Introduction
The last two decades have witnessed a change in the
views towards chemotherapy and carcinogenesis. The discovery that
cancer stem cells can be used to treat solid tumors has changed the
way of thinking with regards to cancer treatment. The self-renewal
capacity of cancer cells remains their most important feature and
function (1–3) as it allows for renewal of cancer cells
and cancer clusters following radiation and chemotherapy treatment
(4).
Salinomycin, a polyketide organic compound extracted
from Streptomyces albus (5,6) and its
derivatives are the first promising compounds that have
demonstrated activity against cancer stem cells (7). They can selectively inhibit seeding,
proliferation and metastasis of breast cancer cells both in
vitro and in vivo (8).
Salinomycin and its derivatives have been reported to possess
anti-cancer properties and they can act against cancer stem cells,
thereby preventing the seeding of previously destroyed cancer cells
(8–10). With a solubility of 17 mg/l,
salinomycin is considered insoluble in water (11). However, such a poor aqueous
solubility can hinder its delivery to the active site. Solubility
is the most important physicochemical property for the successful
delivery of oral drugs. Poor solubility of drug candidates and lead
compounds may cause inefficient absorption at the active site,
resulting in the loss of activity or clinical failure due to poor
pharmacokinetics (12–14). For example, salinomycin is known to
act on the binding site of P-glycoprotein (P-gp), a
well-established plasma membrane protein that is used to facilitate
drug movement out of cells. Binding salinomycin to the P-gp active
site may result in cell sensitivity to drugs (15).
There are a number of methods that improve the
solubility of organic compounds and drugs in order to enhance their
delivery to active sites (16). The
most common methods include using surfactants and co-solvents,
regulating the pH, employing precipitation inhibitors, formation of
complex salts and dissolution in an organic solvent before making a
suspension in water (17). The
majority of these methods pose several drawbacks and may negatively
affect the experiment reproducibility. However, the method of
solubilizing drugs using carriers, such as acids, sugars, polymeric
forms, surfactants and urea has demonstrated better results. The
best carrier is the one in which safety and stability are
confirmed, and it does not interfere with the biological functions
of the drug. Ideally, the carrier must be able to improve drug
solubility and facilitate its delivery to the active site, thereby
improving the activity of the drug. Thus, carriers based on short
peptides or sugars, or a combination of both are considered to be
the best options (18).
To the best of our knowledge, most of the current
biological studies on salinomycin are performed by dissolving it in
dimethyl sulfoxide, and then using the solution to make a
suspension in water (6,9–11,15). In
the present study, a new method was proposed in order to improve
the solubility and enhance the delivery of salinomycin. Several
chemical tools were used in order to improve the delivery of
salinomycin to its active site. However, suspended samples that may
lead to a wide range of precipitation products were not used in the
present study when assessing improved delivery of the compound. In
the present study, the main components of the chemical tools
included a cell-penetrating protein, with or without a sugar
component, and a photo-cleavable linker.
In order to overcome the abundant limitations of the
aforementioned methods and increase the number of tools for
improving the solubility and delivery of a drug to the active site,
a photo-cleavable element, based on a modification of the
salinomycin carboxylic acid, was sought and developed in the
present study (19). Nevertheless,
further work is required to develop novel tools in order to improve
the solubility and facilitate the cell penetration of
salinomycin.
In the present study, salinomycin was conjugated to
the trans-activator of transcription (TAT)-protein, as this
sequence is rich in arginine that forms multiple cationic sites,
under physiological pH (20). The
initial contact between the TAT-protein and the cell surface occurs
via electrostatic binding to the negatively charged
glycosaminoglycans (GAGs) (20). The
design constructed in the present study expected to facilitate cell
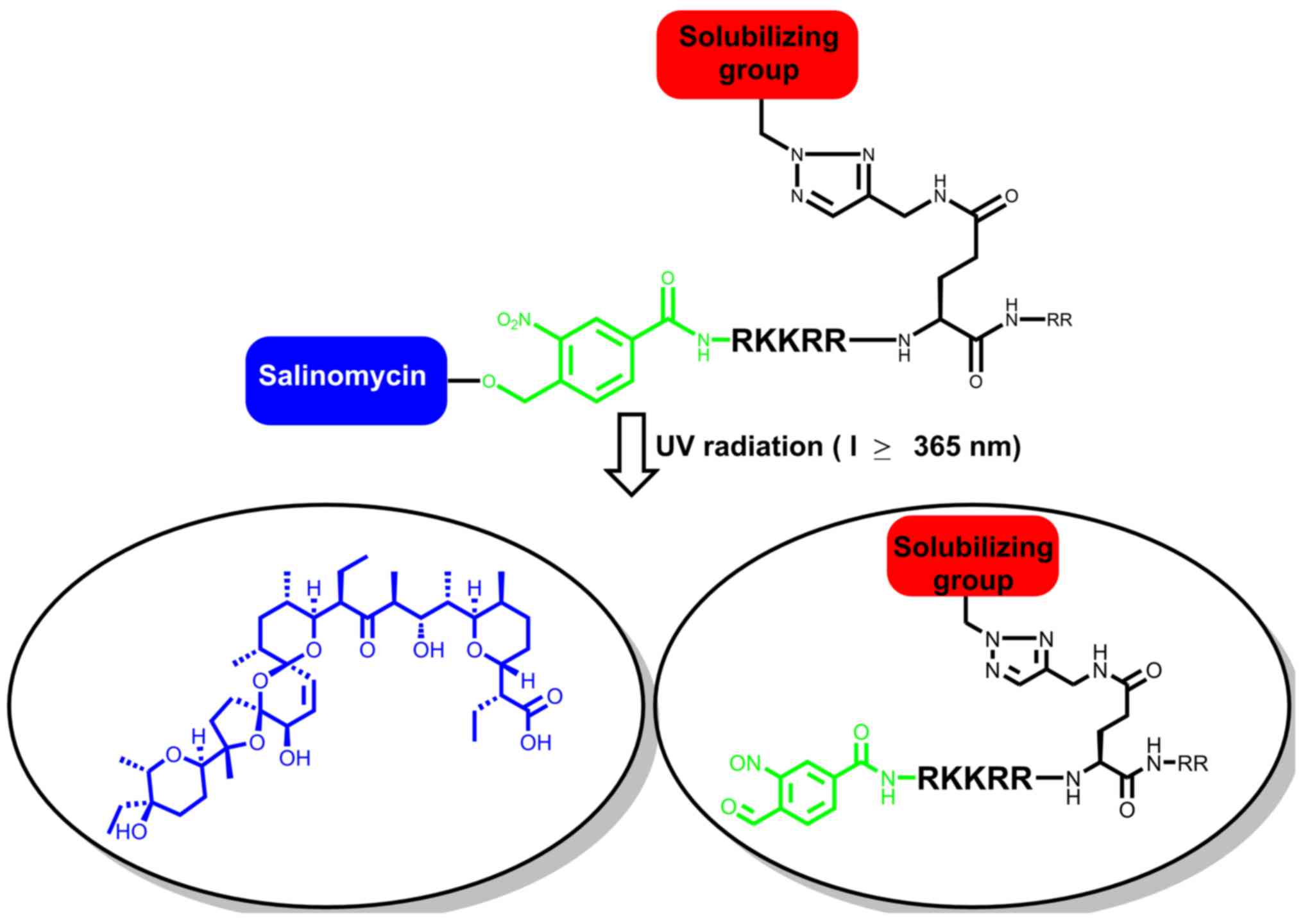
penetration of salinomycin, and is as follows: A novel
photo-cleavable linker-caged salinomycin conjugated to a
TAT-peptide segment (RKKRRQRR). In the system implemented in the
present study, the solubility of the TAT-peptide sequence was
further improved by attaching a sugar moiety to the side chain of
glutamine (Fig. 1). Besides
improving drug solubility, the sugar moiety served as a tool for
penetrating cells (21). The present
study investigated methods to improve the solubility and cell
penetration of salinomycin, in order to improve its activity
against cancer cells. Biological interference was avoided by
introducing a photolinker, 4-(hyroxymethyl)-3-nitro-benzoic acid
(22), between salinomycin and the
N-terminus of the peptide. The chemically stable photolinker, the
sugar moiety and salinomycin were sequentially introduced to the
TAT-protein segment using click chemistry (23,24).
