Introduction
In pathogenesis of myocardial ischemia the coronary
blood flow perfusion declines due to the changes in coronary
circulation, thus reducing the oxygen supply to the heart and
leading to abnormal myocardial energy metabolism, which often
causes myocardial damage. Ischemic heart disease is one of the
major diseases seriously threatening human health in today's
society. Various types of angina and acute myocardial infarction
(AMI) often lead to sudden death of patients, and their morbidity
and mortality rates are high world-wide (1).
Apoptosis is a kind of normal physiological
mechanism of the body, as well as another form of death different
from necrosis. Under physiological conditions, apoptosis is
involved in regulating the renewal of normal cells and clearance of
abnormal cells from the body, and maintaining homeostasis. Under
pathological conditions, apoptosis is related to ischemia,
reperfusion injury, abnormal virus infection, transplant rejection
and tumors (2–4).
Myocardial ischemia is one of the strong inducers of
myocardial apoptosis (5). After MI,
the central areas of infarction mainly present necrosis of
myocardial cells, due to severe ischemia and hypoxia, while in the
infarction border region the myocardial cells mainly present
apoptosis that is reversible due to milder ischemia. Therefore,
reducing myocardial apoptosis in the infarction border region and
non-infarction region is of importance for reducing the damage
after MI. During apoptosis, downstream effector cysteine aspartic
acid-specific protease-3 (Caspase-3) will be eventually activated
by endogenous and exogenous pathways (6), and Caspase-3 plays an important role in
apoptosis after MI (7,8). Moreover, the balance between the
pro-apoptotic proteins [Bid, B-cell lymphoma-2 (Bcl-2) associated X
protein (Bax) and Bak] and anti-apoptotic proteins (Bcl-2 and
Bcl-xL) in the Bcl-2 family plays an important role in initiating
the endogenous apoptotic pathway (9). Bax, induced by myocardial ischemia, can
be transported to the mitochondrial outer membrane to initiate the
endogenous mitochondrial apoptotic pathway. Due to the activated
caspase cascade, the caspase-3 precursors in the cytoplasm are
activated and lysed into activated caspase-3, ultimately leading to
apoptosis. However, the expression of Bcl-2 can stabilize the
mitochondrial membrane and inhibit the occurrence and development
of apoptosis.
The Ras homolog gene family (Rho)/Rho kinase
signaling pathway is composed of Rho protein, Rho kinase, myosin
phosphatase and other signaling molecules. Rho protein is a kind of
small-molecule guanine nucleotide-binding protein, also known as
small G protein, as well as a member of the Ras protein
superfamily, which serves as a signal converter or molecular switch
in the cellular signal transduction pathway, and acts on the
cytoskeleton and its target proteins, thus exerting various
biological effects. Among the Rho family members, Rho member A
(RhoA) plays not only an important role in the regulation of
cytoskeleton, maintenance of cellular morphology, cell migration,
smooth muscle cell contraction and other cell activities, but also
has a key role in various biological activities, such as smooth
muscle cell proliferation, cell adhesion, platelet aggregation,
contact inhibition, growth and apoptosis (10–12).
During cellular metabolism, Rho protein exists in the guanine
trinucleotide phosphate (GTP)- and guanine dinucleotide phosphate
(GDP)-binding forms, and it induces or terminates the cellular
cascade activation through mutual conversion between GDP
phosphorylation and GTP dephosphorylation.
The statins, namely 3-hydroxy-3-methylglutaryl
coenzyme A (HMG-CoA) reductase inhibitors, are a new type of drugs
widely used to lower the cholesterol level. It has been confirmed
in evidence-based medicine that in addition to the lipid-lowering
effect, statins also possess a series of cardiovascular protective
effects, such as anti-inflammation, anti-oxidation, improvement of
endothelial function, inhibition on smooth muscle proliferation,
and inhibition on platelet aggregation and thrombosis (13,14).
Moreover, the non-lipid-lowering effects (such as
anti-inflammatory, anti-oxidation and immunoregulatory effects) of
fluvastatin (Flu), as a kind of classical statin, have attracted
increasing attention. Existing studies have proved that Flu can
improve myocardial apoptosis, ventricular remodeling and cardiac
dysfunction after MI (15,16), but its mechanism of action has not
been clarified yet. In the present study, therefore, the mouse
model of AMI was established to explore the antagonistic effect of
Flu on AMI and myocardial apoptosis, and elucidate its possible
mechanism through the RhoA/Rho-associated coiled-coil protein
kinase 1 (ROCK1) pathway, to lay a theoretical foundation for the
application of Flu in the prevention and treatment of AMI.
Materials and methods
Animals and reagents
This study was approved by the Animal Ethics
Committee of Fujian Medical University Animal Center (Quanzhou,
China). A total of 40 C57BL/6J mice aged 8 weeks and weighing
17.9±0.6 g with normal nutritional status and mental status, were
provided by Laboratory Animal Center of Fujian Medical University.
Bcl-2-associated X protein (Bax), Bcl-2, NF-κB, ROCK1, ROCK2
antibodies were from Sigma-Aldrich; Merck KGaA. TUNEL staining kit
QIA33 was from Shanghai Shiyi Biotechnology Co., Ltd., and the
superoxide dismutase (SOD) and malondialdehyde (MDA) test kits were
from Nanjing Jiancheng Bioengineering Research Institute.
