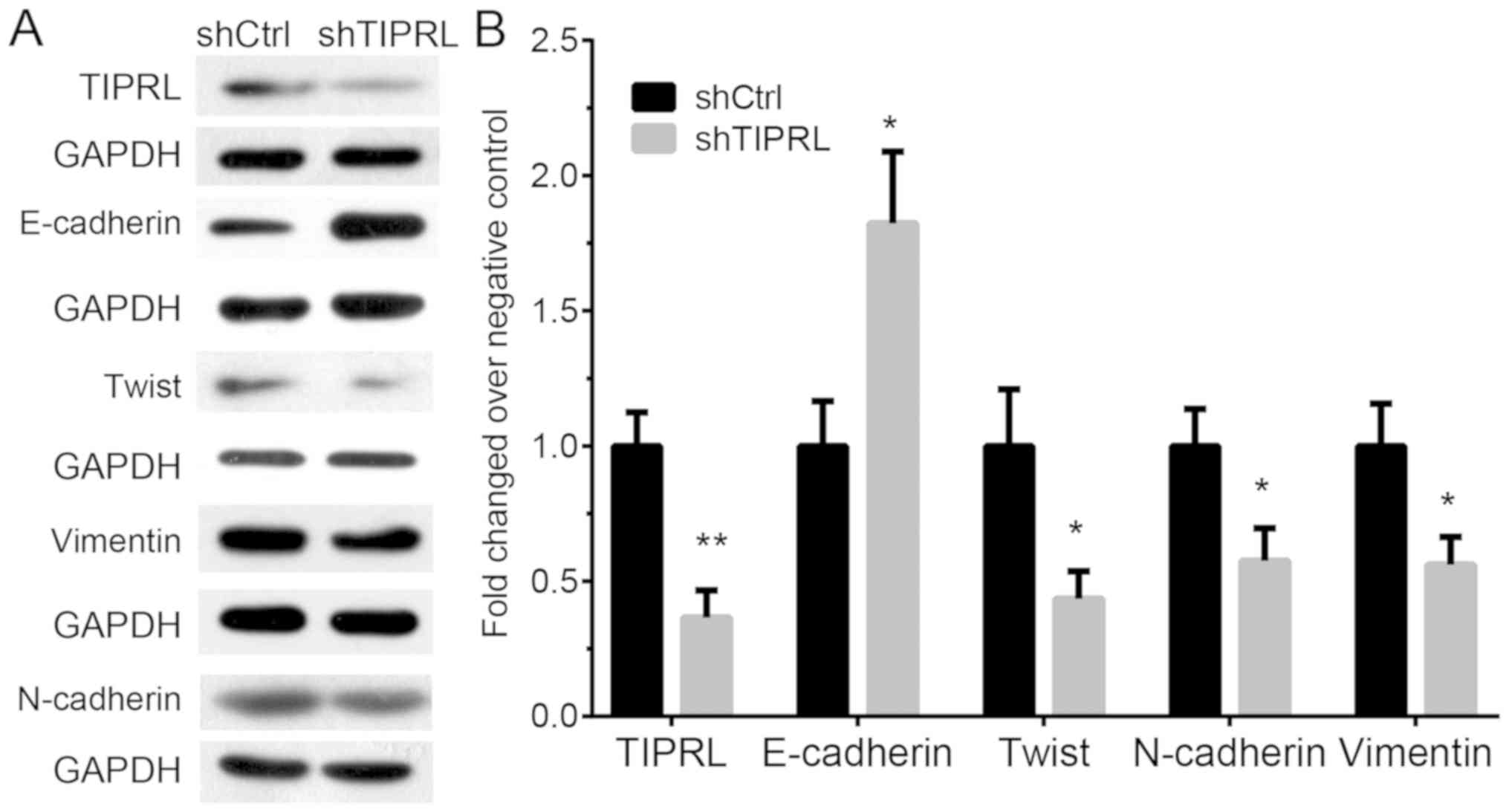
Knockdown of TIPRL inhibits
epithelial-to-mesenchymal transition (EMT) in NSCLC
In order to explore whether TIPRL promoted
metastasis through regulating EMT, the protein levels of
E-cadherin, twist, vimentin and N-cadherin were detected after
TIPRL knockdown in NSCLC. Western blot analysis indicated that
E-cadherin was significantly induced (P<0.05), whereas the
mesenchymal markers twist, vimentin and E-cadherin were
significantly reduced in TIPRL-knockdown A549 cells compared with
those in the control groups (P<0.05; Fig. 5). These results suggested that
knockdown of TIPRL inhibited EMT in NSCLC.