Salinomycin was recovered by mild irradiation at l≥365 nm during
in vitro analysis.
Materials and methods
Preparation of samples and
instruments
Fluorenyl-methyloxycarbonyl (Fmoc)-AA-Wang resin,
(2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexaf
luorophosphate (HBTU) and
(1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium
3-oxid hexafluorophosphate were purchased from Merck KGaA (cat. no.
851006) or AnaSpec (cat. no. AS-21001). All other commercial
reagents, including solvents (HPLC grade or higher), dimethyl
sulfoxide-d6 (6d-DMSO) and chloroform-d
(CDCl3), were purchased from Acros; Sigma Aldrich; Merck
KGaA and VWR; Avantor, and used without further purification. All
solvents used for extraction and chromatography were distilled
prior to use. Anhydrous tetrahydrofuran (THF) (Acros; cat. no.
ACR32697-0010), diethylether (Et2O) (Acros; cat. no.
ACR32686-0010), dichloromethane (DCM) (Merck KGaA; cat. no.
1.06051.1001), methanol (Acros; cat. no. ACR41377-0025) and toluene
(Sigma Aldrich; Merck KGaA; cat. no. 320552-1L) were purified using
a PureSolv filtration system (PureSolv; Sigma Aldrich; Merck KGaA).
Nitromethane (Sigma Aldrich; Merck KGaA; cat. no. 73478-100ML),
potassium hydroxide (Sigma Aldrich; Merck KGaA; cat. no.
P5958-250G), citric acid (Merck KGaA; cat. no. 8.18707.1000),
1,8-Diazabicyclo[5.4.0]undec-7-ene (Sigma Aldrich; Merck KGaA; cat.
no. 139009-100G). Double distilled water was obtained using a
purification system (MilliQ system; Merck Millipore). Glassware was
flame- or oven-dried prior to use. The reactions were followed and
monitored on thin-layer chromatography (TLC) silica gel plates
(Merck KGaA; cat. no. 60F254) and visualized under a short-wave UV
lamp (at 215, 254 and 280 nm), after heating the plates dipped in
ammonium molybdate/cerium (IV) sulfate solution, and/or staining
with a KMnO4 solution (at 100°C for 60 sec). Flash
column chromatography was performed with silica gel (230–400 mesh;
Merck KGaA), using a solvent with a polarity associated with the
TLC mobility. Melting points were measured with an OptiMelt MPA100
melting point apparatus (Stanford Research System; cat. no. MPA100)
and the values were used without correction. Routine electro-spray
spectra were recorded on an 8040-LC-MS (Shimadzu Corporation).
High-resolution mass spectra were recorded on a Micromass-Q-T of
Ultima spectrometer in positive ion mode or on an Axima-CFRTM plus
high-performance linear/reflectron Matrix-Assisted Laser
Desorption/Ionization-Time Of Flight (MALDI-TOF) mass spectrometer
(Shimadzu Corporation). High-performance liquid chromatography
(HPLC) purification was performed with an LC-20AD XR system
(Shimadzu Corporation) and analytical HPLC was performed using an
8040-LC-MS Triple Quadrupole Mass Spectrometer (Shimadzu
Corporation). Peptide synthesis was performed using Biotage-Syro II
(Biotage). Proton nuclear magnetic resonance (1H-NMR)
spectra were recorded on Bruker DPX-400 FT or Bruker ARX-400 FT
spectrometers (Bruker Corporation). All 1H signal
assignments were confirmed by COSY spectra. Carbon-13 nuclear
magnetic resonance (13C-NMR) spectra were recorded on
Bruker DPX-400 FT (100.61 MHz) or Bruker ARX-400 FT (100.61 MHz)
instruments (Bruker Corporation). All 13C signal
assignments were confirmed by heteronuclear single quantum
coherence (HSQC) spectroscopy using CDCl3 (99.9% D) or
6d-DMSO as the solvent (Cambridge Isotope Laboratories,
Inc.). Chemical shifts are referenced to the deuterated solvent
peak and coupling constants are reported in hertz. Photolysis was
performed using an Aicure system (UJ 35; Panasonic
Corporation).
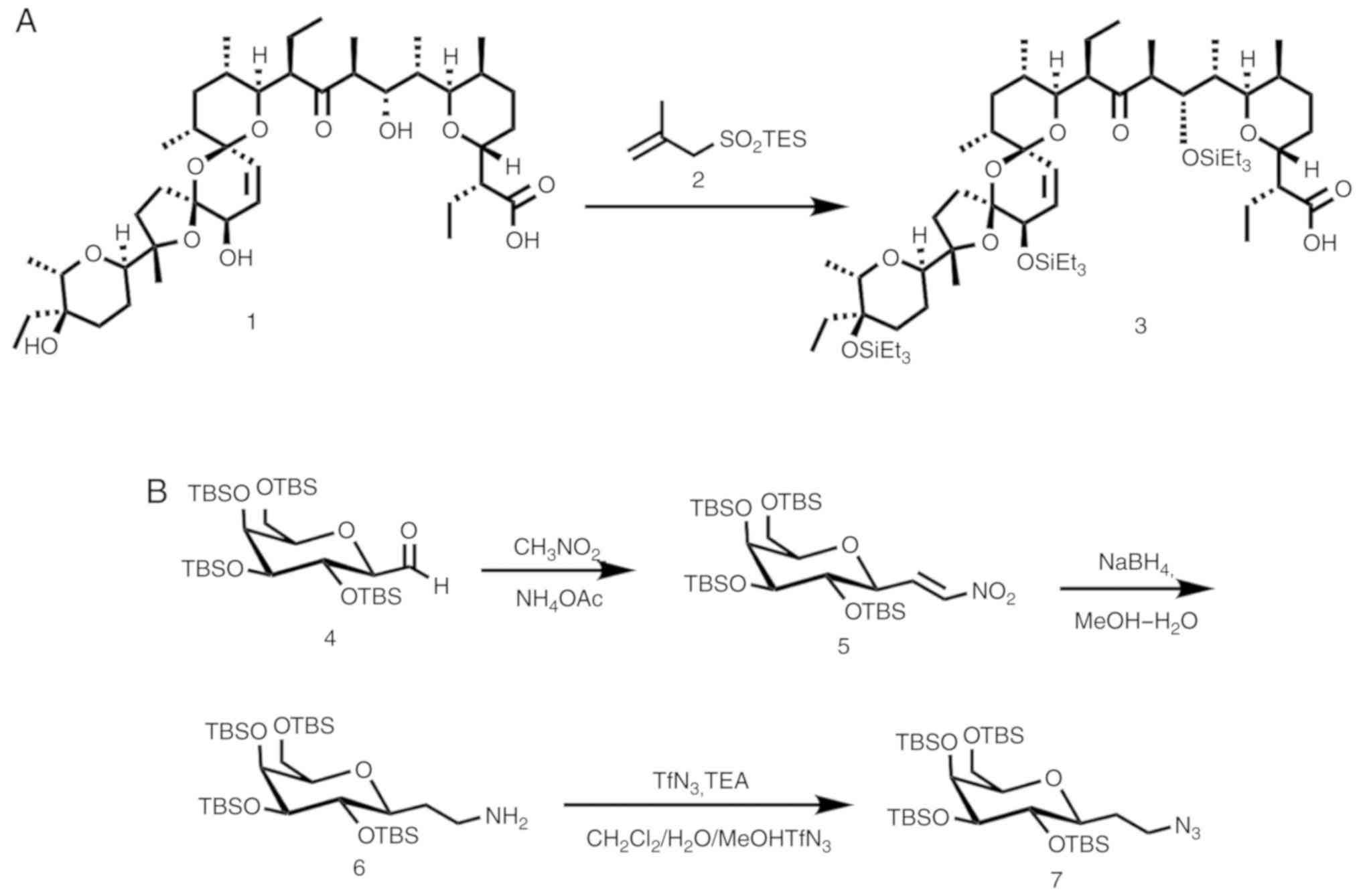
Protected salinomycin
Salinomycin (Lucerna-Chem AG; cat. no.