Model establishment and grouping
Before the operation, mice were weighed,
intraperitoneally injected with avertin solution at a dose of
0.40–0.75 mg/g for anesthesia, and fixed on a rat plate in supine
position. Then, the skin of the neck and precordial region was
disinfected with 70% alcohol, and the hair was shaved. After that,
the skin of the neck was scissored, and the muscle was bluntly
separated, followed by tracheal intubation and connection of a
small animal ventilator (positive pressure ventilation 2–3
ml/cycle, frequency 20 cycles/min). Thereafter, the skin of
precordial region was scissored, the muscle was bluntly separated,
and the chest wall was cut using a pair of scissors at the space
between the third and fourth ribs. For 30 mice in AMI group, Flu
group, Ang II group (group A), a 7–0 suture needle with suture was
used to ligate the left anterior descending coronary artery
(coronary artery) at 3–4 mm below the left auricle. For another 10
mice serving as Sham group (group B), the needle was passed through
the left anterior descending coronary artery, but the artery was
not ligated. Then, the thoracic cavity was closed along the third
and fourth ribs, the chest muscles were sutured layer by layer, and
finally the chest skin was continuously sewn up. After that, the
chest was squeezed to remove thoracic gas to recruit the lungs, the
ventilator was removed, and the neck skin was sewn up. Lastly, mice
were put on a 40°C insulation platform for regaining
consciousness.
Immunohistochemistry
Paraffin-embedded heart tissue sections were
deparaffinized with xylene, dehydrated with graded alcohol, and
incubated with warm deionized water containing 0.3%
H2O2 for 30 min. After endogenous peroxide
was eliminated, sections were blocked with serum and addeded with
primary antibodies for incubation at 4°C overnight. The next day,
sections were incubated with IgG antibody-HRP, and dropwise added
with the mixture prepared in the biotin and ABC kit for culture,
followed by color development with DAB for 10 min. Thereafter,
sections were counterstained with hematoxylin, washed, dehydrated
and permeabilized. Lastly, sections were observed using an optical
microscope.
Terminal deoxynucleotidyl
transferase-mediated deoxyuridine triphosphate (dUTP)-biotin nick
end labeling (TUNEL)
Paraffin-embedded tissue sections were
deparaffinized with xylene, dehydrated with graded alcohol, and
treated with Proteinase K working solution for 15–30 min at 21–37°C
or added with cell-permeable solution for 8 min of reaction. TUNEL
reaction mixture was prepared; 50 µl TdT + 450 µl
fluorescein-labeled dUTP solution was added and mixed in treatment
group, and only 50 µl fluorescein-labeled dUTP solution was added
in negative control group. After slides were dried, 50 µl TUNEL
reaction mixture (only 50 µl fluorescein-labeled dUTP solution was
added in negative control group) was added onto specimens, and
slides were then covered with cover slips or sealing films,
followed by reaction in the dark and humid chamber at 37°C for 60
min. After the slides were dried, 50 µl converter-POD was added
onto specimens, slides were then covered with cover slips or
sealing films, followed by reaction in the dark and humid chamber
at 37°C for 30 min. Thereafter, sections were added with 50–100 µl
DAB substrate for reaction at 15–25°C for 10 min. Then, they were
counterstained with hematoxylin or methyl green for a few seconds
and rinsed with tap water immediately, dehydrated with graded
alcohol, permeabilized with xylene and mounted with neutral gum. A
drop of PBS or glycerol was added, and the optical microscope was
employed for counting (200–500 cells) and photography. Positive
cells were counted, and apoptotic index (AI) = number of positive
cells / total number of cells ×100. The mean calculated in each
group was used as a representative value.
Western blotting
Collected tissues were ground in liquid nitrogen,
diluted with normal saline and placed on ice. The supernatant was
collected and centrifuged at 12,000 × g at 4°C for 5 min, and the
supernatant was discarded. The precipitate was re-suspended in
radioimmunoprecipitation assay (RIPA) lysis buffer containing
phenylmethylsulfonyl fluoride (PMSF) (Beyotime Institute of
Biotechnology), lysed and centrifuged at 4°C and 16,000 × g for 15
min, and the supernatant was taken for protein quantification. The
protein was added with loading buffer, heated for denaturation,
subjected to sodium dodecyl sulphate-polyacrylamide gel
electrophoresis (SDS-PAGE) and transferred onto a membrane. The
membrane was blocked with 5% skim milk for 2 h, added with primary
antibodies for incubation at 4°C overnight and washed with tris
buffered saline-tween (TBST) 3 times (10 min/time), followed by
incubation with corresponding secondary antibodies at room
temperature for 1 h and washing with TBST 3 times (10 min/time).
Lastly, electrochemiluminescence (ECL) assay was performed to
detect the protein expression in different samples.
Statistical analysis
Data were expressed as mean ± standard deviation and
analyzed through paired or unpaired t-test. Comparison between
multiple groups was done using One-way ANOVA test followed by post
hoc test (least significant difference). P<0.05 indicates that
the difference was statistically significant. Data were analyzed
using the Statistical Product and Service Solutions (SPSS) 20.0
statistical software (IBM), and then GraphPad software (GraphPad
Software, Inc.) was used for plotting.