Discussion
Previous studies had demonstrated that TIPRL played
an important role in regulating cancer progression (8,16). TIPRL
acts as an activator of mTORC1 signaling. TIPRL was overexpressed
and served crucial roles in human liver cancer. TIPRL was reported
to be involved in regulating cell apoptosis and cell death through
binding to PP2Ac and PP4. Yoon et al (16) indicated that knockdown of TIPRL in
hepatocellular carcinoma induced tumor-cell apoptosis. Rosales
et al (7) reported that TIPRL
induced cell death in response to genotoxic stress. It has also
been shown that knockdown of TIPRL enhances the apoptosis of
cisplatin-treated cells in lung cancer (19). TIPRL associates with MKK7 and
activates JNK thereby regulating the apoptotic pathway via caspase
3 and 9 release (20). Moreover,
TIPRL acts as an activator of mTORC1 signaling, which is a key
regulator of metabolic pathways and plays a crucial role in
promoting cancer growth (8). The
present study also showed that knockdown of TIPRL suppressed NSCLC
cell proliferation and induced cell apoptosis. To the best of our
knowledge, the present study was the first to report that TIPRL
acts as a metastasis promoter in NSCLC. Knockdown of TIPRL
suppressed NSCLC migration and invasion through regulating EMT
processes. The present study provides insight into a potential
regulatory mechanism involved in NSCLC.
Metastasis is a leading cause of NSCLC-associated
mortality. A series of NSCLC metastasis regulators were identified
in previous studies. For instance, transmembrane protein 106B was
reported to drive lung cancer metastasis through promoting the
synthesis of enlarged vesicular lysosomes (21). A previous study indicated that
GATAD2B promoted KRAS-driven lung cancer metastasis (22). Understanding the detailed mechanisms
underlying NSCLC metastasis may provide novel information to
develop more effective therapeutic strategies. EMT has crucial
roles in NSCLC metastasis (23).
During EMT, the epithelioid cell markers, including E-cadherin, are
downregulated, while interstitial-cell markers, including
N-cadherin and vimentin, are upregulated. Transforming growth
factor-β signaling and Wnt/β-catenin have key roles in the
regulation of EMT processes. In NSCLC, several genes, including
kinesin family member C1 (24), APE1
(25,26) and F-box and WD repeat domain
containing 7 (27), were reported to
participate in the regulation of EMT. The present study
demonstrated that knockdown of TIPRL in A549 cells promoted
E-cadherin expression, whereas suppressed twist and vimentin
expression was observed. Taken together, the present study
demonstrated that TIPRL is a promoter of EMT.
In the past decades, a number of biomarkers,
including carcinoembryonic antigen, cytokeratin-19 fragment and
CYFRA 21-1, were identified in lung cancer (28). However, the sensitivity and
specificity of these markers remain unsatisfactory. To date, the
prognostic value of TIPRL in NSCLC has remained elusive. The
present study indicated that TIPRL was overexpressed in NSLC
tissues. A higher expression of TIPRL was associated with higher
lymphatic metastasis and Tumor-Node-Metastasis stage. Furthermore,
overexpression of TIPRL in NSCLC tissues was associated with
shorter OS and DFS time in NSCLC patients. These results strongly
suggested that TIPRL may serve as a novel biomarker for NSCLC.
In conclusion, the present study suggested that
TIPRL was upregulated and associated with shorter survival time in
NSCLC. Furthermore, silencing of TIPRL inhibited NSCLC migration
and invasion through regulating EMT. The present study presents a
potential novel biomarker for NSCLC, which may be utilized as a
therapeutic target and/or prognostic factor.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors contributions
XX, HZ and GZ designed the experiments. XX, HZ, MY,
EZ, and YZ performed the experiments. XX, JN, RL, ZY and TH
collected and analyzed the data. XX and GZ drafted the manuscript.