MCE-HY-17439-100MG) 1 (0.5 g; 0.666 mmol) (Rf, 0.16 at 3:2:1
Ether/pentane/CH2Cl2 system) was dissolved
with triethylsilyl 2-methylprop-2-ene-1-sulfinate (1 g; 4.300 mmol)
without solvent at −20°C (25). The
mixture was stirred using a teflon-coated magnetic stir bar at 25°C
for 30 min and subsequently cooled to −20°C, following which 5 ml
of methanol (99.8%) was added. The sample was then allowed to reach
25°C following which the volatiles were evaporated in vacuo,
to obtain pure, protected salinomycin 3 (0.72 g, 0.659 mmol, 99%)
(Rf, 0.63 at 3:2:1 Ether/pentane/CH2Cl2
system), without requiring any further purification. TLC indicate
complete conversion and pure product. The structure of protected
salinomycin is presented in Fig.
2A.
(((2S,3S,4R,5S,6R)-2-(2-azidoethyl)-6-(((tert-butyldimethy
lsilyl)oxy)methyl)tetrahydro-2H-pyran-3,4,5-triyl)tris(oxy))
tris(tert-butyldimethylsilane). A total of 15 ml of THF
containing tetra((tert-butyldimethylsilyl)oxy)-carbaldehyde 4 (2 g,
3.1 mmol; Rf, 0.20 at 9:1, light petroleum ether/ether) and
nitromethane (0.625 ml, 11.500 mmol) were added to a stirred
suspension of KOH (56 mg, 1.000 mmol), in 10 ml of THF cooled to
5°C. The mixture was subsequently heated to 20°C and stirred for 3
h. Ethyl acetate (EtOAc) (50 ml) and tap water (50 ml) were added
to the mixture, the pH was adjusted to 3.0 using 10% citric acid
and then extracted with EtOAc (3×30 ml). The combined EtOAc phases
were washed with 4% NaCl (2×30 ml), dried over MgSO4 (10
g) and the volatiles were evaporated in vacuo. The crude
product was subsequently dissolved in CH2Cl2
(2 ml). Subsequently, 1,8-Diazabicyclo[5.4.0]undec-7-ene (DBU) (0.8
ml; 5.000 mmol) was added to the solution of the crude product and
stirred for 30 min, after which the mixture was diluted with
Et2O (50 ml), quenched with 1M HCl (15 ml), washed with
saturated NaHCO3 (10 ml) and resuspended over
MgSO4 (5 g) to dry. The mixture was filtered and
evaporated to give 2.2 g of crude 5 (Rf, 0.60 at 9:1, light
petroleum ether/ether). The crude product was dissolved in 10 ml of
methanol-water (5:1) and sodium borohydride (0.57 g; 15.00 mmol)
was added. The mixture was stirred for 30 min at 0°C, and
subsequently heated to 25°C. A saturated aqueous solution of sodium
potassium tartarate (25 ml) was added and the aqueous phase was
extracted with EtOAc (3×30 ml). The combined organic phase was
dried over MgSO4 (5 g), filtered and concentrated in
vacuo to give 1.8 g of crude 6 (Rf, 0.10 at 9:1, light
petroleum ether/ether). The residue was dissolved in 1 ml of
CH2Cl2 and 3 mg of CuSO4 in 6 ml
of double distilled H2O, followed by the addition of
triethylamine (390 µl; 2.930 mmol) and methanol (18 ml). A freshly
prepared dichloromethane solution of trifluoromethanosulfonyl azide
(3 ml; 1.780 mmol) was immediately added in a single portion. The
reaction mixture was stirred at 0°C until TLC indicated that the
reaction was complete (after ~90 min). The mixture was then
extracted with CH2Cl2 (3×50 ml). The combined
organic phase was dried over MgSO4 (5 g) and
concentrated in vacuo. Flash chromatography (9:1, light
petroleum ether/ether) yielded 1.4 g (68%, 4 steps) of the compound
‘7’ (Fig. 2B; Rf, 0.65 at 9:1, light
petroleum ether/ether), a pure colorless oil (26).
1H NMR (400 MHz, CDCl3), δ
4.17 (d, J=10.0 Hz, 1H); 3.91 (td, J=3.0, 1.3 Hz,
1H); 3.86 (dd, J=10.0, 4.6 Hz, 1H); 3.74–3.71 (m, 1H); 3.69
(d, J=1.2 Hz, 1H); 3.66–3.57 (m, 2H); 3.40 (d, J=11.4
Hz, 2H); 1.94 (dt, J=14.1, 6.9 Hz, 1H); 1.62 (dtd,
J=14.2, 7.0, 5.0 Hz, 1H); 0.93–0.82 (m, 36H); 0.17–0.05 (m,
24H).13C NMR (100.6 MHz, CDCl3), δ 77.60 (d,
1 J (C,H)=160); 77.26 (d, 1 J (C,H)=155); 76.67 (d, 1
J (C,H)=150); 73.15 (d, 1 J (C,H)=165); 72.82 (d, 1
J (C,H)=155); 67.85 (d, 1 J (C,H)=165); 61.44 (t, 1
J (C,H)=150); 48.35 (t, 1 J (C,H)=155); 31.37 (t, 1
J (C,H)=145); 26.10, 25.95, 25.91, 25.44, (4q, 1 J
(C,H)=125; (CH3)3CSi); 19.60, 18.60, 18.40,
18.10 (4s, (CH3)3CSi); −4.05, −4.29, −4.67,
−5.15 (8q, 1 J (C,H)=118, CH3Si); MALDI-HRMS
calculated for (M + Na)
C32H71N3O5Si4Na
712.4368 demonstrated 712.4372. Elementary analysis calculated for
C32H71N3O5Si4
(689.4471): C 55.68, H 10.37; demonstrated: C 55.75, H 10.40. The
structure of (((2S,3S,
4R,5S,6R)-2-(2-azidoethyl)-6-(((tert-butyldimethylsilyl)
oxy)methyl)tetrahydro-2H-pyran- 3,4,5-
triyl)tris(oxy))tris(tert-butyldimethylsilane) is presented in
Fig. 2B.