Results
Protective effect of Flu on myocardial
cells in infarction region in AMI mice
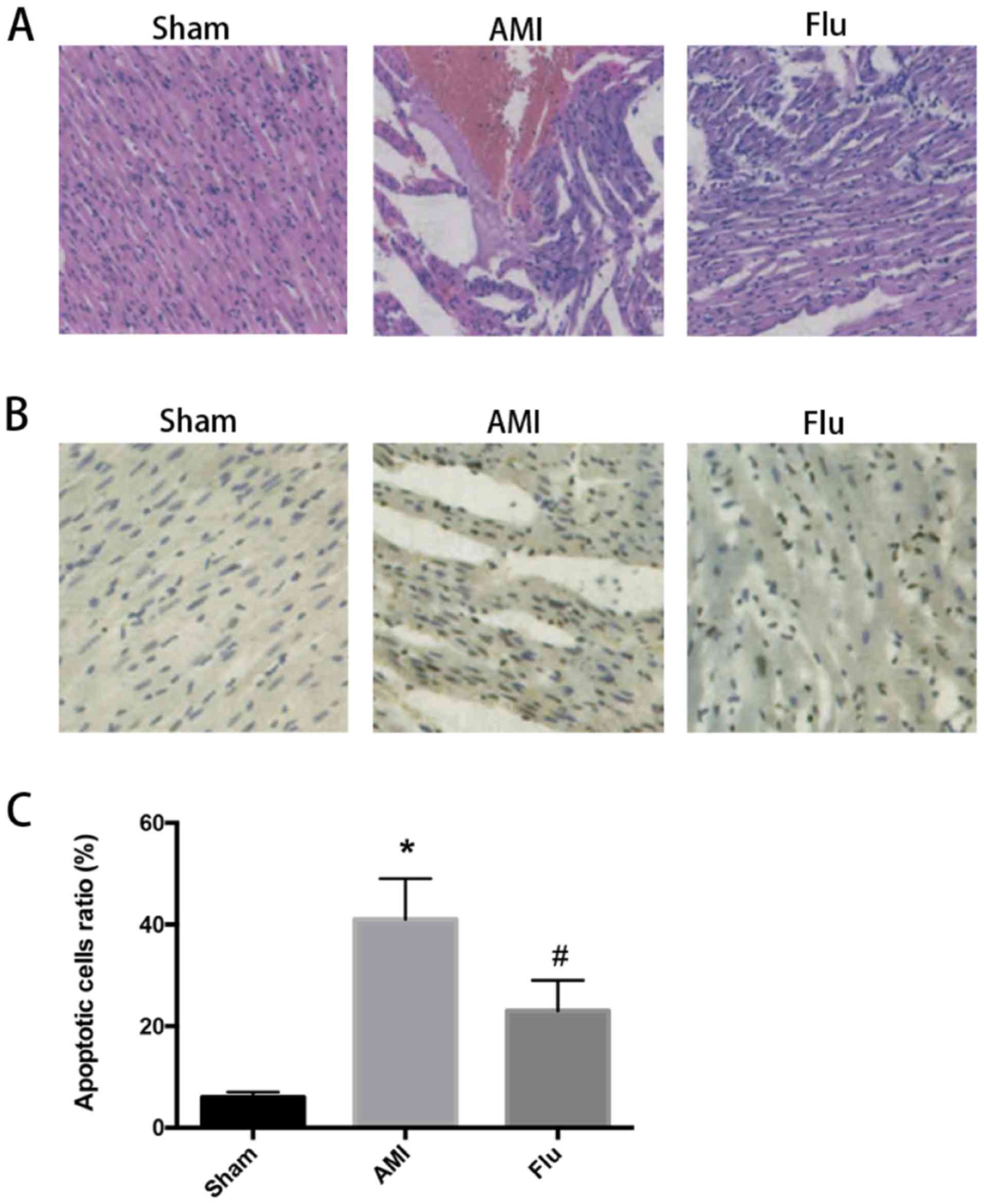
After ischemic preconditioning, MI occurred in AMI
group and Flu group, and it was alleviated in Flu group compared
with that in AMI group (Fig. 1A). In
AMI group, the level of antioxidant molecule superoxide dismutase
(SOD) in serum dramatically declined compared with that in Sham
group, while the level of oxidative damage marker malondialdehyde
(MDA) was significantly higher than that in Sham group (P<0.05).
In Flu group, the level of SOD was significant higher compared with
that in AMI group, and the level of MDA was significantly lower
than that in AMI group (P<0.05), indicating that Flu can exert
an antioxidant effect in the protection of myocardial cells. In
addition, there was no inflammatory cell infiltration, and the
myocardial cells were arranged in an orderly manner in Sham group,
AMI group had more inflammatory cell infiltration in the infarction
region, disorderly myocardial fibers and fewer myocardial cells,
and the inflammatory degree of myocardial cells was low and the
myocardial cells were arranged in an orderly manner in Flu group
(Fig. 1A). The above results suggest
that the degree of myocardial tissue damage in AMI mice declines
after Flu treatment.
Effect of Flu on myocardial apoptosis
in infarction region in AMI mice
The results of terminal deoxynucleotidyl
transferase-mediated dUTP nick end labeling (TUNEL) assay showed
that the number of apoptotic myocardial cells was obviously
increased in AMI group compared with that in Sham group, while it
was obviously decreased in Flu group compared with that in AMI
group (Fig. 1B and C). Compared with
Sham group, AMI group had increased protein expression levels of
Bax and nuclear factor-κB (NF-κB), decreased protein expression of
Bcl-2 and increased Bax/Bcl-2 ratio in myocardial tissues. Compared
with AMI group, Flu group had decreased protein expression levels
of Bax and NF-κB, increased protein expression of Bcl-2 and
decreased Bax/Bcl-2 ratio (Fig. 2A and
B). The above results suggest that Flu can alleviate myocardial
apoptosis in the infarction region in AMI mice.