All authors approved the manuscript for publication.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Zhao B, Han H, Chen J, Zhang Z, Li S, Fang
F, Zheng Q, Ma Y, Zhang J, Wu N and Yang Y: MicroRNA let-7c
inhibits migration and invasion of human non-small cell lung cancer
by targeting ITGB3 and MAP4K3. Cancer Lett. 342:43–51. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gao L, Qiu H, Liu J, Ma Y, Feng J, Qian L,
Zhang J, Liu Y and Bian T: KLF15 promotes the proliferation and
metastasis of lung adenocarcinoma cells and has potential as a
cancer prognostic marker. Oncotarget. 8:109952–109961. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Waqar SN, Samson PP, Robinson CG, Bradley
J, Devarakonda S, Du L, Govindan R, Gao F, Puri V and Morgensztern
D: Non-small-cell lung cancer with brain metastasis at
presentation. Clin Lung Cancer. 19:e373–e379. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Nguyen DX, Chiang AC, Zhang XH, Kim JY,
Kris MG, Ladanyi M, Gerald WL and Massagué J: WNT/TCF signaling
through LEF1 and HOXB9 mediates lung adenocarcinoma metastasis.
Cell. 138:51–62. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Jin L, Chun J, Pan C, Kumar A, Zhang G, Ha
Y, Li D, Alesi GN, Kang Y, Zhou L, et al: The PLAG1-GDH1 axis
promotes anoikis resistance and tumor metastasis through
CamKK2-AMPK signaling in LKB1-deficient lung cancer. Mol Cell.
69:87–99 e7. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ji H, Ramsey MR, Hayes DN, Fan C, McNamara
K, Kozlowski P, Torrice C, Wu MC, Shimamura T, Perera SA, et al:
LKB1 modulates lung cancer differentiation and metastasis. Nature.
448:807–810. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rosales KR, Reid MA, Yang Y, Tran TQ, Wang
WI, Lowman X, Pan M and Kong M: TIPRL inhibits protein phosphatase
4 activity and promotes H2AX phosphorylation in the DNA damage
response. PLoS One. 10:e01459382015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Nakashima A, Tanimura-Ito K, Oshiro N,
Eguchi S, Miyamoto T, Momonami A, Kamada S, Yonezawa K and Kikkawa
U: A positive role of mammalian Tip41-like protein, TIPRL, in the
amino-acid dependent mTORC1-signaling pathway through interaction
with PP2A. FEBS Lett. 587:2924–2929. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chatzimichail E, Matthaios D, Bouros D,
Karakitsos P, Romanidis K, Kakolyris S, Papashinopoulos G and Rigas
A: γ-H2AX: A novel prognostic marker in a prognosis prediction
model of patients with early operable non-small cell lung cancer.
Int J Genomics. 2014:1602362014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Matthaios D, Hountis P, Karakitsos P,
Bouros D and Kakolyris S: H2AX a promising biomarker for lung
cancer: A review. Cancer Invest. 31:582–599. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Roy D, Sin SH, Lucas A, Venkataramanan R,
Wang L, Eason A, Chavakula V, Hilton IB, Tamburro KM, Damania B and
Dittmer DP: mTOR inhibitors block Kaposi sarcoma growth by
inhibiting essential autocrine growth factors and tumor
angiogenesis. Cancer Res. 73:2235–2246. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhou H and Huang S: Role of mTOR signaling
in tumor cell motility, invasion and metastasis. Curr Protein Pept
Sci. 12:30–42. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Paquette M, El-Houjeiri L and Pause A:
mTOR pathways in cancer and autophagy. Cancers (Basel). 10(pii):
E182018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chang L, Graham PH, Ni J, Hao J, Bucci J,
Cozzi PJ and Li Y: Targeting PI3K/Akt/mTOR signaling pathway in the
treatment of prostate cancer radioresistance. Crit Rev Oncol
Hematol. 96:507–517. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tian L, Zhao Z, Xie L and Zhu J:
MiR-361-5p suppresses chemoresistance of gastric cancer cells by
targeting FOXM1 via the PI3K/Akt/mTOR pathway. Oncotarget.