Peptide synthesis
All peptides were synthesized at room temperature
using a 0.20 mmol scale on the Fmoc-Gly-Wang resin (AnaSpec, Inc.;
cat. no. AS-20057) (0.41 mmol/g loading), and an automated CS 336X
peptide synthesizer from CSBio. The Fmoc-NH-Gly-Wang resin (0.41
mmol/g, 100–200 mesh) was used for all peptide syntheses. The
procedure for a typical 0.2 mmol-scale synthesis is described
below.
Resin swelling
Approximately 0.49 g of the Fmoc-NH-Gly-Wang resin
was swollen for 15 min at 25°C in dimethylformamide
(DMF)/CH2Cl2 (1:1).
Fmoc deprotection cycles
Fmoc deprotection was accomplished by adding 8.0 ml
of 20% (v/v) piperidine in DMF to the resin and rocking the mixture
for 20 min (two 10 min cycles).
Coupling cycles
A total of five equivalents of Fmoc-AA-OH and five
equivalents of HBTU were dissolved in 5 ml of DMF, and then six
equivalents of N,N-diisopropylethylamine (DIEA) were added in 3 ml
of DMF. The mixture was added to the resin after shaking for 30 min
at 25°C.
Washing cycles
Washing was performed after each reaction
(deprotection or coupling) with 10 ml of DMF (five times) as
previously described (27). Resin
swelling, Fmoc deprotection, coupling cycle and washing cycle were
performed using an automated Biotage syro II peptide synthesizer
(Biotage) at 25°C.
Protocol for click chemistry
The resin (0.07 mmol) was suspended in DMF (1.5 ml)
and ammonium azide (32 mg, 0.13 mmol) was added followed by the
addition of a suspension of DIEA (113 µl, 0.66 mmol), CuI (24 mg,
10 mmol) and sodium ascorbate (200 mg, 1.00 mmol) in
DMF/H2O (1.25:0.15 v/v). The mixture was shaken at 20°C
for 8 h, and the reaction was monitored by liquid
chromatography-mass spectrometry (LC-MS) (cat. no. LCMS-8040;
Shimadzu Corporation). The resin was filtered and washed
sequentially with: i) 5×10 ml of an imidazole solution in DMF; ii)
5×10 ml of DMF; iii) 5×10 ml of 10% double distilled water in DMF;
iv) 5×10 ml of methanol; v) 20% piperidine in DMF and vi) 5×10 ml
of CH2Cl2.
Washing
The resin was rinsed with DMF, DCM, methanol and
diethyl ether, and subsequently dried under vacuum.
Peptide cleavage
The peptide resin was placed in a 30 ml glass
solid-phase peptide synthesis (SPPS) reactor (Biotage) and was
shaken with 10 ml of the freshly prepared cleavage cocktail for 2 h
at 25°C. The resin cleavage cocktail contained trifluoroacetic acid
(TFA):H2O: thioanisole:1,2-ethanedithiol:anisole
(85:5:5:3:2 v/v). The cleavage solution was subsequently drained
into a 50 ml glass vial and the resin was washed five times with
TFA. The collected washings were then concentrated in vacuo
in a water bath at 24°C for 5 min to reach a volume of 4–5 ml in
order to remove TFA. The deprotected peptide was transferred to a
50 ml falcon tube and the peptide was subsequently precipitated
using 30 ml of cold ether. The milky mixture was centrifuged (5,600
× g; −20°C; 10 min) to a pellet and the ether layer was decanted.
The crude peptide thioester was dissolved in MeCN:H2O
(1:1 v/v) with 0.05% TFA, lyophilized, re-dissolved with
MeCN:H2O (1:1 v/v) with 0.05% TFA, filtered and
lyophilized to provide the crude peptide for HPLC purification.
HPLC conditions
Preparative HPLC
Purification was performed at 25°C with a HPLC
system (600 HPLC system; model no. 6 CE; Waters Corporation) and
the peptides were detected at 215 and 280 nm. The peptides were
eluted from the column using mobile phases A (0.1% TFA in
double distilled H2O) and B (0.1% TFA in MeCN),
at 25°C for 60 min using a Vydac-packed 25×250 mm reversed phase
column (218TP C18 SPRING Col 250×25 mm 10 µm column; cat. no.
DRM-S218TP1025 (BGB Analytik SA) and a flow rate of 10 ml/min were
used.
Analytical HPLC
Analytical HPLC was performed at 25°C for 25 min
with a 8040-LC-MS Triple Quadrupole Mass Spectrometer (Shimadzu
Corporation), with detection from 210–450 nm, using a 4.6×250 mm
Cosmosil C18 column and a flow rate of 0.5 ml/min. Fractions
containing the pure target peptides were collected and lyophilized.
Peptide purity and identity were confirmed by analytical HPLC and
mass spectrometry, either by MALDI-TOF or electrospray
ionization-MS.
Photolysis cleavage
In total, 100 µl of peptide was added to a 1
cm-thick quartz cell that was placed 5 cm away from the UV light
source at ≥365 nm (Aicure UJ30/35 LED Spot Type UV Curing Systems;
Panasonic Corporation). A total of 5 µl was taken from the sample
each 20 sec for 3 min. Then, the samples were taken without any
further treatment and the cleavage was monitored by RP-LC-MS.
RP-HPLC analysis of the photocleavage of the peptide and the
release of salinomycin by photolysis at ≥365 nm were plotted as a
function of time. The compounds were detected using a tandem mass
spectrometry (MS-MS) technique, and quantified by comparing the
spectra with those of standard compounds at the same
concentration.