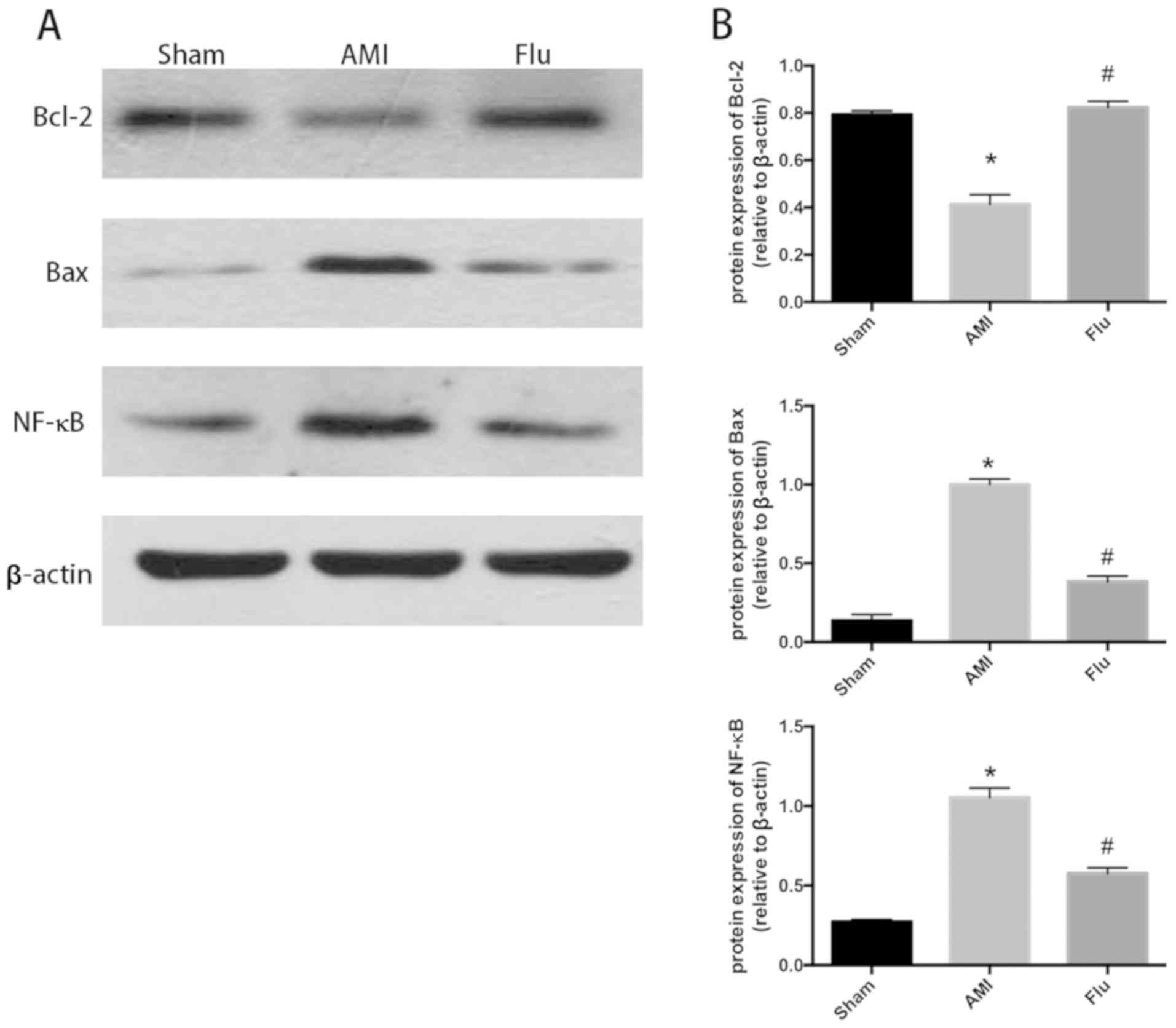 | Figure 2.(A) Bcl-2, Bax and NF-κB protein
content in infarction zones in Sham group, AMI group and
Fluvastatin (Flu) group detected through western blotting. (B)
Quantitatively-analyzed results of Bcl-2, Bax and NF-κB protein
content in infarction zones in Sham group, AMI group and Flu group,
expressed as mean ± standard deviation, *P<0.05 compared with
Sham group, #P<0.05 compared with AMI group. Sham,
sham operation; AMI, acute myocardial infarction; Bcl-2, B-cell
lymphoma-2; Bax, Bcl-2 associated X protein; NF-κB, nuclear
factor-κB. |
Flu protects myocardial cells in
infarction region in AMI mice through RhoA/ROCK pathway
The protein expression levels of ROCK1 and ROCK2
were evidently increased in AMI group compared with those in Sham
group, while they were effectively inhibited by Flu (Fig. 3A and B). After treatment with the
RhoA/ROCK agonist Ang II, the protein expression levels of ROCK1
and ROCK2 increased, the protein expression levels of Bax and NF-κB
increased and the protein expression of Bcl-2 was decreased showing
the mitigation effect of Flu on myocardial apoptosis in the
infarction region in AMI mice was weakened. The results indicated
that the Rho kinase may be the target of Flu (Figs. 3A and B, 4A and B).