9:4886–4896. 2017.PubMed/NCBI
|
|
16
|
Yoon JY, Lee JJ, Gu S, Jung ME, Cho HS,
Lim JH, Jun SY, Ahn JH, Min JS, Choi MH, et al: Novel
indazole-based small compounds enhance TRAIL-induced apoptosis by
inhibiting the MKK7-TIPRL interaction in hepatocellular carcinoma.
Oncotarget. 8:112610–112622. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cui F, Hu J, Ning S, Tan J and Tang H:
Overexpression of MCM10 promotes cell proliferation and predicts
poor prognosis in prostate cancer. Prostate. 78:1299–1310. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kroiss A, Vincent S, Decaussin-Petrucci M,
Meugnier E, Viallet J, Ruffion A, Chalmel F, Samarut J and Allioli
N: Androgen-regulated microRNA-135a decreases prostate cancer cell
migration and invasion through downregulating ROCK1 and ROCK2.
Oncogene. 34:2846–2855. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Park JY and Juhnn YS: cAMP signaling
increases histone deacetylase 8 expression via the Epac2-Rap1A-Akt
pathway in H1299 lung cancer cells. Exp Mol Med. 49:e2972017.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Song IS, Jun SY, Na HJ, Kim HT, Jung SY,
Ha GH, Park YH, Long LZ, Yu DY, Kim JM, et al: Inhibition of
MKK7-JNK by the TOR signaling pathway regulator-like protein
contributes to resistance of HCC cells to TRAIL-induced apoptosis.
Gastroenterology. 143:1341–1351. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kundu ST, Grzeskowiak CL, Fradette JJ,
Gibson LA, Rodriguez LB, Creighton CJ, Scott KL and Gibbons DL:
TMEM106B drives lung cancer metastasis by inducing TFEB-dependent
lysosome synthesis and secretion of cathepsins. Nat Commun.
9:27312018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Grzeskowiak CL, Kundu ST, Mo X, Ivanov AA,
Zagorodna O, Lu H, Chapple RH, Tsang YH, Moreno D, Mosqueda M, et
al: In vivo screening identifies GATAD2B as a metastasis driver in
KRAS-driven lung cancer. Nat Commun. 9:27322018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Brody H: Lung cancer. Nature. 513:S12014.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Takeyama Y, Sato M, Horio M, Hase T,
Yoshida K, Yokoyama T, Nakashima H, Hashimoto N, Sekido Y, Gazdar
AF, et al: Knockdown of ZEB1, a master epithelial-to-mesenchymal
transition (EMT) gene, suppresses anchorage-independent cell growth
of lung cancer cells. Cancer Lett. 296:216–224. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Han J, Wang F, Lan Y, Wang J, Nie C, Liang
Y, Song R, Zheng T, Pan S, Pei T, et al: KIFC1 regulated by
miR-532-3p promotes epithelial-to-mesenchymal transition and
metastasis of hepatocellular carcinoma via gankyrin/AKT signaling.
Oncogene. 38:406–420. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yang X, Peng Y, Jiang X, Lu X, Duan W,
Zhang S, Dai N, Shan J, Feng Y, Li X, et al: The regulatory role of
APE1 in epithelial-to-mesenchymal transition and in determining
EGFR-TKI responsiveness in non-small-cell lung cancer. Cancer Med.
7:4406–4419. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wei X, Li Q, Li Y, Duan W, Huang C, Zheng
X, Sun L, Luo J, Wang D, Zhang S, et al: Prediction of survival
prognosis of non-small cell lung cancer by APE1 through regulation
of Epithelial-Mesenchymal Transition. Oncotarget. 7:28523–28539.
2016.PubMed/NCBI
|
|
28
|
Zhang Y, Zhang X, Ye M, Jing P, Xiong J,
Han Z, Kong J, Li M, Lai X, Chang N, et al: FBW7 loss promotes
epithelial-to-mesenchymal transition in non-small cell lung cancer
through the stabilization of Snail protein. Cancer Lett. 419:75–83.
2018. View Article : Google Scholar : PubMed/NCBI
|