MS-MS conditions
A stock solution of salinomycin (Lucerna-Chem AG;
cat. no. MCE-HY-17439-100MG) was prepared at a concentration of 5
µmol/ml. Linearity was investigated over 10 points of calibrations
with the concentration of the sample ranging between 0.1 and 20
µmol/ml. Samples were injected in increasing order and in
decreasing order with 3 min washing between each injection,
calibration curves solutions were prepared by serial dilution of
acetonitrile. The expected concentration of salinomycin was of ~5
µmol/ml following photolysis cleavage of the peptide; therefore,
the calibration curve samples concentrations were selected based on
the expected concentration of salinomycin. Then, 5 µmol/ml of
peptide was prepared, the photolysis was performed and samples were
collected at various times (at 0, 20, 40, 60, 80 and 100 sec). The
solution was then injected to a LC-MS system, and the MS-MS peaks
were analyzed using a software (LabSolutions LLC; version 2.02;
Shimadzu Corporation) to obtain the corresponding spectra. All
experiments were performed as follows: System, LC-MS-8040; columns,
Column raptor ARC-18 LC (Restek); column temperature, 40°C; mobile
phase, A (0.1% TFA in double distilled water) and B (0.1% TFA in
MeCN); gradients, 0–5 min 95% A, 18–22 min 5% A and 22–25 min 95%
A; flow rate, 0.5 ml/min; injection volume, 30 µl; mass
spectrometer, LCMS-8040 (Shimadzu Corporation; triple quadruple
mass spectrometer); ionization, ESI positive mode; heating block
temperature 400°C; drying gas, nitrogen (15 l/min).
Dose response assay
The dose-response of peptide 1 (with the
solubilizing group), peptide 2 (without the solubilizing group) and
salinomycin was evaluated using the MTT assay. The 50% inhibitory
concentration (IC50) was used in order to assess the
potency. Peptide 1, peptide 2 and salinomycin were initially
dissolved in DMSO (Sigma Aldrich; Merck KGaA; cat. no.
D8418-100ML), and then diluted with PBS to make the concentration
of the three compounds in the range of 0.1–2 µM and the
concentration of DMSO 0.2%. The control sample was prepared with
0.2% DMSO in PBS without adding the compounds (peptide 1, peptide 2
and salinomycin). For the assay, cells were seeded in 96-well
plates (7,500 cells/well for MCF-7 and 5,500 cells/well for JIMT-1,
200 µl). MCF-7 cells were purchased from the American Type Culture
Collection (cat. no. HTB-22). JIMT-1 cells were purchased from DSMZ
(cat. no. ACC 589). The 96-well plates were incubated for 24 h at
37°C, after which peptide 1, peptide 2 and salinomycin were added.
After 72 h of treatment, the plates were removed from the incubator
(37°C) and the samples were exposed to radiation at l≥365 for 3
min. Subsequently, 20 µl of MTT (5 mg/ml) stock solution was added
to each well. Following incubation at 37°C, with a 5%
CO2 overlay for 4 h, the plates were removed from the
incubator and the solutions were aspirated. The purple formazan
product was dissolved by adding 100 µl of DMSO to each well. The
plates were rotated for 5 min to distribute the DMSO evenly, and
the absorbance was measured at 540 nm using a BioTek microplate
reader. For each compound, eight independent dose-response
experiments were performed with five replicates in each experiment.
The software OriginPro (version 8.5; OriginLab Corporation) was
used to plot the dose-response curves.
DEP assay
The DEP assays were conducted on dielectrophoretic
reader (3DEP reader; DEPTech Ltd.), the analysis was performed by
analyzing the radial motion of cells in the well for 10 sec, the
DEP response was measured using 1–45 MHz. Electrical frequencies
applied to the wells induce the cells to move inside the well due
to the phenomenon of dielectrophoresis (28). In total, ~80 µl of prepared cell
suspension was pipetted into a 3DEP disposable chip, inserted into
the reader and exposed to 10–20 MHz to produce a full DEP spectrum.
This process was repeated for each treatment group ≥3 times. Data
were acquired over 15 sec (for JIMT-1) or 20 sec (for MCF-7)
intervals (28).
Results
The synthesis of the caged glutamine model was
designed in the present study using the TAT-protein sequence. In
this sequence, tools for smoothly introducing the solubilizing
groups were used. Another tool was used to attach the
photo-cleavable linker to salinomycin, such that it was released at
the suitable time during the in vitro analysis.
A number of methods have been used in order to
protect salinomycin; however, all the common methods have failed to
protect salinomycin from epimerization or elimination. In the
present study, the triethylsilyl ether (TES) group was successfully
incorporated to fully protect salinomycin, as depicted in (Fig. 2). The structure of salinomycin is
presented in Fig. S1A. The MTT
dose-response curves were analyzed in JIMT-1 and in MCF-7 cells
(Fig. S1B and C). LC-MS-MS was
performed to analyze the purity of salinomycin (Fig. S1D). Commercially available
salinomycin was treated with triethylsilyl
2-methylprop-2-ene-1-sulfinate 2 at 0°C for 30 min in order to
produce the fully silylated salinomycin with a quantifiable yield
(25). Furthermore, the crude
salinomycin-linker 3 was attached to the TAT-protein segment
through a peptide bond (Fig.
2A).
The solubilizing group was selected from among the
sugars of the carbohydrate family in order to avoid any toxicity
interfering with the biological function. The synthesis of the
solubilizing group is outlined in (Fig.
2B). A known aldehyde 4 (25,29–37) was
treated under Henry reaction conditions (38) with nitromethane, in the presence of
ammonium acetate to form the dehydrated product 5. The crude
product 5 was subsequently treated with sodium borohydride in the
presence of double distilled water and methanol for complete
reduction in order to produce the amine form 6, which was treated
with trifluoromethanesulfonyl azide in order to convert it to the
azido sugar 7. The overall yield of the three steps was 55%
(26).
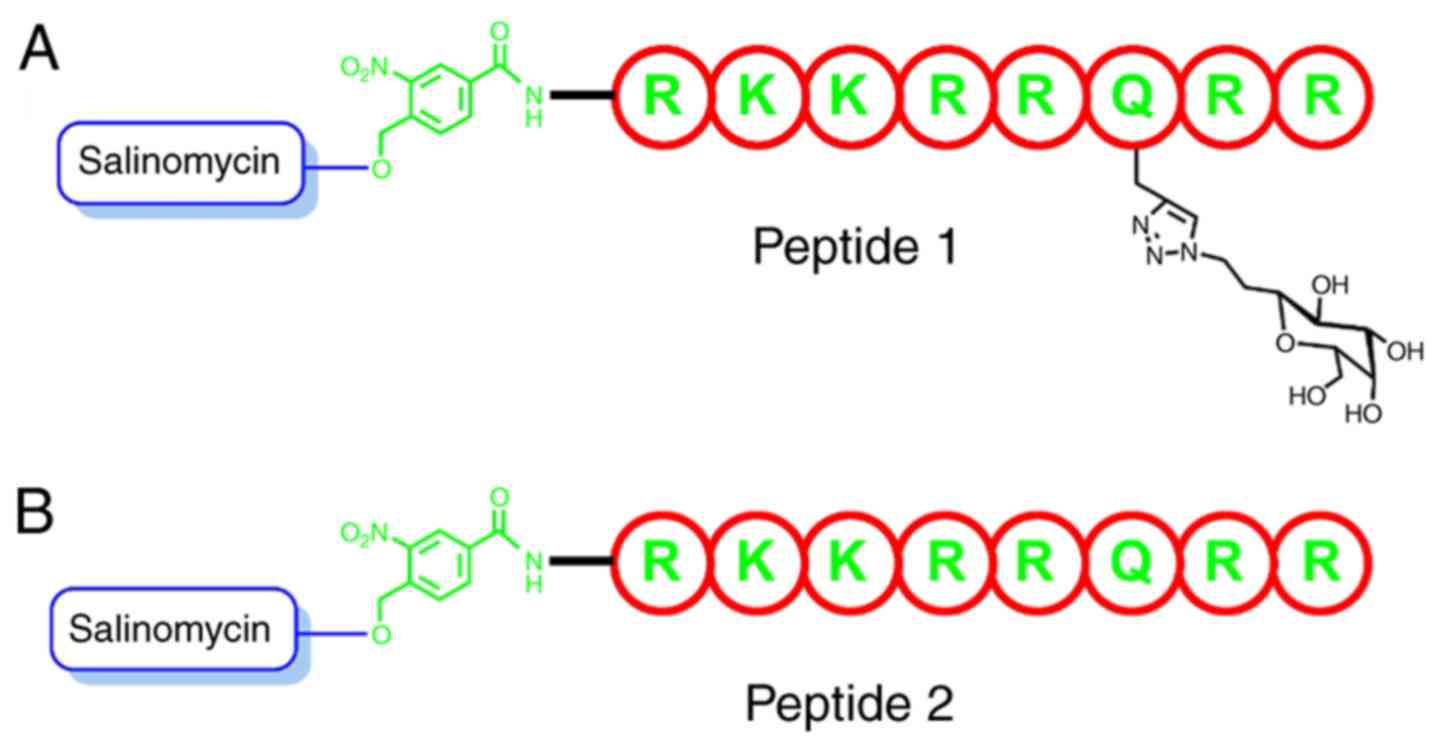
A total of two analogs were prepared in order to
test the assumption and model design of the present study, one
without the solubilizing sugar group and the other with the
solubilizing group (Fig. 3).