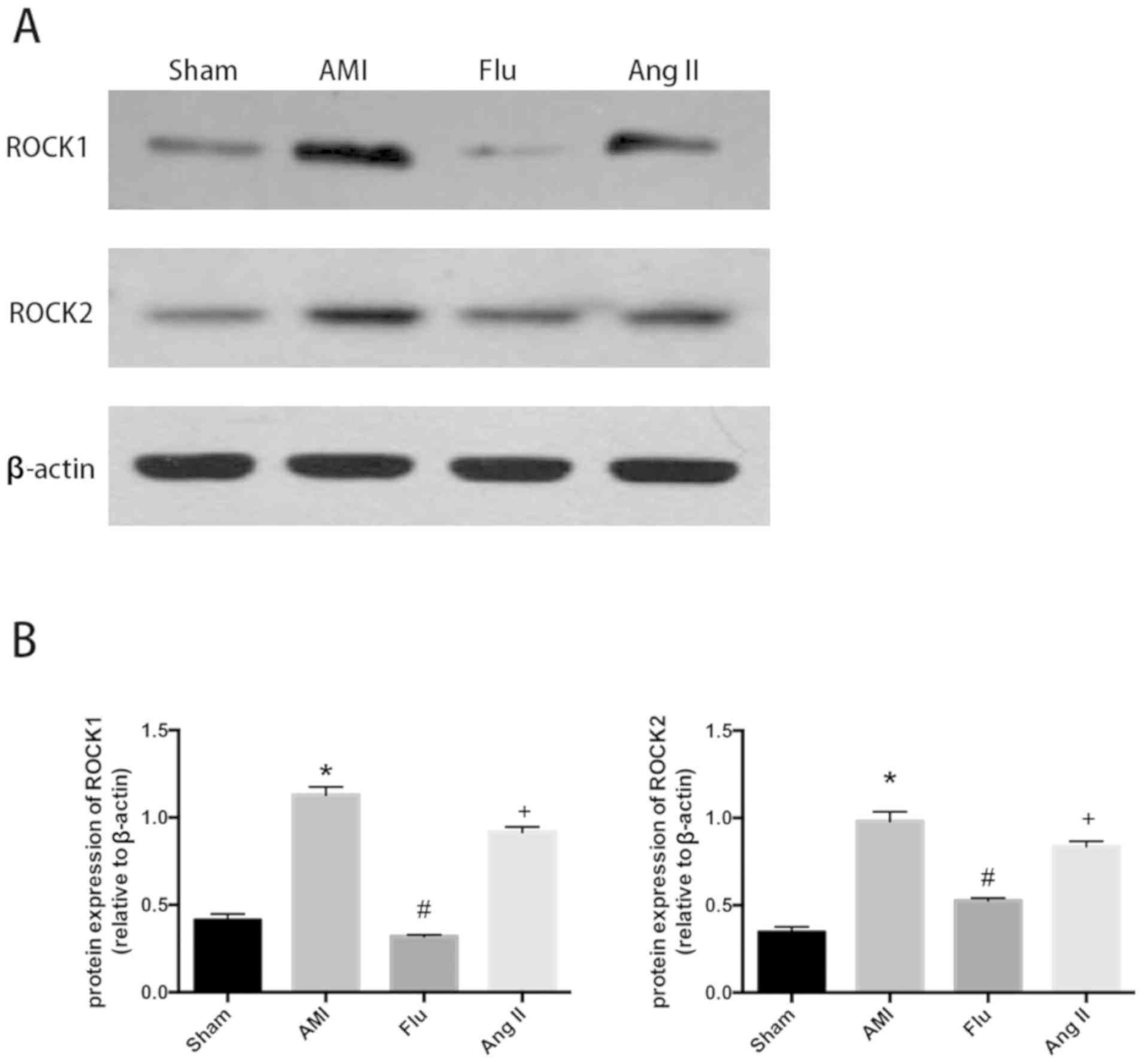 | Figure 3.(A) ROCK1 and ROCK2 protein content in
infarction zones in Sham group, AMI group, Fluvastatin (Flu) group
and Flu+Ang II (Ang II) group detected through western blotting.
(B) Quantitatively analyzed results of ROCK1 and ROCK2 protein
content in infarction zones in Sham group, AMI group, Flu group and
Flu+Ang II (Ang II) group expressed as mean ± standard deviation,
*P<0.05 compared with Sham group, #P<0.05 compared
with AMI group and +P<0.05 compared with Flu group.
Sham, sham operation; AMI, acute myocardial infarction; ROCK1,
(Rho)-associated coiled-coil protein kinase 1; ROCK2,
(Rho)-associated coiled-coil protein kinase 2; Ang II, Angiotensin
II. |
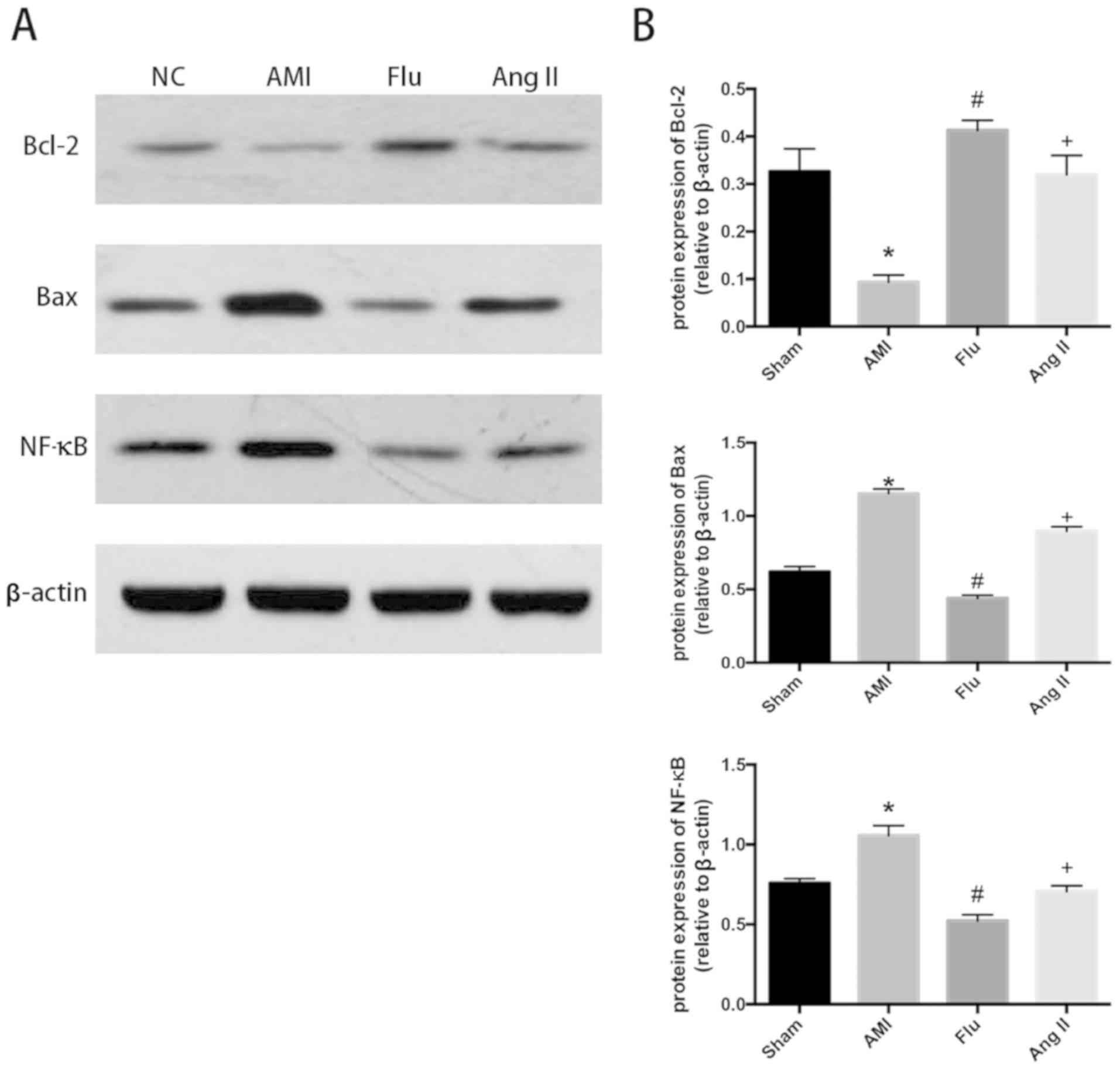 | Figure 4.(A) Bcl-2, Bax and NF-κB protein
content in infarction zones in Sham group, AMI group, Fluvastatin
(Flu) group and Flu+Ang II (Ang II) group detected through western
blotting. (B) Quantitatively-analyzed results of Bcl-2, Bax and
NF-κB protein content in infarction zones in Sham group, AMI group,
Flu group and Flu+Ang II (Ang II) group, expressed as mean ±
standard deviation, *P<0.05 compared with Sham group,
#P<0.05 compared with AMI group and
+P<0.05 compared with Flu group. Sham, sham
operation; AMI, acute myocardial infarction; Bcl-2, B-cell
lymphoma-2; Bax, Bcl-2 associated X protein; NF-κB, nuclear
factor-κB; Ang II, Angiotensin II. |
Discussion
AMI is one of the common diseases seriously
threatening human health. Myocardial dysfunction caused by
myocardial apoptosis is the main cause of heart failure. Studies
have shown that when MI occurs, myocardial apoptosis is increased,
the Bax protein is upregulated and the Bcl-2 protein is
downregulated in the AMI animal model and humans (15–17).
Therefore, inhibiting apoptosis may be an effective method to
prevent MI (18–20). In the present study, the mouse model
of AMI was established to confirm that exogenous injection of Flu
can effectively alleviate ischemia-induced MI, inhibit pathological
changes in myocardial tissues, reduce myocardial apoptosis and
increase Bcl-2 protein expression, which demonstrates that Flu can
relieve ischemic-induced AMI through inhibiting myocardial
apoptosis.
Myocardial apoptosis mediated by oxidative stress
and inflammatory response, and decrease of viable myocardial cells
are the major causes of heart failure in AMI patients.
Mitochondrial dysfunction of ischemic myocardial cells leads to
massive synthesis and secretion of reactive oxygen species (ROS),
induces oxidative stress response in cells, alters mitochondrial
membrane permeability and causes myocardial apoptosis (21–23).