Peptides were synthesized using a standard Fmoc-SPPS protocol
(39). Furthermore, the N-terminus
of the peptide was coupled with (4-tert-butyldimethylsilyl
(hydroxymethyl)-3-nitrobenzoic acid. The silyl group was
subsequently removed using tetra-n-butylammonium fluoride (TBAF),
followed by the esterification of salinomycin using DIC/DMA
(40). The solubilizing group 7 was
subsequently attached to glutamine in the TAT-protein segment using
click chemistry on a solid support (24,41–43).
Finally, the silylated groups were removed using TBAF, the peptides
were cleaved from the solid support and the side chains were
deprotected using dichloromethane/acetic acid/trifluoroethanol
solution (3:1:1) treatment. The peptides were then purified using
HPLC (Figs. S2 and S3).
The nitrobenzyl ester groups of the two peptides are
highly stable under physiological conditions (44). This comes as no surprise given that
the cleavage of this group requires the peptides to be subjected to
conditions of pH 12 and 75°C (45).
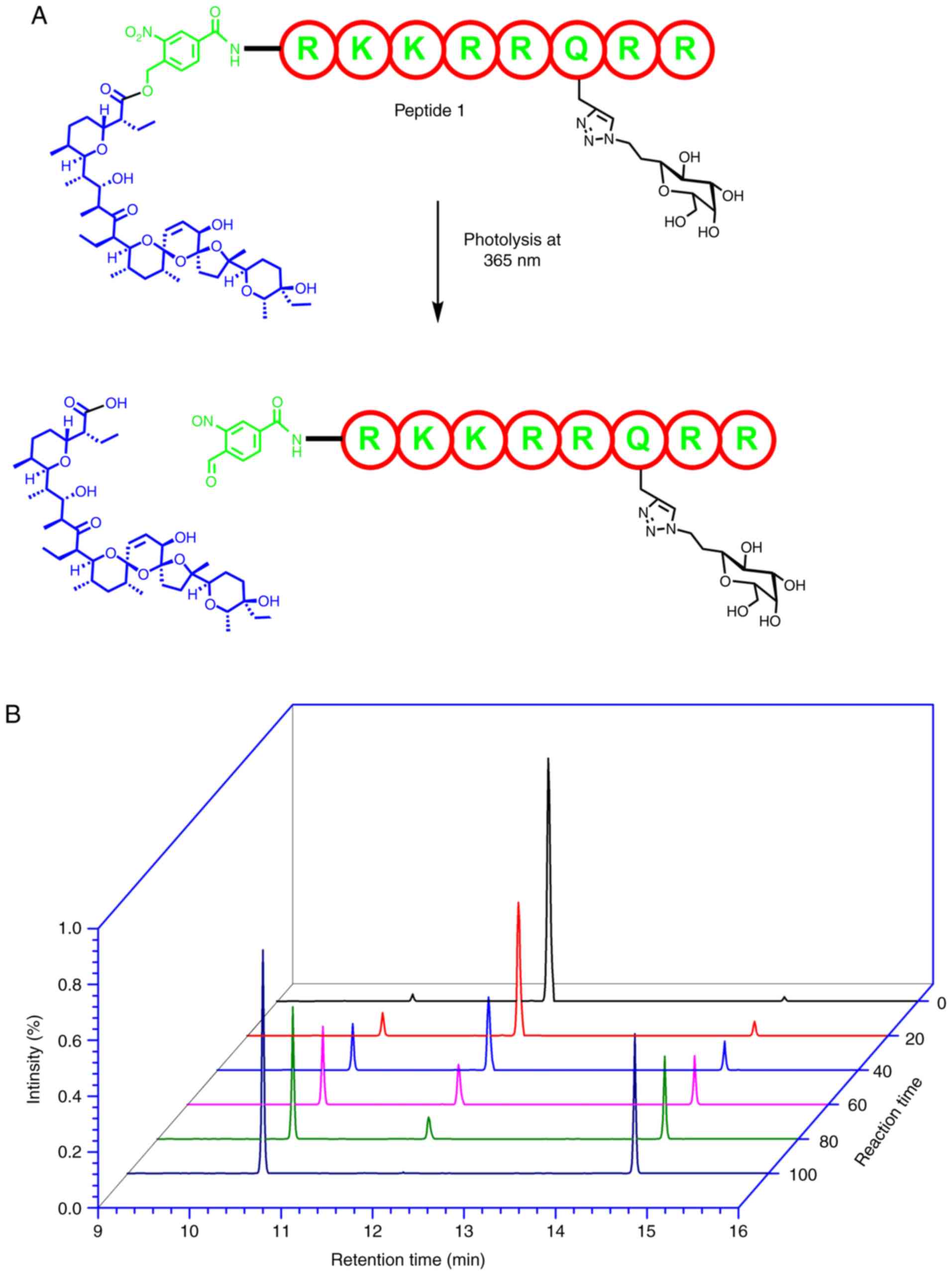
In order to determine whether the cage can be cleaved to release
the drug smoothly, a kinetic analysis was performed in the present
study. The kinetic study was conducted under physiological
conditions in order to control the cleavage conditions and optimize
the procedure. As presented in Fig.
4A, complete and quantifiable conversion was achieved with UV
irradiation at ≥365 nm within 80–100 sec.
The kinetics of the cleavage was analyzed using an
MS-MS technique, and a UV detector was implemented in order to
follow the photolytic cleavage products. As a strong chromophore is
not present, salinomycin does not yield a strong absorbance value
after cleavage and is not well detected by the UV detector. RP-HPLC
analysis of the photocleavage of peptide 1 and the release of
salinomycin after photolysis at ≥365 nm, as a function of time is
presented in (Fig. 4B). The kinetic
study of the cleavage of the designed peptide 1 demonstrated a
quick, clean and smooth cleavage process. The results of the
present study indicate that an in vitro analysis can be
performed conveniently as salinomycin can be released quickly
during biological experiments.
In order to determine the biological effects of the
solubilizing group, the peptide without the sugar stabilizing group
(peptide 2) was synthesized following the same SPPS procedure used
to synthesize peptide 1, and subsequently analyzed. The peptide
without the solubilizing group was evaluated based on the
dose-response curves obtained from the MTT assay.
The antiproliferative activity of the salinomycin
analogs in MCF-7 and JIMT-1 breast cancer cells was evaluated using
an MTT assay (Table I). Salinomycin,
with the protein carrier and solubilizing group (peptide 1),
exhibited an IC50 value three times lower than that of
salinomycin in both cell lines. Similarly, the second analog, with
the protein carrier but without the solubilizing group (peptide 2),
exhibited an IC50 value lower than that of salinomycin
in both cell lines. However, the IC50 value of peptide 2
was slightly higher than that of peptide 1. The results of the
present study confirm that conjugating salinomycin to a protein
carrier increases the activity of salinomycin, most likely due to
an increase in cell penetration. Increasing the solubility, by
attaching a sugar to the peptide, also yields increased activity
(46). In order to quantify the cell
viability, MTT was used, the cell viability change is presented in
(Fig. S4A, C and E). In order to
quantify apoptosis progression, the dielectrophoretic (DEP) method
was used, according to the approach by Henslee et al
(28), the progress in cancer cells
is presented in (Fig. S4B, D and
F).
 | Table I.Antiproliferative activity of the
peptide-conjugated analogs of salinomycin evaluated by an MTT-based
dose-response assay. |
Table I.
Antiproliferative activity of the
peptide-conjugated analogs of salinomycin evaluated by an MTT-based
dose-response assay.
| Group | Cell,
IC50, mean ± SE, µM |
|---|
| Salinomycin SA,
1 | JIMI-1,
0.442±0.079 |
|
| MCF-7,
0.577±0.033 |
| Peptide 1, with
sugar solubilizing | JIMI-1,
0.195±0.032 |
| group | MCF-7,
0.134±0.016 |
| Peptide 2, without
sugar solubi | JIMI-1,
0.382±0.052 |
| lizing group | MCF-7,
0.308±0.041 |
Discussion
Many previous studies tried to improve salinomycin
activity against cancer cells. Borgström et al (47) showed
promising results, following the construction of a library of
salinomycin analogs. Borgström et al (47) selectively
modified alcohols on salinomycin. According to this previous study
(47), the modifications of the allylic alcohol on C20 to smaller
and less bulky groups of ester, carbonates and carbamate improved
significantly the activity of native salinomycin. In addition, most
of the groups used to modify salinomycin can be cleaved via
esterases enzyme in the cells. This previous study demonstrated
that these groups are temporary groups and can act as carriers by
enhancing the lipophilic penetration, and the activity in the cell
derives from the native salinomycin form.
The results of the present study confirm that the
activity of salinomycin alone suffers due to its poor penetration
of cancer cells. Salinomycin exhibits an improved activity level
when conjugated to a protein carrier, such as TAT-protein, and a
solubilizing group that allows greater penetration of the cells. In
order to ensure that the maximum quantity of salinomycin reached
the active site, the technique used in the present study employed
photolysis in order to release salinomycin following cell
penetration. As the ester bond is sensitive to X-ray radiation,
this method can be used in cells, in vivo and in humans
during radiotherapy of cancer cells. When machines producing mild
X-rays, such as when a linear accelerator or cyberknife are used,
exposure of the cancer cells to radiation is expected to release
salinomycin inside the tumor cells. The same idea can be applied to
other drugs that suffer from low solubility or the inability to
penetrate cells.
Collectively, the present study improved the
activity of salinomycin by increasing the penetration and the
solubility using small TAT-peptides, which allowed the release of
salinomycin inside the cells by photolysis. Using peptide carrier
to improve salinomycin penetration resulted in IC50 values against
cancer cells that were >4 times lower compared with the native
salinomycin.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was funded by the Deanship of
Scientific Research at Imam Abdulrahman Bin Faisal University
(grant no. 2014255).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
LA designed, synthesized, purified and analyzed the
chemical compounds, and wrote and reviewed the published
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The author declares that he has no competing
interests.
References
|
1
|
Reya T, Morrison SJ, Clarke MF and
Weissman IL: Stem cells, cancer, and cancer stem cells. Nature.
414:105–111. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Al-Hajj M, Becker MW, Wichal M, Weissman I
and Clarke MF: Therapeutic implications of cancer stem cells. Curr
Opin Genet Dev. 14:43–47. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Morrison SJ, Wandycz AM, Hemmati HD,
Wright DE and Weissman IL: Identification of a lineage of
multipotent hematopoietic progenitors. Development. 124:1929–1939.
1997.PubMed/NCBI
|
|
4
|
Dean M, Fojo T and Bates S: Tumour stem
cells and drug resistance. Nat Rev Cancer. 5:275–284. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ruben GC: The iso-competition point, a new
concept for characterizing multivalent versus monovalent counterion
competition, successfully describes cation binding to DNA. Biophys
J. 77:1–2. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mitani M, Yamanishi T, Miyazaki Y and
Ōtake N: Salinomycin effects on mitochondrial ion translocation and
respiration. Antimicrob Agents Chemother. 9:655–660. 1976.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Daugschies A, Gässlein U and Rommel M:
Comparative efficacy of anticoccidials under the conditions of
commercial broiler production and in battery trials. Veterinary
Parasitology. 76:163–171. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gupta PB, Onder TT, Jiang G, Tao K,
Kuperwasser C, Weinberg RA and Lander ES: Identification of
selective inhibitors of cancer stem cells by high-throughput
screening. Cell. 138:645–659. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhou S, Wang F, Wong ET, Fonkem E, Hsieh
TC, Wu JM and Wu E: Salinomycin: A novel anti-cancer agent with
known anti-coccidial activities. Curr Med Chem. 20:4095–4101. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Antoszczak M and Huczyński A: Anticancer
activity of polyether ionophore-salinomycin. Anticancer Agents Med
Chem. 15:575–591. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Huczynski A: Salinomycin: A new cancer
drug candidate. Chem Biol Drug Des. 79:235–238. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Caldwell GW, Ritchie DM, Masucci JA,
Hageman W and Yan Z: The new pre-preclinical paradigm: Compound
optimization in early and late phase drug discovery. Curr Top Med
Chem. 1:353–366. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hartmann T, Schmitt J, Röhring C, Nimptsch
D, Nöller J and Mohr C: ADME related profiling in 96 and 384 well
plate format-a novel and robust HT-assay for the determination of
lipophilicity and serum albumin binding. Curr Drug Deliv.
3:181–192. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Di L, Kerns EH, Li SQ and Petusky SL: High
throughput microsomal stability assay for insoluble compounds. Int
J Pharm. 317:54–60. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Dewangan J, Srivastava S and Rath SK:
Salinomycin: A new paradigm in cancer therapy. Tumor Biol.