Inhibiting the expression of inflammatory factors in myocardial
tissues can effectively alleviate AMI-induced cardiac dysfunction.
In the present study, the expression levels of CRP, tumor necrosis
factor-α (TNF-α) and interleukin-6 (IL-6) in serum and ischemic
myocardial tissues after Flu treatment were detected, and it was
proven that Flu exerts a protective effect on ischemic myocardial
tissues through reducing oxidative stress and inflammatory
responses of myocardial tissues.
The Rho kinase belongs to the serine/threonine
kinase of the small G protein family (protein kinase A, G and C),
and it is involved in various physiological functions of cells
through the downstream effector ROCK, which is considered to be
related to myocardial apoptosis and MI (24–26).
Studies have shown that after myocardial ischemia-reperfusion, the
expression levels of RhoA and ROCK are upregulated, the
phosphorylation of I-κB is enhanced, and NF-κB is activated and
enters the nucleus to induce expression of inflammatory factors,
thereby promoting inflammatory cell infiltration and adhesion.
Inhibiting ROCK activity or knocking out the ROCK gene can
effectively reduce the leukocyte recruitment and adhesion, and
inhibit the level of various inflammatory factors (27,28).
ROCK is also involved in regulating the oxidative stress response,
and inhibiting ROCK activation can lower the expression and
activity of NAD(P)H in endothelial cells, and reduce the production
of ROS (29). Rho kinase activation
can upregulate the expression levels of TNF-α and IL-6, and its
inhibitors have a protective effect on the myocardium (30). In addition, studies have manifested
that apoptosis can be induced through a variety of pathways after
Rho kinase is activated, such as exogenous myocardial apoptosis
through regulating the TNF-α expression (16), and myocardial apoptosis by regulating
the mitochondrial pathway (31) and
through regulating caspase-3 activity (32). In this study, the expression levels
of ROCK1 and ROCK2 as well as the key transcription factor for
inflammatory response, NF-κB, in myocardial tissues obviously
declined after Flu treatment, suggesting that Flu exerts a
protective effect against ischemia-induced AMI through regulating
RhoA/ROCK.
In conclusion, the AMI model was established through
acute myocardial ischemia in this study, to explore the effect of
Flu on myocardial apoptosis and its protective mechanism in
myocardial tissues. The results revealed that after Flu treatment,
the MI severity and myocardial tissue lesions were effectively
ameliorated in mice, the protein expression levels of Bax and NF-κB
significantly declined, and the apoptotic myocardial cells were
significantly reduced, whose mechanism may be that the activation
of RhoA/ROCK signaling pathway is inhibited, thus suppressing the
NF-κB-mediated inflammatory response, ROS-mediated oxidative stress
and myocardial apoptosis. This study provides preclinical
experimental support for the application of Flu and other statins
in the prevention and treatment of AMI.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
ZY, JK and KC designed the study and performed the
experiments, ZY and YW collected the data, JK and YW analyzed the
data, ZY, JK and KC prepared the manuscript. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
The study was approved by the Animal Ethics
Committee of Fujian Medical University Animal Center (Quanzhou,
China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Shiny KS, Kumar SH, Farvin KH, Anandan R
and Devadasan K: Protective effect of taurine on myocardial
antioxidant status in isoprenaline-induced myocardial infarction in
rats. J Pharm Pharmacol. 57:1313–1317. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Carson DA and Ribeiro JM: Apoptosis and
disease. Lancet. 341:1251–1254. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wyllie AH, Kerr JF and Currie AR: Cell
death: The significance of apoptosis. Int Rev Cytol. 68:251–306.
1980. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yi J and An Y: Circulating miR-379 as a
potential novel biomarker for diagnosis of acute myocardial
infarction. Eur Rev Med Pharmacol Sci. 22:540–546. 2018.PubMed/NCBI
|
|
5
|
Baldi A, Abbate A, Bussani R, Patti G,
Melfi R, Angelini A, Dobrina A, Rossiello R, Silvestri F, Baldi F,
et al: Apoptosis and post-infarction left ventricular remodeling. J
Mol Cell Cardiol. 34:165–174. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Slee EA, Harte MT, Kluck RM, Wolf BB,
Casiano CA, Newmeyer DD, Wang HG, Reed JC, Nicholson DW, Alnemri
ES, et al: Ordering the cytochrome c-initiated caspase cascade:
Hierarchical activation of caspases-2, −3, −6, −7, −8, and −10 in a
caspase-9-dependent manner. J Cell Biol. 144:281–292. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sam F, Sawyer DB, Chang DL, Eberli FR,
Ngoy S, Jain M, Amin J, Apstein CS and Colucci WS: Progressive left
ventricular remodeling and apoptosis late after myocardial
infarction in mouse heart. Am J Physiol Heart Circ Physiol.
279:H422–H428. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wu J, Guo Z, Wang LL and Zhang RL:
Degeneration of sensory afferent nerves enhances pulmonary
inflammatory alterations in acute myocardial infarction in rats.
Cardiovasc Pathol. 21:149–157. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Youle RJ and Strasser A: The BCL-2 protein
family: Opposing activities that mediate cell death. Nat Rev Mol
Cell Biol. 9:47–59. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Burridge K and Wennerberg K: Rho and Rac
take center stage. Cell. 116:167–179. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Etienne-Manneville S and Hall A: Rho
GTPases in cell biology. Nature. 420:629–635. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Shimokawa H: Rho-kinase as a novel
therapeutic target in treatment of cardiovascular diseases. J
Cardiovasc Pharmacol. 39:319–327. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yamakawa T, Tanaka S, Kamei J, Kadonosono
K and Okuda K: Pitavastatin inhibits vascular smooth muscle cell
proliferation by inactivating extracellular signal-regulated
kinases 1/2. J Atheroscler Thromb. 10:37–42. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wiesbauer F, Kaun C, Zorn G, Maurer G,
Huber K and Wojta J: HMG CoA reductase inhibitors affect the
fibrinolytic system of human vascular cells in vitro: A comparative
study using different statins. Br J Pharmacol. 135:284–292. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kitano K, Usui S, Ootsuji H, Takashima S,
Kobayashi D, Murai H, Furusho H, Nomura A, Kaneko S and Takamura M:
Rho-kinase activation in leukocytes plays a pivotal role in
myocardial ischemia/reperfusion injury. PLoS One. 9:e922422014.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chang CC, Wang SS, Hsieh HG, Lee WS,
Chuang CL, Lin HC, Lee FY, Lee SD and Huang HC: Rosuvastatin
improves hepatopulmonary syndrome through inhibition of
inflammatory angiogenesis of lung. Clin Sci (Lond). 129:449–460.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang Y, Zhang H, Chai F, Liu X and Berk M:
The effects of escitalopram on myocardial apoptosis and the
expression of Bax and Bcl-2 during myocardial ischemia/reperfusion
in a model of rats with depression. BMC Psychiatry. 14:3492014.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chen TL, Zhu GL, He XL, Wang JA, Wang Y
and Qi GA: Short-term pretreatment with atorvastatin attenuates
left ventricular dysfunction, reduces infarct size and apoptosis in
acute myocardial infarction rats. Int J Clin Exp Med. 7:4799–4808.
2014.PubMed/NCBI
|
|
19
|
Malick M, Gilbert K, Barry M, Godbout R
and Rousseau G: Desvenlafaxine reduces apoptosis in amygdala after
myocardial infarction. Brain Res Bull. 109:158–163. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liu Q: Lentivirus mediated interference of
Caspase-3 expression ameliorates the heart function on rats with
acute myocardial infarction. Eur Rev Med Pharmacol Sci.
18:1852–1858. 2014.PubMed/NCBI
|
|
21
|
Kumphune S, Surinkaew S, Chattipakorn SC
and Chattipakorn N: Inhibition of p38 MAPK activation protects
cardiac mitochondria from ischemia/reperfusion injury. Pharm Biol.
53:1831–1841. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Motloch LJ, Hu J and Akar FG: The
mitochondrial translocator protein and arrhythmogenesis in ischemic
heart disease. Oxid Med Cell Longev. 2015:2341042015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang Q, Wang H, Yang YJ, Dong QT, Wang
TJ, Qian HY, Li N, Wang XM and Jin C: Atorvastatin treatment
improves the effects of mesenchymal stem cell transplantation on
acute myocardial infarction: The role of the RhoA/ROCK/ERK pathway.
Int J Cardiol. 176:670–679. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Loirand G, Guérin P and Pacaud P: Rho
kinases in cardiovascular physiology and pathophysiology. Circ Res.
98:322–334. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Shi J and Wei L: Rho kinases in
cardiovascular physiology and pathophysiology: The effect of
fasudil. J Cardiovasc Pharmacol. 62:341–354. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hung CN, Huang HP, Wang CJ, Liu KL and Lii
CK: Sulforaphane inhibits TNF-α-induced adhesion molecule
expression through the Rho A/ROCK/NF-κB signaling pathway. J Med
Food. 17:1095–1102. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bao W, Hu E, Tao L, Boyce R, Mirabile R,
Thudium DT, Ma XL, Willette RN and Yue TL: Inhibition of Rho-kinase
protects the heart against ischemia/reperfusion injury. Cardiovasc
Res. 61:548–558. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Shimokawa H and Takeshita A: Rho-kinase is
an important therapeutic target in cardiovascular medicine.
Arterioscler Thromb Vasc Biol. 25:1767–1775. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Higashi M, Shimokawa H, Hattori T, Hiroki
J, Mukai Y, Morikawa K, Ichiki T, Takahashi S and Takeshita A:
Long-term inhibition of Rho-kinase suppresses angiotensin
II-induced cardiovascular hypertrophy in rats in vivo: Effect on
endothelial NAD(P)H oxidase system. Circ Res. 93:767–775. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Petrache I, Crow MT, Neuss M and Garcia
JG: Central involvement of Rho family GTPases in TNF-alpha-mediated
bovine pulmonary endothelial cell apoptosis. Biochem Biophys Res
Commun. 306:244–249. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Del Re DP, Miyamoto S and Brown JH:
RhoA/Rho kinase up-regulate Bax to activate a mitochondrial death
pathway and induce cardiomyocyte apoptosis. J Biol Chem.
282:8069–8078. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chang J, Xie M, Shah VR, Schneider MD,
Entman ML, Wei L and Schwartz RJ: Activation of Rho-associated
coiled-coil protein kinase 1 (ROCK-1) by caspase-3 cleavage plays
an essential role in cardiac myocyte apoptosis. Proc Natl Acad Sci
USA. 103:14495–14500. 2006. View Article : Google Scholar : PubMed/NCBI
|