39:10104283176950352017. View Article : Google Scholar
|
|
16
|
Savjani KT, Gajjar AK and Savjani JK: Drug
solubility: Importance and enhancement techniques. ISRN Pharm.
2012:1957272012.PubMed/NCBI
|
|
17
|
Fasinu P, Pillay V, Ndesendo VMK, du Toit
LC and Choonara YE: Diverse approaches for the enhancement of oral
drug bioavailability. Biopharm Drug Dispos. 32:185–209. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Patel SG, Sayers EJ, He L, Narayan R,
Williams TL, Mills EM, Allemann RK, Luk LYP, Jones AT and Tsai YH:
Cell-penetrating peptide sequence and modification dependent uptake
and subcellular distribution of green florescent protein in
different cell lines. Sci Rep. 9:62982019. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Löwik DW, Meijer JT, Minten IJ, van
Kalkeren H, Heckenmüller L, Schulten I, Sliepen K, Smittenaar P and
van Hest JC: Controlled disassembly of peptide amphiphile fibres. J
Pept Sci. 14:127–133. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Borrelli A, Tornesello A, Tornesello M and
Buonaguro F: Cell penetrating peptides as molecular carriers for
anti-cancer agents. Molecules. 23:E2952018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gallego I, Rioboo A, Reina JJ, Díaz B,
Canales Á, Cañada FJ, Guerra-Varela J, Sánchez L and Montenegro J:
Glycosylated cell-penetrating peptides (GCPPs). ChemBioChem.
20:1400–1409. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Abramova TV and Silnikov VN:
4-Aminometyl-3-nitrobenzoic acid-a photocleavable linker for
oligonucleotides containing combinatorial libraries. Nucleosides
Nucleotides Nucleic Acids. 24:1333–1343. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kolb HC, Finn MG and Sharpless KB: Click
chemistry: Diverse chemical function from a few good reactions.
Angew Chem Int Ed Engl. 40:2004–2021. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sharpless KB and Kolb HC: Click chemistry
A concept for merging process and discovery chemistry. Abstr Pap Am
Chem Soc. 217:U95–U. 1999.
|
|
25
|
Huang X, Craita C, Awad L and Vogel P:
Silyl methallylsulfinates: Efficient and powerful agents for the
chemoselective silylation of alcohols, polyols, phenols and
carboxylic acids. Chem Commun (Camb). 1297–1299. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Levy DE and Tang C: The Chemistry of
C-glycosides. Elsevier; 1995
|
|
27
|
Amblard M, Fehrentz JA, Martinez J and
Subra G: Methods and protocols of modern solid phase peptide
synthesis. Mol Biotechnol. 33:239–254. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Henslee EA, Torcal Serrano RM, Labeed FH,
Jabr RI, Fry CH, Hughes MP and Hoettges KF: Accurate quantification
of apoptosis progression and toxicity using a dielectrophoretic
approach. Analyst. 141:6408–6415. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Vogel P and Awad LK: Disaccharide mimics
as drugs against cancer and epitopes for anti-cancer vaccine
candidates. Immun Res. 12:572016.
|
|
30
|
Awad L, Madani R, Gillig A, Kolympadi M,
Philgren M, Muhs A, Gérard C and Vogel P: A C-linked disaccharide
analogue of thomsen-friedenreich epitope induces a strong immune
response in mice. Chemistry. 18:8578–8582. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Vogel P, Gerber-Lemaire S, Awad L, Bello
C, Fiaux H, Juillerat-Jeanneret L, Gillig A and Kolympadi M; CARB
29-C-Disaccharides and Analogs, : The Search for Anticancer Agents.
Amer Chemical Soc; Washington, DC: pp. 493. 2007
|
|
32
|
Awad L, Demange R, Zhu YH and Vogel P: The
use of levoglucosenone and isolevoglucosenone as templates for the
construction of C-linked disaccharides. Carbohydr Res.
341:1235–1252. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Awad L, Riedner J and Vogel P: C-linked
disaccharide analogue of the Thomsen-Friedenreich (T)-epitope
alpha-O-conjugated to L-serine. Chemistry. 11:3565–3573. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Awad L: Synthesis of a C-linked
disaccharide analogue of the Thomsen Friedenreich (TF)-epitope
a-O-conjugated to L-serine and formation of a cluster as potential
anticancer vaccine. 2005.
|
|
35
|
Loay A: Synthesis of a C-linked
disaccharide analogue of the Thomsen Friedenreich (TF)-epitope
α-O-conjugated to l-serine and formation of a cluster as potential
anticancer vaccine. École Polytechnique Fédérale De Lausanne.
2005.
|
|
36
|
Demange R, Awad L and Vogel P: Synthesis
of C-linked analogues of
β-d-galactopyranosyl-(1→3)-d-galactopyranosides and of
β-d-galactopyranosyl-(1→3)-d-galactal. Tetrahedron: Asymmetry.
15:3573–3585. 2004. View Article : Google Scholar
|
|
37
|
Awad L: Synthesis of C-linked and
glycopeptide towards non-hydrozable T epitopes and artificial
Anticancer Vaccines. DEA. 2001.
|
|
38
|
Hallinan EA, Kramer SW, Houdek SC, Moore
WM, Jerome GM, Spangler DP, Stevens AM, Shieh HS, Manning PT and
Pitzele BS: 4-Fluorinated L-lysine analogs as selective i-NOS
inhibitors: Methodology for introducing fluorine into the lysine
side chain. Org Biomol Chem. 1:3527–3534. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Hansen PR: Antimicrobial peptides. Methods
Mol Biol. Humana Press; New York, NY: 2017, View Article : Google Scholar
|
|
40
|
Lécaillon J, Gilles P, Subra G, Martinez J
and Amblard M: Synthesis of cyclic peptides via O-N-acyl migration.
Tetrahedron Lett. 49:4674–4676. 2008. View Article : Google Scholar
|
|
41
|
Awad L, Jejelava N, Burai R and Lashuel
HA: A new caged-glutamine derivative as a tool to control the
assembly of glutamine-containing amyloidogenic peptides.
ChemBioChem. 17:2353–2360. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Butterfield S, Hejjaoui M, Fauvet B, Awad
L and Lashuel HA: Chemical strategies for controlling protein
folding and elucidating the molecular mechanisms of amyloid
formation and toxicity. J Mol Biol. 421:204–236. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Awad L, Jejelava N, Brik A and Lashuel H:
Novel chemical tools to facilitate the synthesis and control the
folding and Self-assembly of amyloid-forming polypeptides.
Biopolymers. 2011.
|
|
44
|
Klán P, Šolomek T, Bochet CG, Blanc A,
Givens R, Rubina M, Popik V, Kostikov A and Wirz J: Photoremovable
protecting groups in chemistry and biology: Reaction mechanisms and
efficacy. Chem Rev. 113:119–191. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Greene TW and Wuts PG: Protective groups
in organic synthesis. Wiley; 1999, View Article : Google Scholar
|
|
46
|
Borgström B, Huang X, Pošta M, Hegardt C,
Oredsson S and Strand D: Synthetic modification of salinomycin:
Selective O-acylation and biological evaluation. Chem Commun
(Camb). 49:9944–9946. 2013. View Article : Google Scholar : PubMed/NCBI
|